

Abstract
In this unit, we describe the detailed protocol of a three-plasmid transfection method for rAAV production, its advantages, limitations, and troubleshooting techniques. We further discuss the rAAV purification process using CsCl gradients and the subsequent quality control steps by SDS-PAGE and rtPCR to ensure vector purity and efficient packaging, as well as to ensure viral titer. Finally, we elaborate on a PCR-based strategy to discover novel AAV capsid sequences from primate tissue, which can be used to develop newer generation rAAVs with a greater diversity of tissue tropism for clinical gene therapy.
INTRODUCTION
Recombinant adeno-associated viral vectors (rAAV) are exceptionally versatile and powerful gene delivery vehicles (tools) for use in translational science and basic biology. Strategies have been developed to use rAAVs for gene augmentation, miRNA and shRNA mediated-knockdown, endogenous miRNA antagonism and genome editing both by the expression of zinc finger nucleases and the use of rAAV to induce homologous recombination (Asuri, Bartel et al.; Ellis, Hirsch et al.; Gorbatyuk, Li et al.; Handel, Gellhaus et al.; Li, Haurigot et al.; Mueller, Tang et al.); Xie, Ameres et al. (); (Egan, Flotte et al. 1992; Cruz, Mueller et al. 2007; Cideciyan, Aleman et al. 2008; Sun, Yan et al. 2008; Brantly, Chulay et al. 2009). For the most part AAV vectors lead to robust, long-term in vivo transgene expression in both dividing and non-dividing cells in a myriad cell types and animal models. To date several protocols for rAAV production have been described for both research and clinical grade manufacturing. Methods that easily permit large scale production include; recombinant baculovirus based systems (Urabe, Ding et al. 2002), a recombinant herpes simplex virus based method (Conway, Zolotukhin et al. 1997; Clement, Knop et al. 2009) and a protocol based on adenovirus-AAV hybrid using a stable cell line expressing rep/cap (Gao, Qu et al. 1998). However despite some difficulties with its scalability, the classic helper-free triple transfection method using HEK-293 cells remains one of the more versatile and easily adaptable protocols for making rAAV in a research laboratory setting. In this chapter we will describe in detail the process for generating, purifying and quantifying rAAV vectors. In addition we provide a PCR-based method for discovery of novel AAV cap sequences to generate novel rAAV vectors.
The basic requirements for all rAAV vector production are essentially the same; they all rely on a set of factors acting in trans with respect to the viral genome. These include both the AAV nonstructural components (ie. Rep proteins); AAV structural cap gene products (ie. VP1, VP2 and VP3) and adenonvirus trans-acting helper factors (ie. E1a, E2a, E4 and VA RNA). The viral genome and the recombinant vector genome are both composed of linear single stranded DNA with interrupted terminal palindromes at each end. These are often referred to as the inverted terminal repeated (ITRs) and these sequences are the only cis elements required for packaging of rAAV. The helper-free triple transfection system simply provides all of these trans and cis elements in three plasmids that will be transfected into HEK-293 cells where rAAV DNA rescue from the plasmid occurs and is followed by its replication and packaging. Specifically, vector production is achieved by using the following three plasmids: (i) the vector plasmid (pCIS) carrying the an expression cassette with the transgene of interest flanked by the ITRs; (ii) the packaging plasmid (pTRAN) expressing the AAV Rep and Cap genes for vector genome rescue, replication and packaging; and (iii) the adenovirus helper plasmid expressing the necessary trans-acting factors mentioned above with the exception of E1 which is inherently expressed in HEK-293 cells. An overview of this methodology is summarized in Figure 1.
Figure 1.

Schematic illustration of the process for production of recombinant adeno-associated virus by 293 cell–triple-transfection method
The triple transfection results in the harvest of crude cell lysates with rAAV vector particles that can be subsequently purified by a variety of methods such as gradient sedimentation, column chromatography, or a combination of both. We describe a gradient sedimentation based on cesium chloride that has the advantage of being robust and applicable to all rAAV serotypes, and can further separate virion particles that are empty from those containing the packaged recombinant genome. The recombinant vectors can be further characterized for titer and purity by real-time PCR and SDS-PAGE analysis, respectively, as described below. These series of protocols starting from the triple plasmid transfection to the final high-titer, high-purity vector stock can be used to produce novel rAAV vectors with newly identified capsids (pseudotypes). The naturally abundant diversity of AAV cap genes has considerably expanded the versatility and utility of AAV vectors. Importantly, the ITR and rep genes from AAV2 can be kept constant while cap genes from different serotypes or isolates are used to derive “pseudotyped” vectors that only differ by the nature of their capsid (Rabinowitz, Rolling et al. 2002). A pseudotype can be easily produced using an AAV2-ITR containing transgene vector (pCis) and replacing the cap gene in the pTRANS plasmid with a novel cap sequence identified with the PCR-based AAV cap discovery protocol at the end of the unit.
Basic Protocol 1: Production of AAV vector by transient transfection
The most commonly used method for production of AAV vectors in research laboratories consists of a two or three plasmid transfection of adherent HEK-293 cells. The triple transfection method described below uses the AAV cis- and trans-plasmids, along with a helper plasmid providing the adenoviral gene products necessary for replication and packaging. This protocol describes the processes for splitting cells, transfecting the three plasmids, harvesting the crude cell lysate, and preparing the lysate for viral vector purification by cesium chloride gradients as described in the subsequent protocol.
MATERIALS
See Appendix A for reagent recipes.
Reagents
Dulbecco’s Modified Eagle’s Medium (DMEM)
Dulbecco’s Phosphate Buffered Saline Solution (D-PBS)
Fetal bovine serum (FBS)
Sterile tissue culture plates (150 mm)
Sterile disposable pipettes
Penicillin-streptomycin (P/S)
TRANS plasmids of different serotypes (Gao, Alvira et al. 2002; Gao, Alvira et al. 2003; Gao, Vandenberghe et al. 2004) (available at the Penn Vector Core, vector@mailmed.upenn.edu)
ΔF6 Adeno helper plasmid (available at the Penn Vector Core, vector@mailmed.upenn.edu)
CIS plasmid with the gene of interest
2.5 M CaCl2 solution
2x HBS solution
Sterile 125 mL bottle
Sterile 50 mL conical tubes
Sterile aspirating pipettes
Sterile 500 mL centrifuge bottles
Equipment
37°C water bath
Refrigerated tabletop centrifuge
Sterile cell scarpers
Humidified cell culture incubator set to 37°C and 5% CO2 atmosphere
Preparation of 293 cells
-
1
Prepare regular cell growth medium (DMEM supplemented with 10% FBS and 1% P/S). A volume of 20 mL of medium is needed for each 150 mm plate. For large scale AAV vector production, prepare 800 mL of growth medium for 40 plates of 293 cells.
-
2
Seed the cells one day prior to transfection at 2×107 293 cells per 150 mm plate. The cells should be approximately 70–80% confluent at the time of transfection.
Note: 293 are suitable for AAV vector production until passage 100–130 (293 cells obtained from ATCC are usually around passage 40). -
3
On the day of transfection, change medium 2 hours before transfection with fresh growth medium. Carefully add 20 mL of medium per plate without disturbing the cell monolayer.
Note: Maintaining an intact cell monolayer is critical to the efficiency of transfection. If the outcome of this step is not satisfactory, please see Troubleshooting.
Preparation of transfection cocktail
-
4
Prepare the following DNA mix in sterile 125 mL bottle:
Sterile MiliQ water to make a final volume of 54 mL
5.2 mL 2.5M CaCl2
1040 ug<microgram> ΔF6 Adeno helper plasmid
520 ug<microgram> TRANS plasmid
520 μg<microgram> CIS plasmid
These numbers are for transfection of 40 plates -
5
Prepare four 50 mL conical tubes with 12.5 mL 2X HBS each. To make the transfection cocktail, add drop-wise 12.5 mL of DNA mixture to each 2xHBS tube while vortexing it. Incubate the transfection cocktail at room temperature for 5 min.
Note: After 5 min incubation, the transfection cocktail should show a uniform whitish cloudiness. If it is not cloudy at all or has large precipitates, please see Troubleshooting.
Transfection of 293 cells
-
6
Add drop-wise 2.5 mL of the transfection cocktail to each plate. Gently rock the plate to evenly distribute the cocktail over the entire cell monolayer. Place the transfected plates back in the 37°C, 5% CO2 incubator. Sixteen hours after transfection, aspirate the transfection medium from the plates and replace with fresh growth medium.
When changing the medium, take no more than 12 plates out of the incubator at a time and add the medium slowly from the side of the plate without disturbing the cell monolayer.
Harvesting transfected cells
-
7
At approximately 72 h after transfection, scrape the cells into the medium using a sterile cell scraper. Transfer the cell suspension from all plates to two 500 mL sterile centrifuge bottles using a 25 mL pipette.
-
8
Centrifuge in a refrigerated tabletop centrifuge at 4,000 rpm for 20 min at 4°C. Decant the supernatant.
-
9
Resuspend the cell pellet in each of the 500 ml centrifuge bottles in ~14 ml of 50 mM Tris (pH 7.4), 1 mM MgCl2 buffer. Transfer the cell suspensions to a sterile 50 mL conical centrifuge tube. Use 7 ml of the same buffer to rinse the 500 ml bottles, combine the rinsing buffer with the rest of cell suspension in the 50 mL conical centrifuge tube and then store at −80°C until time of processing.
Basic Protocol 2: Purification of AAV vectors by cesium chloride gradient sedimentation
Cesium chloride (CsCl) is widely used as a medium for separation of viral vectors by gradient centrifugation. This is known as isopycnic gradient ultra-centrifugation and it is used to separate and purify viral vectors based on their isopycnic point (i.e. buoyant density). By spinning the virus preparations in an ultra-centrifuge at high G-forces in solutions containing various CsCl concentrations one can establish a density gradient which allows separation of empty, partially packaged, and fully packaged viral vector particles from cellular debris, proteins, and nucleic acids in the crude viral lysate. This protocol describes the purification, desalting, and final formulation of AAV vectors from crude viral lysate using CsCl gradient sedimentation.
MATERIALS
See Appendix A for reagent recipes
Reagents
50 mM Tris (pH 7.4), 1 mM MgCl2 buffer
Benzonase® (EMD Chemicals, Gibbstown, NJ, USA)
10% deoxycholic acid (DOC)
Light CsCl for AAV purification
Heavy CsCl for AAV purification
Ultra-pure cesium chloride
Dulbeco’s Phosphate Buffered Saline (DPBS)
Sterile 100% glycerol
70% Ethanol
SW32 centrifuge tubes
18G x 1½″ needle
16G needle
70 Ti quick seal tubes
10 mL syringe
1L glass beaker, autoclaved
Glass stir bar, autoclaved
3 mL syringe
Equipment
Refrigerated tabletop centrifuge
Beckman ultracentrifuge
SW 32 rotor
70.1 Ti rotor
Refractometer (Milton Roy, USA)
Sonicator
Processing of transfected cell suspension
-
1
Thaw the 50 mL conical centrifuge tube with 35 mL of frozen cell suspension (from protocol 1) in a 37°C water bath for 10 min. Sonicate cell lysate for 1 min at 25% output in ice/water bath. Repeat two times with a 2-min interval each time. Always keep the tube on ice.
-
2
Add 150 μL<microliter> of Benzonase® to the sonicated cell lysate, and invert gently to mix. Incubate the samples at 37°C for 20 minutes; invert the tube every 5 min. Then add 1.25 mL of 10% deoxycholic acid (DOC), mix by gently inverting the tube and extend the incubation at 37°C for 10 more min. Immediately place the tube on ice for 10–20 min. Clarify the lysate by centrifugation at 4,000 rpm for 30 min at 4°C in a refrigerated tabletop centrifuge. Transfer the supernatant to a sterile 50 mL conical tube.
Benzonase® is a genetically engineered endonuclease from Serratia marcescens. It has no proteolytic activity but completely degrades all forms of DNA and RNA (single stranded, double stranded, linear and circular) to 5′-monophosphate terminated oligonucleotides 2 to 5 bases in length. It is ideal for removal of nucleic acids (of both cellular and plasmid origins) from crude viral lysates for viscosity reduction and improvement of virus purification.
Sodium deoxycholate, the sodium salt of deoxycholic acid, is used as a biological detergent here to help lyse the 293 cells and free rAAV virions from cellular and membrane components with which the virions are usually associated.
First cesium chloride gradient centrifugation
-
3
Add 0.454 gram of ultra-pure CsCl for every mL of clarified lysate. Mix well by gentle inversion. The final volume of the viral lysate should be ~40 mL.
-
4
Add 9 mL of Light-CsCl solution to two SW28/32 centrifuge tubes. Fill a 10 mL pipette with 9 mL of Heavy-CsCl solution, and carefully lower the pipette to the bottom of the tube through the L-CsCl layer. Dispense H-CsCl solution slowly to create a sharp interface without intermixing of the two solutions. Slowly add 18–20 mL of clarified lysate on top of the 2-layered CsCl gradient using a 10 mL pipette, avoiding any mixing of lysate and L-CsCl layer.
-
5
Very carefully, so as not to disturb the gradient, place the tubes in the rotor buckets and balance them. Place the buckets on the SW28/32 rotor and centrifuge at 25,000 rpm for 18-20 h in an ultracentrifuge at 15°C. Be sure to align the buckets numerically in the appropriate position on the rotor.
-
6
For each SW28/32 centrifuge tube, set up 16 sterile 1.5 mL microcentrifuge tubes on a rack and two 15 mL conical tubes in order to collect gradient fractions from the first spin.
-
7
Remove the centrifuge tube from the bucket without disturbing the gradient. Secure the tube to a holder. Carefully insert an 18G x 1½″ needle at the bottom of the tube without disturbing the gradient (Fig. 2a), collect the first 11 mL from the tube into a 15 mL conical tube, and discard it.
-
8
Collect the next 16 mL in 1 mL fractions into the 1.5 mL microcentrifuge tubes. Label each tube with the fraction number. Take 5μL<microliter> of each fraction and read the refraction index using a refractometer. The fractions containing AAV virions have a refractive index from 1.3650 to 1.3760. Discard the fractions that are outside of this range.
Figure 2.
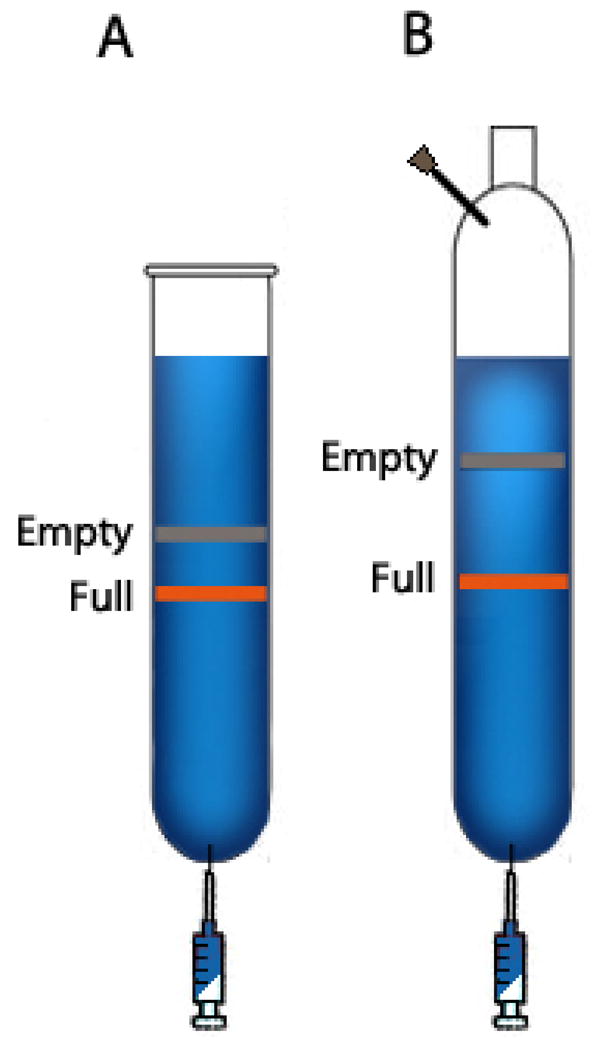
Representative CsCl gradients after the first (A), second (B), CsCl gradient centrifugation.
Second cesium chloride gradient centrifugation
-
9
Combine all fractions with the appropriate refractive index, and transfer into two 70.1 Ti Quick-seal tubes using a 10 mL syringe with an 18G x 1½″ needle. Fill the remaining space in each tube with 1.41 g/mL CsCl solution and seal the tube using a cordless tube topper (Beckman Coulter, Fullerton, CA, USA). Centrifuge at 60,000 rpm in a Beckman 70.1 Ti rotor for 20-24 h at 15°C.
-
10
To collect fractions from the second centrifugation, set up 12 sterile 1.5 mL microcentrifuge tubes on a tube rack for each 70.1 Ti tube. Carefully remove the centrifuge tube from the rotor without disturbing the gradient. Secure the tube on a tube holder. Insert a 16G needle at the top of the tube first, and then an 18G x 1½″ needle at the bottom of the tube (Fig. 2B).
-
11
Collect 13 mL in 1 mL fractions from each 70.1 Ti tube into 1.5 mL microcentrifuge tubes. Label each tube with fraction number. Take 5μL<microliter> of each fraction and read the refraction index using a refractometer. The fractions containing AAV virions have a refractive index from 1.3650 to 1.3760. Discard the fractions that are outside of this range.
Third cesium chloride gradient centrifugation
-
12
Combine the AAV-containing fractions collected from two 70.1 Ti tubes and transfer into one new 70.1 Ti Quick-seal tube using a 10 mL syringe with an 18G needle. Fill the remaining space in the tube with 1.41 g/mL CsCl solution, seal the tube and centrifuge as in step 9.
-
13
Repeat step 10, and collect 13 mL in 0.5 mL fractions. Read the refractive indexes as in Step 11. After this final centrifugation, the fractions with refractive indexes in the range of 1.3670–1.3740 contain AAV virions.
Desalting and formulation
-
14
Use a 5 mL syringe with a 18G x 1½″ needle to transfer the vector into a 0.5–3 mL or 3–12 mL Slide-a-Lyzer® cassette (10,000 MWCO, Pierce, USA), depending on the volume of collected vector, and dialyze in a 4 L plastic beaker with a stirring bar and 3 L of cold PBS, pH 7.6 on a magnetic stirring plate at 4°C with slow continuous stirring. Replace with 3 L of fresh cold PBS every 3 h twice, and then one more time for overnight dialysis. Use cold 5% sorbitol/PBS, pH 7.6 for the final overnight dialysis to formulate the vector.
-
15
Use a 5 mL syringe with a 18G × 1½ ″ needle to carefully transfer the desalted virus from the dialysis cassette to a sterile 15 conical centrifuge tube. Take 20 μL<microliter> aliquots from the conical tube for genome copy titration (Protocol 3), and silver staining of SDS-PAGE (Protocol 4) and store the remaining AAV vector at 4°C temporarily until the completion of vector characterization.
Depending on the viral capsid and virus particle concentration in vector preparations, sometimes, whitish precipitates may appears in purified rAAVs, which is usually indicative of virus aggregation, and will negatively impact gene-transfer efficiency, particularly in the case of systemic delivery. Please see Troubleshooting for strategies to prevent aggregation of AAV vectors.
Vector Storage, Freezing, and Thawing
-
16
For long-term storage, AAV vectors should aliquoted in cryovials and stored at −80°C. If it is found that the vector genome titer of a purified and desalted vector preparation is higher than 1×1013 genome copies/ml, it is recommended the vector be diluted to 1×1013 GC/ml before freezing for storage. Once thawed on ice, it is best to store AAV vectors at 4°C for at least several weeks and avoid multiple freeze-thaw cycles.
Basic Protocol 3: Vector genome copy titration by real-time PCR
The objective of this assay is to determine the concentration of DNase-resistant vector genomes (i.e. packaged in the capsid) in purified AAV vector preparations. The assay begins with treatment of the vector stock with DNase I to eliminate non-encapsidated AAV genome DNA, and/or contaminating plasmid DNA. This is followed by a heat treatment to heat-inactivate DNase I and to disrupt viral capsid to release the packaged vector genomes for quantification by real-time PCR using a set of standards (linearized pCIS plasmid used for vector production) of known copy numbers. In order to accomplish high-throughput titration, the primer and probe sets used in real-time PCR are usually designed to target common elements present in most AAV vector genomes, such as promoters and poly A signals. This strategy significantly cuts down on PCR reactions, controls, and turn-around time. Several important controls should be included in the assay. The first two controls should have a known copy number of the vector genome plasmid with and without DNase I to gauge the effectiveness of DNase treatment. To control for potential cross-contamination between samples during the preparation process, a blank control containing nuclease-free water only should go through the sample treatment and titration. A validation vector sample with a known titer should be included in every assay to monitor inter-assay variability. Finally, for the PCR run, a no-template control (NTC) is included to indicate cross-contamination during PCR set-up.
MATERIALS
Reagents
Nuclease-free water
10x GeneAmp® PCR Buffer (Applied Biosystems, CA, USA)
Sheared salmon sperm (SSS) DNA (Applied Biosystems, CA, USA)
Vortex mixer
Forward primer working stock solution (9 μM<micromolar>)
Reverse primer working stock solution (9 μM<micromolar>)
6FAM fluorescent probe working stock solution (2 μM<micromolar>)
Vector genome standard set
TaqMan® Universal PCR Mix, No UNG (Applied Biosystems, CA, USA)
Equipment
Real-time PCR station from any manufacturer
PCR-dedicated hood
Sample Preparation
-
1
Set up 0.65 mL microcentrifuge tubes on a tube rack, and label the tubes as 10-fold dilutions for each test article (vector preparations to be assayed), validation vector sample, (+) and (−) DNase treatment controls, and sample preparation blank control.
-
2
Prepare the DNase digestion reaction master mix in a 1.7 mL microcentrifuge tube as follows:
Reagent Vol. per sample Nuclease-free water 38 μL<microliter> 10x DNase buffer 5 μL<microliter> DNase I, RNase-free 2 μL<microliter> (20 units) Calculate the total volume of each reagent to add to the reaction master mix by multiplying the volume per sample (above) by the total number of samples (vector dilutions and controls). Before adding DNase I to the reaction mixture, add 2 μL<microliter> of nuclease-free water, and 43 μL<microliter> DNase digestion reaction mixture without DNase I to (−) DNase control tube to make up 45 μL<microliter> total volume. Mix well by vortexing. Next, add DNase I to the remaining DNase digestion reaction mix (minus one volume, i.e., 2 μL<microliter>), vortex, and add 45 μL<microliter> to all other tubes.
-
3
In a PCR-dedicated hood, add 5 μL<microliter> of test article and controls to dedicated tubes in the following order: 1) control plasmid (5×107 GCs/ul) to (+) DNase control tube; 2) test article; 3) nuclease-free water to sample preparation blank control; 4) control plasmid (5×107 GCs/ul) to (-) DNase control tube; 5) AAV validation sample. Incubate at 37°C for 30 minutes.
-
4
Meanwhile, set up microcentrifuge tubes in a tube rack to prepare the following 10-fold serial dilutions:
1). Test articles: 100- to 10,000-fold 2). Sample preparation blank control: 100-fold 3). (+) DNase treatment control: 100-fold 4). (−) DNase control: 100- to 1000-fold 5). Validation vector sample: Extent of dilution is dependent on the known concentration (e.g. 1000-fold dilution for the validation vector with a titer in the range of 1012 GC/ml) -
5
Thaw the sample dilution buffer (1x PCR Buffer with 20 ng/μL SSS DNA) on ice. Dispense 45 μL<microliter> of this buffer to the serial dilution tubes in the second microcentrifuge tube rack. Keep these tubes at room temperature, and proceed to the next step below.
Real-time PCR Set-Up
-
6
Thaw the primers and probe in advance of sample preparation. The TaqMan® Universal PCR Mix-No UNG should be kept at 4°C at all times. Thaw the standard set immediately before use (please see Appendix B for preparation of standard set).
-
7
While the DNase digestion is in progress, and after dispensing the sample dilution buffer into the dilution tubes, prepare the real-time PCR master mix as follows:
Reagent Vol. per reaction TaqMan® Universal PCR Mix (2X) 25 μL<microliter> Forward primer (9 μM<micromolar>) 5 μL<microliter> Reverse primer (9 μM<micromolar>) 5 μL<microliter> Fluorescent probe (2 μM<micromolar>) 5 μL<microliter> Nuclease-free water 5 μL<microliter> Sample DNA 5 μL<microliter> Multiply the volumes of each reagent per reaction (with the exception of the sample DNA) by three (for triplicate sets) and by the total number of samples. Include an additional two volumes to compensate for pipetting errors. Prepare the master mix in a 15 mL conical tube. Mix by vortexing and keep it on ice.
Note: To minimize cross-contamination, while dispensing reagents or samples, keep all containers and microcentrifuge tubes closed at all times when not in use. Also, keep pipette tip rack lids closed when not in use. -
8
Dispense 45 μL<microliter> of PCR master mix into triplicate wells for each of NTC, standards (1 – 108 copies), test articles, plasmid DNA without DNase I (two dilutions), plasmid DNA treated with DNase I, sample preparation blank control, and validation vector sample in a 96-well plate. Keep the plate on ice.
-
9
After the DNase digestion is complete, prepare 10-fold serial dilutions of DNase I treated test articles, validation vector sample, and controls using the microcentrifuge tubes prepared in step 4 in a PCR-dedicated hood.
-
10
Dispense 5 μL<microliter> each of nuclease-free water for NTC, standards, test articles, controls, and validation sample, at appropriate dilutions to triplicate wells in 96-well plates, each well containing 45 ul of PCR master mix in the following order:
Standards (1–108 genome copies in 10-fold serial dilutions in ascending order)
DNase I-treated test articles (10,000-fold dilution)
Plasmid control without DNase I (1000-fold dilution)
Plasmid control without DNase I (100-fold dilution)
DNase I-treated plasmid control (100-fold dilution)
Sample preparation blank control (100-fold dilution)
DNase I-treated AAV vector validation control
Nuclease-free water for NTC
-
11
Properly seal the 96-well plate using the ultra-clear cap strips and the Cap-It tool (or film), and place the plate in the thermocycler block. Start the PCR run with the following PCR program: one cycle of 2′ at 50°C, and 10′ at 95°C to release the viral DNA from the capsid, and 40 cycles of 15″ at 95°C, and 1′ at 60°C. After the PCR run, analyze the data following manufacturer’s instructions. Calculate the final vector genome copy titer after considering dilution and conversion factors, and the single-stranded nature of the AAV genome using the following formula:
Basic Protocol 4: Analysis of rAAV purity by silver-stained SDS-PAGE
AAV virions consist of 3 major capsid proteins: VP1, VP2, and VP3 at a ratio of 1:1:8. On a silver-stained SDS-polyacrylamide gel (SDS-PAGE), VP1, VP2, and VP3 should be the only visible bands in a highly purified AAV vector preparation, migrating at approximately 87 kDa, 73 kDa, and 62 kDa, respectively. This protocol describes the process of sample preparation, SDS-gel electrophoresis, and silver staining to determine the purity of an AAV preparation. By including a highly purified rAAV preparation with known virus titer as a reference standard, the assay also serve to estimate virus particle concentration. Note this titer is semi-quantitative and is reported as ‘vector particle’ whereas the real-time PCR titer is reported as DNase resistant ‘vector genome’(Fig 3).
Figure 3.
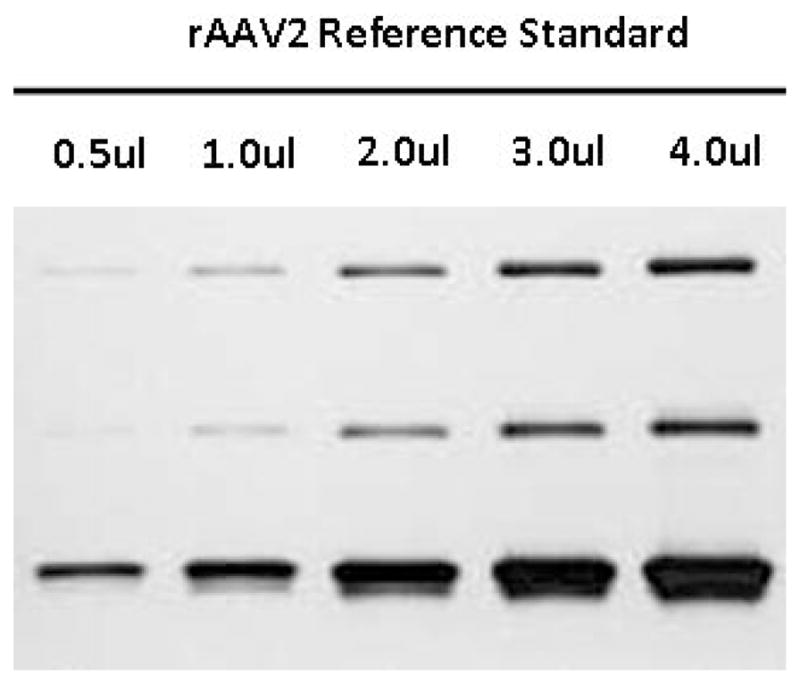
Silver-stained SDS-PAGE of rAAV2 Reference standard. 0.5, 1, 2, 3, and 4 uL each of an AAV2 vector with a concentration of 1x1013 virus particles/mL were loaded onto a SDS-PAGE gel and silver stained for total protein.
MATERIALS
Reagents
Protein molecular weight marker in the range of 20–220 kD
Novex® 15-well 10% Tris-Glycine Gels (Invitrogen, CA USA) or equivalent
Novex® Tris-Glycine SDS Running Buffer (10x, Invitrogen)
Tris-glycine SDS sample buffer (2x, Invitrogen)
NuPage® Reducing Agent (10x, Invitrogen)
SilverXpress Silver Staining Kit (Invitrogen) or equivalent
Equipment
-
Corresponding gel electrophoresis apparatus
Prepare 1x gel running buffer from 10x stock.
-
Prepare reference standard and test articles as follows:
Reference standard with 1×1013 virus particles/mL of rAAV
0.5, 1, 2, 3, and 4 μL<microliter> each in 0.65 mL microcentrifuge tubes
5 μL<microliter> 2x tris-glycine SDS sample buffer
1 μL<microliter> 110x NuPage® Reducing Agent
Add dH2O to a final volume of 10 μL<microliter>
Test articles
2 and 6 μL<microliter> each in 0.5 ml microcentrifuge tubes
7.5 μL<microliter> 2x tris-glycine SDS sample buffer
1.5 μL<microliter> 10x NuPage® Reducing Agent
Add dH2O to a final volume of 15 μL<microliter>
Use of NuPage® Reducing Agent in sample preparation helps disassociate and stabilize VP1, VP2 and VP3 viral capsid proteins. Prepare appropriate amount of protein molecular weight standard marker according to manufacturer’s instructions.
-
Mix the samples well, heat at 95°C for 5 minutes, and pulse-spin.
The reference standard is selected from AAV vector preparations that are first titrated for GC titer/mL, and then subjected to EM analysis and silver-stained SDS-PAGE analyses. The vector preparation that should be used as the reference standard has a GC titer 1×1013 GC/mL and no more than 10% empty particles, and shows only VP1, VP2, and VP3 bands. Set up the gel apparatus. Load 10 μl of reference standards, and 12 μl of test samples into the wells. Run gel at 125 V for 1.5–2 hours.
Perform silver staining of the gel using the silver staining kit according to manufacturer’s instruction.
-
Scan the silver-stained gel on a gel station (e.g. BioRad) and perform semi-quantitative analysis of the test articles (Fig. 3).
The main purpose of this analysis is to assess the purity of vector preparations. Virus particle titer estimation based on a single loading of test article sometimes is unreliable. Please see Troubleshooting for alternative strategy.
Basic Protocol 5: PCR-based method for Novel AAV discovery
AAV2-based recombinant genomes have been packaged in dozens of different capsid types, resulting in a wide array of “pseudotyped vectors” that constitute a rich resource for the development of gene therapy clinical trials. Analysis of novel AAV cap sequences has revealed a rapidly evolving proviral genome both by accumulation of mutations and recombination, primarily in the capsid gene. This protocol describes a PCR-based method to rescue AAV cap genes from tissues. It is based on primers designed to recognize the highly conserved regions in known AAV isolates to generate amplicons across the hyper-variable regions of naturally divergent AAV genomes present in the sample of choice.
Strategic Planning
PCR-based method for Novel AAV discovery
The isolation of novel AAV cap genes from tissues can be accomplished by simple PCR amplification provided the appropriate primers are available. cap sequences of different AAVs are naturally segregated into 12 hypervariable regions which are distinct from each other but interspersed with conserved regions (Gao, Alvira et al. 2003). This kind of molecular structure allows the design of PCR primers specific for conserved regions to amplify across the hypervariable regions for molecular retrieval of novel cap sequences.
As an initial attempt for detecting and isolating novel AAV cap sequences from tissues of interest, one can start by performing a “signature PCR” (Gao, Alvira et al. 2002). This highly sensitive method of retrieving a short fragment of AAV cap sequence is called “signature PCR” because the 255 bp PCR fragment spans a hyper-variable region. Signature PCR is very useful and reliable for high throughput screening and identification of endogenous AAVs present in tissues of interest. Sequencing or restriction analysis of this PCR product with a panel of selected restriction enzymes can inform on AAV cap sequence diversity in the desired tissue, and guide subsequent efforts to isolate the extended proviral sequences (Gao, Alvira et al. 2002).
To isolate full-length cap sequences, two different strategies can be employed. First, one can generate two overlapping PCR amplicons using primers in the conserved regions and then fuse them into a full length cap gene sequence (Gao, Alvira et al. 2002). The second strategy is to directly amplify a 3.1 kb region spanning 3′ of rep (about 800 bp) and the entire cap gene sequence. The primers used for the full length cap gene amplification are located in a highly conserved sequence of rep and an untranslated region in the 3′ end of AAV genomes (Gao, Alvira et al. 2003; Gao, Vandenberghe et al. 2004).
A potential limitation of the PCR based method for rescue of proviral sequences is the possibility of sequence artifacts. To rule out PCR artifacts such as PCR-mediated gene splicing by overlap extension between different partial DNA templates with homologous sequences or by possible recombination events in bacteria, extensive analyses have been performed. Results of those experiments confirmed that sequence variation captured by the molecular rescue method indeed reflected the natural diversity of primate AAVs (Gao, Alvira et al. 2003).
PCR Primer Design
Perform sequence alignment of the cap genes from different AAV serotypes and variants using ClustalX and identify the conserved regions for primer design. For selection of conserved oligonucleotide sequences of your choice, make sure that the first 3–5 nucleotides at the 3′ end of each primer are perfectly matched with those in most AAV cap sequences. Alternatively, the following pairs of conserved primers have proven to work effectively for isolating over 120 novel AAV sequences and should be considered for your applications. 1). For signature PCR with an amplicon size of approximately 250 bp, use AV19s 5′AGGTAATGCCTCAGGAAATTGGCATT3′ as the 5′ primer and AV18as 5′GAATCCCCAGTTGTTGTTGATGAGTC3′ as the 3′ primer; 2). To amplify the entire cap sequence and a part of rep sequence by two separate but overlapping amplicons (an 1.7 kb upstream fragment spanning 800 bp of rep and 700 bp of cap gene and an 1.5 kb downstream fragment of cap gene), use AV1ns 5′ GCTGCGTCAACTGGACCAATGAGAAC 3′ as the 5′ primer, AV18as 5′GAATCCCCAGTTGTTGTTGATGAGTC3′ as the 3′ primer for the 1.7 kb fragment. For the 1.5 kb fragment, use AV19s 5′ AGGTAATGCCTCAGGAAATTGGCATT 3′ as the 5′ primer, and AV2cas 5′ CGCAGAGACCAAAGTTCAACTGAAACGA 3′ as the 3′ primer; 3). To amplify the entire cap gene and a portion of rep gene as one 3.1 kb fragment, use AV1ns 5′ GCTGCGTCAACTGGACCAATGAGAAC 3′ as 5′ primer and AV2cas 5′ CGCAGAGACCAAAGTTCAACTGAAACGA 3′ as the 3′ primer.
MATERIALS
Reagents
Preparation of tissue DNA
QIAamp Tissue DNA mini Kit (Qiagen, Valencia, CA)
Disposable Scalpels (Fisher, Pittsburg, PA)
TissueLyser (Qiagen, Valencia, CA)
Stainless steel beads, 5mm (Qiagen, Valencia, CA)
Proteinase K 20mg/mL stock (Qiagen, Valencia, CA)
RNaseA (Qiagen, Valencia, CA)
PCR reagents
Platinum PCR SuperMix High Fidelity (InVitrogen, Carlsbad, CA)
Nuclease-free water (Qiagen, Valencia, CA)
Primers of choices in 10 μM concentration
Agarose gel electrophoresis and Topo-cloning
1–1.2 % of agarose gel (Fisher, Pittsburg, PA)
1X TAE buffer (Fisher, Pittsburg, PA)
Topo TA cloning kit for sequencing (InVitrogen, Carlsbad, CA)
QIAprep Spin Miniprep kit (Qiagen, Valencia, CA)
Vectorology of novel AAV serotypes
Molecular cloning
Appropriate restriction endonucleases and corresponding buffers
Alkaline phosphatase, Calf intestinal (CIP) (New England BioLab, Ipswich, MA)
T4 DNA Ligase (New England BioLab, Ipswich, MA)
QIAquick gel extraction kit (Qiagen, Valencia, CA)
MAX Efficiency DH5α Competent cell (InVitrogen, Carlsbad, CA)
Equipment
PCR Equipment
Corresponding gel electrophoresis apparatus
Tissue Sample DNA Extraction
-
1
For non-human primates (NHP) tissues, apply for local IACUC approval for tissue collection from NHP.
-
2
For collection of human tissues from either surgical procedures or postmortem examination or organ donors, seek the approval of IRB protocols by local Institutional Review Board.
-
3
Ideally, each tissue sample should be dissected with a single disposable scalpel and snap-frozen in individual sterilized container.
-
4
For tissue DNA extraction, prepare work space in a Biosafety Level II Tissue Culture Hood first by UV irradiating all surfaces for 15 minutes, and then wiping all surfaces with Lysol followed by 70% ethanol.
-
5
Take the frozen tissues out of freezer and keep the tissues on dry ice.
-
6
Place one 5mm steel bead in each of the labeled sterile 2mL microcentrifuge tubes.
-
7
Thaw no more than 3 tissues at a time in the hood at room temperature.
-
8
Quickly cut about 25mg each of the partially thawed tissues and place in the correspondingly labeled tubes.
-
9
Immediately return the remaining tissues to dry ice and then freezer.
-
10
Repeat for remaining tissue samples.
-
11
Pulse spin (30 seconds, max speed) all samples in a microcentrifuge such that tissues are at the bottom of the tubes.
-
12
To each sample, add 180μl Buffer ATL from the QIAamp Tissue DNA mini Kit and homogenize tissues in Qiagen Tissue Lyser (20 seconds at frequency 30) and pulse spin (30 seconds, max speed) all samples again.
-
13
Perform tissue DNA extraction, following the user manual of the QIAamp Tissue DNA mini Kit.
-
14
Finally, dilute each DNA samples to a concentration of 50 ng/μl in nuclease-free water and store at 4 °C.
PCR Reaction
-
15
Prepare work space in a BSL II hood as described above.
-
16
Thaw 1X High Fidelity Platinum PCR SuperMix on ice.
-
17
For each 20 μl PCR reaction, use 18 μl of 1X SuperMix, 0.6 = μl each of primers (10 uM) and 1 μl of DNA template.
-
18To prepare PCR master mix containing both 1X SuperMix and primers, count the total numbers (n) of tissue DNA samples and prepare the PCR master mix in a 1.5 ml sterile eppendorf tube according to the following formula:
-
19
Mix the PCR master mix by repeated pipeting up and down.
-
20
Label 0.2 ml micro-PCR tubes and add 19.2 μl PCR master mix to each tube.
-
21
Add 1 μl of nuclease-free water to the first PCR tube as the no template control and close the tube.
-
22
Subsequently, add 1 μl of one tissue DNA (50 ng/μl) into a PCR tube, mix by pipeting and close the tube immediately after mixing.
-
23
Repeat for other tissue samples.
-
24
Finally, add 10 ng of pAAVrep2/cap2 plasmid (10 ng/μl) to the positive control PCR tube on the regular lab bench and load the PCR tubes onto a PCR device.
-
25
PCR runs with the following conditions should result in satisfactory outcomes.
For signature PCR, 1 minute at 97 °C, followed by 35-42 cycles (optimization may be required) of 15 seconds at 97 °C, 10 seconds at 60 °C, 45 seconds at 68 – 72 °C (optimization may be required) and ended with a 10 minute extension at 68 – 72 °C (optimization may be required).
To retrieve large cap fragments, 1 minute at 97 °C, followed by 35-45 cycles (optimization may be required) of 15 seconds at 97 °C, 10 seconds at 55-60 °C (optimization may be required), 2 – 4 minutes at 68 – 72 °C (optimization may be required)and ended with a 10 minute extension at 68 – 72 °C (optimization may be required). However, depending on the quality/integrity of template DNA, conservativeness of the novel AAV sequences in the primer regions and abundance of AAV proviral DNA in the tissue, more extensive optimization for PCR conditions may be required in some cases.
-
26
At the end of PCR, take 5 μl each reaction from PCR tubes and load onto a 1 (large cap PCR fragment)-1.2% (Signature PCR fragment) agarose gel to perform gel electrophoresis in 1X TAE buffer and examine the results of PCR rescue of AAV sequences from tissue samples.
PCR Product Cloning and Analysis
-
27
Perform Topo-cloning following manufacturer’s instruction.
-
28
Identify the PCR-insert containing clones by Eco RI digestions by agarose gel electrophoresis. For each PCR fragment, analyze at least 6 clones by sequencing.
-
29
For sequence characterization, align the newly isolated VP1 sequences with those of known AAV serotypes/variants available from GeneBank using ClustalX and further analyze with the Vector NTI software package (Informax, Inc, Bethesda, MD, USA).
Reagents and Solutions
2.5M CaCl2 solution
Dissolve 183.74 g of calcium chloride (CaCl2) in 500 mL of H2O. Sterilize by passing through a 0.22 μm<micrometer> filter and store at room temperature.
2X HBS
REAGENT QUANTITY (FOR 1L) FINAL CONCENTRATION NaCl 16.4 g 280 mM Hepes (C8H18N2O4S) 11.9 g 50 mM Na2HPO4.7H2O 0.38 g 1.42 mM H20 to 1 L Adjust pH to 7.05 with 10 M NaOH. Sterilize by passing through a 0.22 μm<micrometer> filter and store at room temperature.
Cell re-suspension buffer for CsCl gradient purification
REAGENT QUANTITY (FOR 1L) FINAL CONCENTRATION Tris-Cl (1 M, pH 7.4) 50 mL 50 mM MgCl2 (1M) 1 mL 1 mM H20 to 1 L Mix well, sterilize by passing through a 0.22 μm<micrometer> filter and store at room temperature.
Light CsCl for AAV purification (Density = 1.41 g/mL)
Dissolve 513.89 g of biological grade Cesium chloride (CsCl) in 786 mL of 10 mM Tris-Cl, pH 8.0, sterilize by passing through a 0.22 μm<micrometer> filter and store at room temperature.
Heavy CsCl for AAV Purification (Density = 1.61 g/mL)
Dissolve 672.1 g of biological grade Cesium chloride (CsCl) in 672.9 mL of 10 mM Tris-Cl, pH 8.0, sterilize by passing through a 0.22 μm<micrometer> filter and store at room temperature.
5% D-Sorbitol/PBS, pH 7.6
Dissolve 25 g of D-sorbitol in 5 Ls of D-PBS, pH 7.6. Sterilize by passing through a 0.22 μm<micrometer> filter and store at 4 °C
Sample dilution buffer (1X PCR Buffer/20 ng/μL SSS DNA)
REAGENT QUANTITY (FOR 10 mL) FINAL CONCENTRATION 10 x GeneAmp PCR Buffer 1 mL 1 x Sheared Salmon Sperm DNA (1 mg/mL) 0.2 mL 0.02 mg/mL Nuclease-free water 8.8 ml Mix well, aliquot into 1.5 eppendorf tubes and store at − 20 °C.
Commentary
Background Information
Adeno-associated viruses (AAVs) are parvovirus with a 4.7 kb single-stranded DNA genome that belong to the Dependovirus genus of the family Parvovirinae in the order Parvoviridae of single-stranded DNA viruses(Carter, Khoury et al. 1975) (Hermonat, Labow et al. 1984). The original isolate, AAV2 was discovered as a laboratory contaminant of adenovirus cultures (Atchison, Casto et al. 1966) (Hoggan, Blacklow et al. 1966) and was subsequently found to replicate only in the presence of adenovirus, herpes virus or other helper virus, which supply a number of early functions required for AAV gene expression and replication (Blacklow, Hoggan et al. 1968; Blacklow, Hoggan et al. 1968). AAV are non-enveloped icosahedral virions of approximately 20 to 25nm in diameter. AAV serotypes 1,4,6,7,8,10 and 11 are found in non human primates, and AAV2, AAV3, and AAV5 are particularly common in humans (Blacklow, Hoggan et al. 1967) (Blacklow, Hoggan et al. 1968; Blacklow, Dolin et al. 1971). To date, no AAV has been associated with any human disease, despite the fact that 90% of adult humans are seropositive for one or more of the known AAV serotypes (Afione, Conrad et al. 1995). The AAV genome consists of two 145-nucleotide inverted terminal repeat (ITR) sequences, each an identical palindrome at either terminus of the genome, flanking the two AAV genes, rep and cap (Senapathy and Carter 1984; Senapathy, Tratschin et al. 1984). The rep gene is transcribed from the p5 and p19 promoters, with the latter embedded within the coding sequence of the longer Rep proteins. Transcripts from these promoters are alternatively spliced to encode four different Rep proteins, Rep78, Rep68, Rep52, and Rep40. Rep78 and Rep68 are multifunctional DNA binding proteins which possess helicase activity and site-specific, strand-specific nickase activity, both of which are required for terminal resolution of replicating AAV genomes(Im and Muzyczka 1990). These viral proteins also bind the chromosomal target sequence for AAV integration in the human genome, the AAVS1 site, and these proteins are required for normal integration into this site. Finally, Rep78/68 are also potent bi-functional transcription regulators that generally activate transcription from AAV promoters during productive infection and repress their transcription during latent infection(Pereira, McCarty et al. 1997; Pereira and Muzyczka 1997; Pereira and Muzyczka 1997). The shorter Rep proteins, Rep52 and Rep40 act as modifier proteins for the transcriptional activities of Rep 68/78, and are required for sequestration of single-stranded AAV genomes into capsids during productive infection. The AAV cap gene is transcribed from the p40 promoter generating two mRNAs via alternative splicing to encode for the three viral capsid proteins. The longer of the two mRNAs encodes for VP1, the largest of the three AAV capsid proteins (87kDa). The alternatively spliced transcript can initiate translation using either a non-canonical ACG start codon, which results in the translation of VP2 (73kDa), or an ATG codon further downstream, which gives rise to VP3 (61kDa)(Trempe and Carter 1988). VP3 is the shortest and most abundant of the AAV capsid proteins, comprising about 90% of the capsid’s protein. The less abundant VP1 and VP2 are also required for capsid formation. In total, 60 copies of the three viral proteins (VP1:VP2:VP3) in the ratio 1:1:8 are required for the synthesis of a subunit T=1 icosahedral particle.
TROUBLESHOOTING
Tissue Culture and Transfection Problems
HEK-293 cells are permissive to latent infection by wild-type AAV which may result in wtAAV contamination in the purified vector stocks. For this reason we suggest periodic assessment of HEK-293 cell lines for wt-AAV contamination by PCR for repa and cap sequences. This has been largely overcome by the helper-virus free AAV production protocol described here which employs a helper plasmid to provide all necessary helper functions of adenovirus. This protocol can be further streamlined into a two plasmid transfection by using a single plasmid to express the Ad helper genes along with the AAV rep and cap genes. The single most important variable in vector yield is the transfection efficiency of the HEK-293 cells. One of the key components to achieve high transfection efficiencies (> 70%) is careful preparation and testing of 2x HBS solution with the correct pH. Optimal calcium phosphate transfections have a narrow tolerance for pH variation, it is critical to ensure that the pH value of 2X HBS is 7.05. When making this solution, ensure that the pH meter is properly calibrated. Each batch of the transfection reagents should be tested for transfection efficiency in 293 cells using an EGFP expressing plasmid before their use for AAV vector production. Quality and lot-to-lot variation of commercial fetal bovine serum could also have a significant impact on transfection efficiency. Testing different lots and manufacturers of fetal bovine serum for their effect on transfection by calcium phosphate precipitation is strongly recommended. In addition, the pH of the tissue culture medium in the transfected cell plates is also an influential factor in the efficiency of transfection. It is best to take no more than 12 plates out of the incubator right before the transfection, and to return the plates to the incubator immediately after adding the transfection cocktail.
Plasmid purity and integrity are also important factors to consider, all plasmids should be prepared by the triton alkaline lysis-CsCl ethidium bromide density centrifugation or similar method. AAV-ITR sequences can be very unstable, frequently undergoing deletion or rearrangement. Therefore, it is of utmost importance to propagate ITR-containing plasmids in bacteria that are deficient of certain rec genes. A reliable rec- E. coli host strain is E. coli SURE2 cells from Stratagene or STABL2 cells from Invitrogen. Even when these rec- strains are used, it is important to monitor each DNA preparation for deletion of ITR sequences. The ITR sequences in the pCIS plasmids serve as not only the recognition and cleavage sites for Rep proteins to release the viral genome from the plasmid backbone, but also as replication origin and packaging signal. Therefore, ensuring the integrity of the ITR sequences in the pCIS plasmids is a key step in rAAV production. Checking for ITR integrity can be easily accomplished by restriction digestion of the pCIS plasmid with Sma I, for which multiple recognition sites are present in the ITRs.
Recombinant AAV Purification
In contrast to adenoviral vector preps which are characterized by the milky hue, for the most part AAV vector preparations purified by a CsCl gradient should be colorless solutions. AAV2 vectors tend to aggregate at high concentrations, which ultimately have a negative impact on transduction efficiency. The factors that contribute to AAV2 vector aggregation, and formulations that prevent it have been thoroughly investigated (Wright, Le et al. 2005). Formulation of AAV2 vectors in high-ionic-strength buffers prevents aggregation. Because the biological properties of any AAV vector are determined by the amino acids that are exposed on the surface of the viral capsid, it seems reasonable to speculate that different AAV capsids have different surface composition, and possibly different overall charge. These changes are likely to lead to differences in solubility and susceptibility to aggregation, but this remains to be investigated for AAV capsids other than AAV2. Nonetheless, as a general precaution, it is best to store other serotypes of AAV vectors at a virus particle concentration or genome copy titer no higher than 2–3 x 1013 (virus particles/mL or GC/mL). In order to reduce the likelihood of aggregation as a precautionary step it is helpful to perform a quick SDS-PAGE silver staining analysis on the pooled AAV-containing CsCl gradient fractions to estimate viral particle concentration (see protocol 5) before performing the dialysis. If the concentration is higher than 2–3 x 1013 vector particles/ml, an equal volume of PBS (i.e. 1:1 dilution) should be added before proceeding with dialysis. In addition if subsequently it is found that the vector genome titer of a purified and desalted vector preparation is higher than 1x1013 genome copies/ml, it is recommended the vector be diluted to 1x1013 GC/ml before freezing for storage. For long-term storage, AAV vectors should be kept at −80°C. Once thawed, it is best to store AAV vectors at 4°C for at least several weeks.
Vector Titering
Silver-stained SDS-PAGE analysis of rAAV preparations aims to analyze the test articles for purity, and to reveal if there are any other cellular or transgene protein contaminants. Nonetheless, it cannot differentiate empty virions from fully packaged virions. When performing the semi-quantitative SDS-PAGE gel it is recommended that the test articles be prepared in duplicates and that the reference standard be loaded in the middle of the wells flanked by the unknown test sample on each side.
PCR-based method for novel AAV discovery
The most common problem with this assay is environmental contamination of AAV genomes; this is especially the case when these assays are run in space that is shared with laboratories actively producing rAAV. This in combination with the prevalence of AAV in humans requires stringent precautions to minimize environmental or operator contamination of the PCR reaction. It is recommended that this work be carried out in a room specifically designated for PCR where there is minimal possibility of contamination by pTRANS plasmids. In addition, all work should be carried out in a BSL II hood. All surfaces and pipettors should be cleaned with Lysol followed by 70% EtOH, and the hood and tubes to be used should be irradiated by UV light for 15 minutes. It is recommended that the operator wear a mask and disposable barrier clothing when possible such as a Tyvek coverall or a similar product.
Anticipated Results
Viral vector yields performed with this protocol typically yield highly pure viral vector preparations with few empty capsids (>10%) as determined by electron microscopy analysis and with titers around 1.0x1013 vector particles in total final volume that ranges between 2-3 mls.
Time Considerations
Starting from splitting cells (protocol 1), the entire transfection procedure takes 6 days. Logistically if the investigator splits cells on Friday, they will be ready for transfection on Monday, and on Thursday they will be ready for harvesting. The purification process can be completed in approximately one week, and subsequent vector genome titration by rtPCR takes 1-2 days. rAAV purity analysis by SDS-PAGE also requires 1-2 days. In total, protocols 1-4 can be completed in 15-17 days under optimal conditions.
The discovery of novel AAVs depends on the time necessary to obtain and harvest tissues, the number of tissue samples to be analyzed, and other logistic issues that will vary. The actual PCR step takes 1-2 days, followed by analysis.
Acknowledgments
The authors would like to acknowledge the technical assistance of Qin Su and Ran He of University of Massachusetts Medical School, and Julio Sanmiguel and Mauricio Alvira of the University of Pennsylvania in the development of this protocol.
Footnotes
Basic Protocols 1–4 have been adapted from Chapter 16 of Molecular Cloning 4th edition, 2012, by Green and Sambrook with permission from Cold Spring Harbor Laboratory Press.
Basic Protocol 5 has been adapted from Adeno-Associated Virus Methods and Protocols, Chapter 4 Exploiting Natural Diversity of AAV for the Design of Vectors with Novel Properties with permission from Springer.
The background information has been adapted with permission from Chapter 1 in the book “Regenerative Therapy for the Musculoskeletal System Using Recombinant Adeno-Associated Viral Vectors”, 2010: 1–21 ISBN: 978-81-308-0423-1.
Literature Cited
- Afione SA, Conrad CK, et al. Gene therapy vectors as drug delivery systems. Clin Pharmacokinet. 1995;28(3):181–189. doi: 10.2165/00003088-199528030-00001. [DOI] [PubMed] [Google Scholar]
- Asuri P, Bartel MA, et al. Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol Ther. 20(2):329–338. doi: 10.1038/mt.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchison RW, Casto BC, et al. Electron microscopy of adenovirus-associated virus (AAV) in cell cultures. Virology. 1966;29(2):353–357. doi: 10.1016/0042-6822(66)90045-6. [DOI] [PubMed] [Google Scholar]
- Blacklow NR, Dolin R, et al. Studies of the enhancement of an adenovirus-associated virus by herpes simplex virus. J Gen Virol. 1971;10(1):29–36. doi: 10.1099/0022-1317-10-1-29. [DOI] [PubMed] [Google Scholar]
- Blacklow NR, Hoggan MD, et al. Epidemiology of adenovirus-associated virus infection in a nursery population. Am J Epidemiol. 1968;88(3):368–378. doi: 10.1093/oxfordjournals.aje.a120897. [DOI] [PubMed] [Google Scholar]
- Blacklow NR, Hoggan MD, et al. Isolation of adenovirus-associated viruses from man. Proc Natl Acad Sci U S A. 1967;58(4):1410–1415. doi: 10.1073/pnas.58.4.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacklow NR, Hoggan MD, et al. Serologic evidence for human infection with adenovirus-associated viruses. J Natl Cancer Inst. 1968;40(2):319–327. [PubMed] [Google Scholar]
- Brantly ML, Chulay JD, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci U S A. 2009;106(38):16363–16368. doi: 10.1073/pnas.0904514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BJ, Khoury G, et al. Physical map and strand polarity of specific fragments of adenovirus- associated virus DNA produced by endonuclease R-EcoRI. J Virol. 1975;16(3):559–568. doi: 10.1128/jvi.16.3.559-568.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105(39):15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement N, Knop DR, et al. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum Gene Ther. 2009;20(8):796–806. doi: 10.1089/hum.2009.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JE, Zolotukhin S, et al. Recombinant adeno-associated virus type 2 replication and packaging is entirely supported by a herpes simplex virus type 1 amplicon expressing Rep and Cap. J Virol. 1997;71(11):8780–8789. doi: 10.1128/jvi.71.11.8780-8789.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz PE, Mueller C, et al. In vivo post-transcriptional gene silencing of alpha-1 antitrypsin by adeno-associated virus vectors expressing siRNA. Lab Invest. 2007;87(9):893–902. doi: 10.1038/labinvest.3700629. [DOI] [PubMed] [Google Scholar]
- Egan M, Flotte T, et al. Defective regulation of outwardly rectifying Cl- channels by protein kinase A corrected by insertion of CFTR. Nature. 1992;358(6387):581–584. doi: 10.1038/358581a0. [DOI] [PubMed] [Google Scholar]
- Ellis BL, Hirsch ML, et al. Zinc-finger nuclease-mediated gene correction using single AAV vector transduction and enhancement by Food and Drug Administration-approved drugs. Gene Ther. doi: 10.1038/gt.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Alvira MR, et al. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc Natl Acad Sci U S A. 2003;100(10):6081–6086. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Vandenberghe LH, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78(12):6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(18):11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GP, Qu G, et al. High-titer adeno–associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum Gene Ther. 1998;9(16):2353–2362. doi: 10.1089/hum.1998.9.16-2353. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol Ther. 18(8):1450–1457. doi: 10.1038/mt.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel EM, Gellhaus K, et al. Versatile and Efficient Genome Editing in Human Cells by Combining Zinc-Finger Nucleases With Adeno-Associated Viral Vectors. Hum Gene Ther. doi: 10.1089/hum.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermonat PL, Labow MA, et al. Genetics of adeno-associated virus: isolation and preliminary characterization of adeno-associated virus type 2 mutants. J Virol. 1984;51(2):329–339. doi: 10.1128/jvi.51.2.329-339.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggan MD, Blacklow NR, et al. Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc Natl Acad Sci U S A. 1966;55(6):1467–1474. doi: 10.1073/pnas.55.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im DS, Muzyczka N. The AAV origin binding protein Rep68 is an ATP-dependent site-specific endonuclease with DNA helicase activity. Cell. 1990;61(3):447–457. doi: 10.1016/0092-8674(90)90526-k. [DOI] [PubMed] [Google Scholar]
- Li H, Haurigot V, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 475(7355):217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller C, Tang Q, et al. Sustained miRNA-mediated Knockdown of Mutant AAT With Simultaneous Augmentation of Wild-type AAT Has Minimal Effect on Global Liver miRNA Profiles. Mol Ther. 20(3):590–600. doi: 10.1038/mt.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira DJ, McCarty DM, et al. The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J Virol. 1997;71(2):1079–1088. doi: 10.1128/jvi.71.2.1079-1088.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira DJ, Muzyczka N. The adeno-associated virus type 2 p40 promoter requires a proximal Sp1 interaction and a p19 CArG-like element to facilitate Rep transactivation. J Virol. 1997;71(6):4300–4309. doi: 10.1128/jvi.71.6.4300-4309.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira DJ, Muzyczka N. The cellular transcription factor SP1 and an unknown cellular protein are required to mediate Rep protein activation of the adeno-associated virus p19 promoter. J Virol. 1997;71(3):1747–1756. doi: 10.1128/jvi.71.3.1747-1756.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JE, Rolling F, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76(2):791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senapathy P, Carter BJ. Molecular cloning of adeno-associated virus variant genomes and generation of infectious virus by recombination in mammalian cells. J Biol Chem. 1984;259(7):4661–4666. [PubMed] [Google Scholar]
- Senapathy P, Tratschin JD, et al. Replication of adeno-associated virus DNA. Complementation of naturally occurring rep- mutants by a wild-type genome or an ori-mutant and correction of terminal palindrome deletions. J Mol Biol. 1984;179(1):1–20. doi: 10.1016/0022-2836(84)90303-6. [DOI] [PubMed] [Google Scholar]
- Sun X, Yan Z, et al. Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J Clin Invest. 2008;118(4):1578–1583. doi: 10.1172/JCI34599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JP, Carter BJ. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J Virol. 1988;62(9):3356–3363. doi: 10.1128/jvi.62.9.3356-3363.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urabe M, Ding C, et al. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13(16):1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- Wright JF, Le T, et al. Identification of factors that contribute to recombinant AAV2 particle aggregation and methods to prevent its occurrence during vector purification and formulation. Mol Ther. 2005;12(1):171–178. doi: 10.1016/j.ymthe.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Xie J, Ameres SL, et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods. doi: 10.1038/nmeth.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]