

Abstract
Background: Although osteoarthritis (OA) is the commonest joint disorder and has a rising prevalence as the population ages, no drugs are available that prevent or delay the onset and progression of disease. Recent studies identified the DIO2 gene encoding type 2 deiodinase (D2) as a susceptibility locus for OA, and further data suggest deiodinase-regulated local availability of triiodothyronine (T3) in the joint plays an important role in cartilage maintenance and repair. To investigate the hypothesis that reduced tissue T3 availability protects joints from development of OA, the joint phenotypes of adult mice lacking D2 (D2KO) or lacking both D1 and D2 (D1D2KO), the only enzymes that catalyze conversion of the prohormone thyroxine to active T3, were determined.
Methods: Knee joints were prepared from male 16-week-old adult wild type (WT; n=9), D2KO (n=5), and D1D2KO (n=3) mice. Articular cartilage pathology was scored using the Osteoarthritis Research Society International (OARSI) histopathology scale for murine OA to determine the severity and extent of disease. Digital X-ray microradiography was used to determine the area and mineral content of subchondral bone immediately beneath the articular cartilage surface.
Results: There were no differences in maximum and standardized OA scores, cartilage erosion indices, or articular cartilage cellularity among WT, D2KO, and D1D2KO mice. Subchondral bone area did not differ among genotypes, but mineral content was markedly increased in both D2KO and D1D2KO mice compared to WT.
Conclusions: Although adult D2KO mice have normal articular cartilage and no other features of spontaneous joint damage, they exhibit increased subchondral bone mineral content.
Introduction
Osteoarthritis (OA) is the commonest joint disorder worldwide, and its prevalence is increasing in the ageing population (1). The disease affects all tissues of the joint (2), leading to pain, progressive dysfunction, and disability. OA is a major cause of time off work, second only to ischemic heart disease in the United States (1). Despite this increasing health burden, no drugs are currently available that prevent or delay the onset and progression of joint damage. Characteristic pathological changes in OA include damage to cartilage lining the joint surface, sclerosis of underlying subchondral bone (3), osteophyte formation, and synovial thickening. Articular chondrocytes undergo abnormal terminal differentiation, and the balance between synthesis and destruction of cartilage matrix favors loss of matrix due to increased production of degrading enzymes (2). Although understanding of the pathogenesis of OA has progressed in recent years, its complex etiology is multifactorial with important genetic and environmental contributions (2). The heritability of OA is between 40% and 65%, although known disease susceptibility loci account for only a small fraction (4). Currently, these loci have mostly identified regulatory rather than structural proteins as modulators of OA risk.
Recent data suggest that altered thyroid hormone availability in articular cartilage or joint tissues may play a key role in predisposition to OA (5–9). Normal euthyroid status is maintained by the hypothalamic-pituitary-thyroid axis via a negative feedback loop in which thyroid hormones produced by the thyroid gland inhibit synthesis and secretion of thyrotropin (TSH) from the anterior pituitary. The main hormone secreted by the thyroid gland is the prohormone thyroxine (T4), which is converted to the physiologically active metabolite 3,5,3′-L-triiodothyronine (T3) by removal of an outer ring iodine atom. Thyroid hormones enter peripheral tissues, including bone and cartilage (10), via specific membrane transporters. In target cells, the type 2 iodothyronine deiodinase (D2) converts T4 to T3, whereas the type 3 enzyme (D3) irreversibly inactivates T3 and prevents activation of T4 by removal of an inner ring iodine. Thus, the balance of D2 and D3 activities determines local T3 availability and tissue thyroid hormone responsiveness (11–13).
The D2 enzyme is labile due to substrate-induced ubiquitination and proteasome-mediated degradation (12,13), and its specific activity is difficult to measure, especially at low levels. Nevertheless, in-depth studies of iodothyronine metabolism in mouse skeletal tissues, cell lysates, and intact primary cell cultures have shown that specific D2 enzyme activity is restricted to mature bone-forming osteoblasts and not detectable in bone-resorbing osteoclasts or growth plate chondrocytes (10). A recent study using quantitative reverse transcription polymerase chain reaction (RT-qPCR) identified Dio2 mRNA expression in rat articular cartilage at levels comparable to brown adipose tissue, although D2 enzyme activity was not measured (14). By contrast, functional D3 enzyme, a much more stable protein with readily detectable activity, was identified in osteoblasts, osteoclasts, and growth plate chondrocytes, with activity highest in cells derived from developing and younger animals (10). It is unknown whether D3 is expressed in articular cartilage or whether D2 expression in articular cartilage is regulated during joint development. The type 1 deiodinase (D1), which converts T4 to T3 mainly in the liver and kidney and generates a large proportion of the circulating T3, is not expressed in bone or cartilage and appears to have no physiological role in the skeleton (10).
T3 regulates cartilage maturation and is essential for endochondral ossification, bone modeling, and longitudinal growth (15). A role for thyroid hormones in the pathogenesis of OA has been proposed. Although chondrocytes resist terminal differentiation in healthy articular cartilage, a process resembling endochondral ossification occurs during the progression of OA in which articular chondrocytes undergo hypertrophic differentiation with acceleration of cartilage mineralization (16,17). Increased activities of ADAMTS5 (a disintegrin and metalloproteinase with thrombospondin motifs-5) (18,19) and MMP13 (matrix metalloproteinase-13) (20) in OA cause degradation of cartilage matrix, and these enzymes are regulated in the growth plate by T3 (21–24), which also induces hypertrophic chondrocyte differentiation at this location (14,21,25).
A genome-wide linkage analysis of siblings with generalized OA found a nonsynonymous coding variant (rs225014; Thr92Ala) that identified DIO2 as a disease-susceptibility locus (6). Replication studies in Asian and European cohorts confirmed this by identifying a DIO2 haplotype containing the minor allele of rs225014 and major allele of rs12885300 that was associated with symptomatic OA (6). The relationship between this haplotype and OA, however, was not replicated in the Rotterdam Study population (26), and DIO2 was not identified as a susceptibility locus in a more recent meta-analysis (27). Nevertheless, further data suggest the risk allele of rs225014 may be expressed at higher levels than the protective allele in OA cartilage from heterozygous patients (28), possibly because of epigenetic modifications (29). Increased DIO2 mRNA and protein expression has also been documented by RT-qPCR and immunohistochemical staining in cartilage obtained from joints affected by end-stage OA (14,28). Additionally, the rs12885300 DIO2 polymorphism has been suggested to influence the association between hip shape and OA susceptibility by possibly increasing vulnerability of articular cartilage to abnormal hip morphology (30). Furthermore, studies in transgenic rats that overexpress human D2 in chondrocytes indicate transgenic animals have an increased susceptibility to OA following surgical destabilization of the knee (14). Finally, a recent meta-analysis studying factors that regulate thyroid hormone metabolism and action identified a possible role for DIO3 as a disease-modifying locus in OA (5).
Together, these data suggest that deiodinase-regulated local availability of T3 in joint tissues may play an important role in articular cartilage renewal and repair, particularly as the polymorphisms in DIO2 and DIO3 that are associated with OA do not influence systemic thyroid status (31). Overall, current findings suggest that increased T3 availability in OA may be detrimental to joint maintenance and articular cartilage homeostasis. Thus, it was hypothesized that reduced tissue T3 availability protects joints from damage and the development of OA. To investigate, the joint phenotypes of adult mice lacking D2 (D2KO) or lacking both D1 and D2 (D1D2KO) were determined.
Materials and Methods
Mice
D1KO or D2KO mice were generated after targeted disruption of Dio1 or Dio2 in 129/SvJ embryonic stem cells. Mice were bred and maintained as homozygous colonies in a C57BL/6 background at Dartmouth Medical School animal facility using protocols approved by the Institutional Review Board (32,33). D1KO and D2KO mice were crossed to obtain double heterozygous F1 progeny, which were used to generate a colony of D1D2KO homozygous double mutants (12,34). Mice were housed under conditions of controlled lighting and temperature and sacrificed at 16 weeks of age. Bone samples were shipped to Imperial College London for phenotyping studies in which nine wild type (WT), five D2KO, and three D1D2KO male mice were used for detailed histological analysis of the joints. Histological sections from four wild type, four D2KO, and three D1D2KO mice were prepared at directly comparable levels for subchondral bone analysis.
Preparation of histological sections
Lower limbs were fixed in 10% neutral buffered formalin for 24 h and decalcified in 10% EDTA, pH7.4, for 7–10 days. Knee joints were embedded in paraffin and 5 μm thick midline sections were cut at 80 μm intervals. Slides were stained for cartilage proteoglycans using Safranin-O Fast-Green (35) and imaged using a Leica MZ75 binocular microscope (Leica AG, Heerbrugg, Switzerland), KL1500 light source, and DFC320 digital camera.
Analysis of articular cartilage
Articular cartilage pathology was scored by a blinded observer using the Osteoarthritis Research Society International (OARSI) histopathology scale for murine OA (35). A score of 0 represents normal cartilage; 0.5 represents loss of Safranin-O staining without structural changes; 1 represents small fibrillations without loss of cartilage; and 2 represents vertical clefts down to the layer immediately below the superficial layer and some loss of surface lamina. Vertical clefts or erosions to the calcified cartilage extending to <25%, 25–50%, 50–75%, or >75% of the articular surface are assigned scores of 3, 4, 5, or 6 respectively. Using this system, the severity and extent of osteoarthritis is expressed as a maximum score for each joint and as a cumulative score over the whole joint (35). Because the numbers of histological sections obtained differed between individual mice, a standardized OA score was determined. Thus, a reference histological atlas of a 16-week-old male C57BL/6 WT mouse knee was constructed using 5 μm thick coronal sections cut at 80 μm intervals through the intact joint to yield 16 levels of sectioning, of which nine passed directly through the joint cavity (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com.thy). The total articular surface was determined for the reference atlas and each experimental animal (Fig. 1A and B). A standardized OA score for each experimental animal was defined as: cumulative OA score from all histological sections×(reference articular surface/experimental animal articular surface). A validated erosion index was also determined to assess the extent of cartilage lesions as a measure of OA progression (36,37): the length of eroded articular cartilage was measured and divided by the total length of articular cartilage surface and expressed as a percentage (Fig. 1C). The articular cartilage cellularity was also determined and defined as the mean number of chondrocytes per mm2.
FIG. 1.

Articular cartilage and subchondral bone analysis in wild type (WT) C57BL/6 mice. (A) Right knee mid-joint coronal section from a 16-week-old male C57BL/6 mouse stained with Safranin-O Fast-Green. Numbered blue lines (1–4) delineate the articular surfaces. Scale bar=0.5 mm. (B) Magnified view of medial joint compartment showing normal articular cartilage (red) and subchondral bone (green). Scale bar=0.5 mm. (C) Erosion index is calculated by measuring the eroded surface (3) divided by total articular surface (1+2+3). Scale bar=0.5 mm. (D) High power view of the eroded surface shown in panel (C). The intact surface lamina in undamaged articular cartilage is indicated; area number 1 shows loss of safranin O staining, and area number 2 shows fibrillations without loss of cartilage. Scale bar=0.25 mm. (E) Subchondral bone region of interest enclosed by yellow line. Scale bar=0.5 mm. (F) Articular cartilage and marrow space after thresholding in Image J to allow the subchondral bone area in the region of interest to be determined automatically.
Histological analysis of subchondral bone area
Histological sections were imaged in 8-bit Tiff format. The subchondral bone region of interest (ROI) between the articular cartilage and epiphyseal growth plate of the tibia was contoured using ImageJ (http://rsb.info.nih.gov/ij/), and the subchondral bone area relative to tissue area (BA/TA) determined (Fig. 1D and E).
Subchondral bone mineral content
Quantitative digital X-ray microradiography was used to determine relative bone mineral content in tibial subchondral bone as described (38,39). Increasing gradations of mineralization density were represented in 16 equal intervals by a pseudocolor scheme (Fig. 2). The bone compartment proximal to the tibial growth plate was selected as the subchondral bone ROI. Relative and cumulative frequency distributions of gray levels in ROIs were determined and compared among genotypes.
FIG. 2.
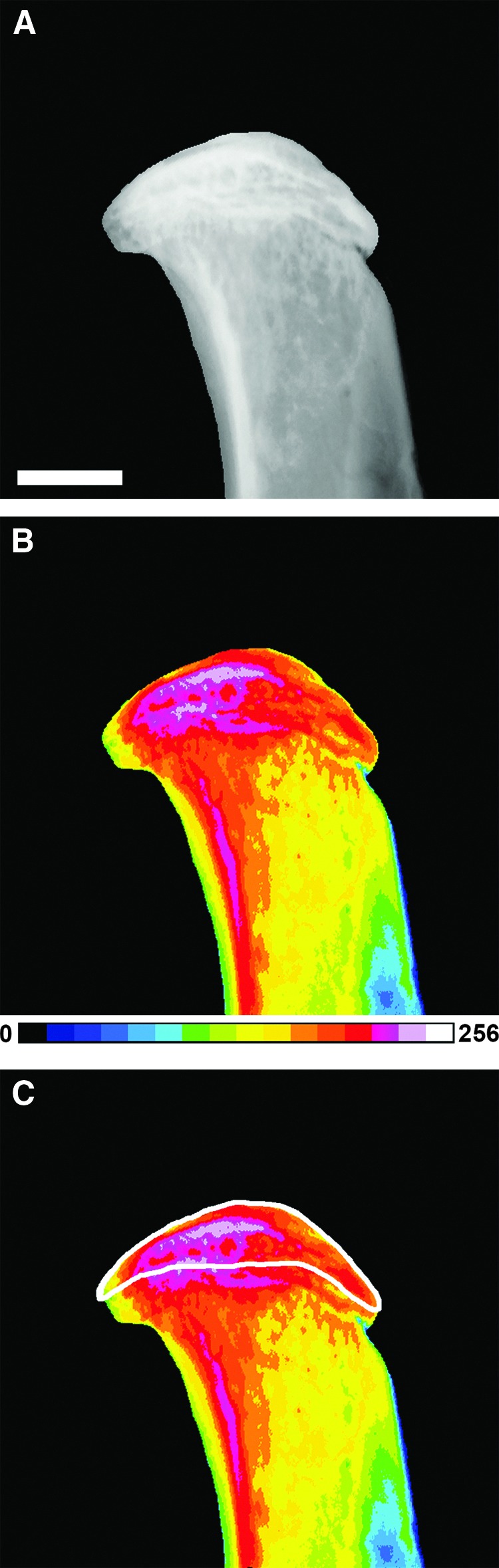
Analysis of subchondral bone mineral content in WT C57BL/6 mice. (A) Gray-scale digital X-ray microradiograph of proximal tibia. (B) Pseudocolored images representing gradations of relative bone mineral content (BMC) in which blue-green is low BMC and pink-white is high BMC. (C) Subchondral bone region of interest is indicated within the white boundary.
Statistics
Individual comparisons between WT and mutant mice for the maximum OA score and range were analyzed using the Mann–Whitney U-test. Comparisons between WT and mutant mice for mean standardized OA score, erosion index, and subchondral bone area were analyzed by unpaired Student's t-test. p-Values<0.05 were considered significant. The Kolmogorov–Smirnov test was used to compare gray-level cumulative frequency distributions in digital X-ray micrographs between WT and mutant mice. p-Values for the D statistic were D>6.01, p<0.05; D≥7.20, p<0.01; and D≥8.62, p<0.001 (39).
Results
Length of articular cartilage
The articular cartilage surface in the reference atlas was determined in sections from the nine levels that passed directly through the joint cavity (Supplementary Fig. S1 and Fig. 1). The measured complete articular cartilage surface in the reference joint was 27.4 mm, and this length was used to calculate standardized OA scores. In sections from experimental samples, the measured articular cartilage surface was: WT 5.0±1.2, n=9; D2KO 6.0±1.1, n=5; D1D2KO 10.99±2.99, n=3 (mm±SEM; ANOVA; not significant).
Articular cartilage is not altered in D2KO mice
There were no differences in maximum and standardized OA scores or erosion indices between WT and D2KO mice (Fig. 3). The maximum OA scores were: WT 3 (0–3), n=9; D2KO 4 (0.5–4), n=5 (maximum score (range); Mann–Whitney U-test; not significant). The standardized OA scores were: WT 7.8±2.1, n=9; D2KO 10.9±3.6, n=5 (mean standardized score±SEM; Student's t-test; not significant). The erosion indices were WT 2.7±1.4, n=9; D2KO 6.1±2.7, n=5 (mean %±SEM; Student's t-test; not significant). There were also no differences in these parameters in D1D2KO mice: maximum OA score (0–2), n=3; standardized OA score 9.6±5.4, n=3; erosion index 5.5±3.3, n=3 (Mann–Whitney U-test and Student's t-tests vs. WT or D2KO mice; not significant).
FIG. 3.

Osteoarthritis (OA) pathology and scores in WT and D2KO mice. (A) High-powered views showing normal articular cartilage with an OARSI Score of zero in WT and D2KO mice. Scale bar=50 μm. sl, surface lamina; hac, hyaline articular cartilage; mf, mineralizing front; cc, calcified cartilage; scb, subchondral bone. (B) Maximum OA scores for WT (3=vertical clefts or erosions to the calcified cartilage extending to <25% of articular surface) and D2KO (4=vertical clefts or erosions to the calcified cartilage extending to 25–50% of articular surface) mice. (C) Graphs showing maximum OA scores in WT and D2KO mice; measures of cartilage damage (mean standardized OA score in red bars, mean erosion index in green bars); and mean subchondral bone area (BA/TA). Error bars represent standard error of the mean, with no significant differences among genotypes for all parameters. (D) Subchondral bone regions of interest in WT and D2KO mice.
Articular cartilage cellularity also did not differ between WT and D2KO mice: WT 1412±82, n=9; D2KO 1705±161, n=5 (mean number of chondrocytes per mm2±SEM; Student's t-test; not significant). Articular cartilage cellularity also did not differ in D1D2KO mice: 1493±251, n=3 (Student's t-test vs. WT or D2KO mice; not significant).
Subchondral bone area is unchanged but bone mineral content is increased in D2KO mice
The subchondral bone area did not differ between WT and D2KO mice: WT 64±7.6%, n=4; D2KO 56±10.3%, n=4 (mean BA/TA; Student's t-test; not significant; Fig. 3) and was also not different in D1D2KO mice: 52±3.8%, n=3 (Student's t-test vs. WT or D2KO mice; not significant). Despite this, subchondral bone mineral content was markedly increased in D2KO mice (n=4) compared to WT (n=5), Kolmogorov–Smirnov test (p<0.001; Fig. 4), and a similar increase was evident in D1D2KO mice (n=2; p<0.001).
FIG. 4.

Subchondral bone mineral content in WT and D2KO mice. (A) Pseudocolored images of proximal tibias from WT and D2KO mice in which blue-green is low BMC, and pink-white is high BMC. Subchondral bone region of interest is indicated within the white boundary. (B) Relative and cumulative frequency histograms demonstrating increased subchondral bone mineral content in D2KO mice. Kolmogorow–Smirnov test; ***p<0.001 (WT n=5, D2KO n=4).
Discussion
It is demonstrated that 16-week-old adult male D2KO mice, which have a normal circulating T3 concentration (33) but lack the D2 enzyme that regulates intracellular T3 availability, have increased subchondral bone mineral content but normal articular cartilage and no other features of OA. Additional deletion of D1 in D1D2KO mice, which also have a normal circulating T3 concentration (12), did not affect the phenotype observed in D2KO mice, supporting the notion that D1 has no physiological role in the skeleton (10).
D2KO mice have a generalized increase in bone mass and mineralization resulting in bones with reduced toughness and increased susceptibility to fracture (34). The abnormality results from disruption of a critical function for D2 in osteoblasts, in which a higher T3 concentration relative to other skeletal cells is maintained in order to optimize bone mineralization (34). In this study, it is demonstrated that adult D2KO mice also have increased mineral content in subchondral bone but no abnormalities of articular cartilage or other pathological features of spontaneous OA.
It is important to consider whether the subchondral bone abnormality in D2KO mice reflects a potential role for D2 in the pathogenesis of OA. This is a complex question because OA may result from primary abnormalities in articular chondrocytes, changes in adjacent subchondral bone that lead to secondary cartilage damage and degeneration, or from defects in both tissues. Although it is well established that both subchondral bone remodeling and articular cartilage damage occur in OA, it is not clear whether these alterations occur simultaneously or whether subchondral bone changes result in cartilage damage or vice versa (3,9,40,41). For example, cathepsin K knockout mice have increased subchondral bone mineralization and are protected from OA (42), whereas rabbits treated with parathyroid hormone have increased subchondral bone mineral density but worsened articular cartilage integrity (43). The current findings demonstrate abnormal subchondral bone in the absence of articular cartilage damage, suggesting that any effect of D2 deletion on OA pathogenesis would likely be mediated via primary effects in subchondral bone osteoblasts.
In this respect, it is essential to consider the cells in which D2 enzyme activity is actually present; chondrocytes, osteoblasts, or both cell types. In detailed studies demonstrating that functional D2 enzyme is restricted to bone-forming osteoblasts, it was shown that measurable D2 mRNA expression does not necessarily correlate with detectable enzyme activity (10). An important reason for this discrepancy is that D2 protein is labile, undergoing rapid degradation following exposure to increasing concentrations of T4 in a local negative feedback loop (13,44). A further difficulty is that available antibodies for detection of D2 by immunohistochemistry or Western blotting are insufficiently sensitive or unreliable. It has not been established, therefore, whether functional D2 is genuinely expressed in cartilage. Thus, Dio2 mRNA expression was demonstrated in growth plate cartilage but measureable enzyme activity using a high sensitivity assay could not be detected (10), whereas others have shown Dio2 mRNA expression in rat articular cartilage (14) and increased levels of Dio2 mRNA (14) and protein (28) in human articular cartilage from joints resected for end-stage OA, although in each case enzyme activity was not determined. The significance of apparently increased D2 expression in end-stage OA cartilage is therefore uncertain; it is also not known whether increased D2 expression would represent a secondary response to joint destruction or whether it might precede cartilage damage and be a causative factor in disease progression.
In this context, transgenic rats overexpressing Dio2 in chondrocytes had increased susceptibility to OA following provocation by surgical destabilization of the knee (14). Nevertheless, a causal relationship between increased D2 expression and OA susceptibility was not established because D2 activity was not determined in transgenic rats overexpressing the enzyme (14). This is an important point because in a previous study, transgenic mice overexpressing D2 in the heart surprisingly displayed only mild thyrotoxic changes in cardiomyocytes (45), presumably because the phenotype was mitigated by T4-induced degradation of the overexpressed enzyme (44). Furthermore, in a recent study, siRNA-mediated inhibition of DIO2 mRNA expression in primary human chondrocytes resulted in decreased expression of liver X receptor α (LXRα) and increased expression of interleukin-1β (IL1β) and IL1β-induced cyclooxygenase-2 (COX2) expression (46). At odds with the notion that deletion or inhibition of D2 might protect against OA, these findings indicate that suppression of D2 results in a pro-inflammatory response in cartilage. The authors suggest that D2 has an anti-inflammatory role in chondrocytes resulting in inactivation of LXRα and suppression of IL-1β and COX2 (46). Thus, the apparent increase in D2 expression observed in end-stage human OA (14,28) could represent a consequence of disease progression resulting from activation of pro-inflammatory pathways such as the nuclear factor κB (NFκB) pathway, which is known to stimulate D2 expression (47,48).
In light of these conflicting findings, it is prudent to reconsider the original hypothesis that reduced tissue T3 availability protects joints from damage and the development of OA. Genetic studies proposed DIO2 as a susceptibility gene for OA based on identification of the rs225014 (Thr92Ala) polymorphism. The functional effect of this polymorphism on enzyme activity is, however, uncertain. Initial in vitro transfection studies demonstrated the Thr92Ala substitution in D2 did not affect protein half-life, enzyme activity, or its sensitivity to substrate-mediated inhibition (49). Nevertheless, analysis of thyroid and skeletal muscle samples from subjects homozygous for the Ala risk allele revealed D2 enzyme velocity was reduced by 50% compared to heterozygous individuals or subjects homozygous for the protective Thr allele (49), although it was subsequently suggested this finding may have resulted from a methodological artifact (50,51). The initial genetic studies suggesting that reduced D2 activity is associated with increased susceptibility to OA (6) were based on the assumption that the Thr92Ala polymorphism results in reduced enzyme activity (49). However, studies showing that transgenic rats overexpressing D2 in articular cartilage had increased susceptibility to OA (14) were not consistent with this conclusion, and recent findings of allelic imbalance and increased expression of D2 protein in OA cartilage (28,29) further suggest that increased D2 activity may in fact be detrimental for articular cartilage maintenance. On the other hand, a large genome-wide association study and a recent meta-analysis both failed to identify DIO2 as a disease-susceptibility locus for OA (26,27), while a recent study in primary human chondrocytes actually suggests an anti-inflammatory role for D2 in cartilage (46). Currently, it remains unclear, therefore, whether variation in DIO2 plays a role in osteoarthritis pathogenesis, although the independent identification of DIO3 as a disease susceptibility locus (5) strongly suggests a role for control of local tissue T3 availability in the regulation of joint homeostasis.
Overall, it is not certain whether the original hypothesis that reduced tissue T3 availability protects joints from damage and the development of OA is likely to be correct or whether the reverse is more probable. Alternatively, it is possible that tissue T3 deficiency or excess are each detrimental for optimal articular cartilage homeostasis, thus accounting for the apparently contradictory literature. Indeed, OA pathophysiology is complex, with examples of increased or decreased signaling in both the Wnt/β-catenin (52,53) and transforming growth factor-β pathways (54,55) having deleterious effects on joint integrity. The finding of increased subchondral bone mineral content in D2KO mice will thus require careful study to determine its pathophysiological significance; that is, whether loss of D2 protects joints from OA or whether it results in increased disease susceptibility. Future studies will require the use of an OA provocation model, such as surgical destabilization of the medial meniscus (56), or study of the consequences of ageing to determine whether deletion of D2 accelerates or delays the onset and progression of predictable joint destruction. Additional studies to investigate the temporo-spatial specific role of D2 in cartilage could also be performed by crossing mice harboring floxed Dio2 alleles (57) with Agc1-CreERT2 mice (58) that conditionally express Cre-recombinase in cartilage following treatment with tamoxifen.
Supplementary Material
Acknowledgments
The authors thank Professors Valerie Galton (Dartmouth Medical School, Lebanon, NH) and Donald St. Germain (Maine Medical Center Research Institute, Scarborough, ME) for kindly providing samples from D2KO and D1D2KO mice, and Mahrokh Nohadani, Ann Sandison and Philippe Donatien (Imperial College London) for their generous help with histology. J.A.W. received funding for this work form an Arthritis Research UK Clinical Research Fellowship.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Arden N, Nevitt MC.2006Osteoarthritis: epidemiology. Best Pract Res Clin Rheumatol 20:3–25 [DOI] [PubMed] [Google Scholar]
- 2.Lories RJ, Luyten FP.2011The bone-cartilage unit in osteoarthritis. Nat Rev Rheumatol 7:43–49 [DOI] [PubMed] [Google Scholar]
- 3.Burr DB, Gallant MA.2012Bone remodelling in osteoarthritis. Nat Rev Rheumatol 8:665–673 [DOI] [PubMed] [Google Scholar]
- 4.Reynard LN, Loughlin J.2013Insights from human genetic studies into the pathways involved in osteoarthritis. Nat Rev Rheumatol 9:573–583 [DOI] [PubMed] [Google Scholar]
- 5.Meulenbelt I, Bos SD, Chapman K, van der Breggen R, Houwing-Duistermaat JJ, Kremer D, Kloppenburg M, Carr A, Tsezou A, Gonzalez A, Loughlin J, Slagboom PE.2011Meta-analyses of genes modulating intracellular T3 bio-availability reveal a possible role for the DIO3 gene in osteoarthritis susceptibility. Ann Rheum Dis 70:164–167 [DOI] [PubMed] [Google Scholar]
- 6.Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing-Duistermaat JJ, van der Wijk HJ, Kroon HM, Nakajima M, Ikegawa S, Uitterlinden AG, van Meurs JB, van der Deure WM, Visser TJ, Seymour AB, Lakenberg N, van der Breggen R, Kremer D, van Duijn CM, Kloppenburg M, Loughlin J, Slagboom PE.2008Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet 17:1867–1875 [DOI] [PubMed] [Google Scholar]
- 7.Loughlin J.2011Osteoarthritis year 2010 in review: genetics. Osteoarthritis Cartilage 19:342–345 [DOI] [PubMed] [Google Scholar]
- 8.Bos SD, Slagboom PE, Meulenbelt I.2008New insights into osteoarthritis: early developmental features of an ageing-related disease. Curr Opinion Rheum 20:553–559 [DOI] [PubMed] [Google Scholar]
- 9.Goldring MB.2013Insight into the function of DIO2, a susceptibility gene in human osteoarthritis, as an inducer of cartilage damage in a rat model: is there a role for chondrocyte hypertrophy? Osteoarthritis Cartilage 21:643–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams AJ, Robson H, Kester MH, van Leeuwen JP, Shalet SM, Visser TJ, Williams GR.2008Iodothyronine deiodinase enzyme activities in bone. Bone 43:126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng SY, Leonard JL, Davis PJ.2010Molecular aspects of thyroid hormone actions. Endocr Rev 31:139–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galton VA, Schneider MJ, Clark AS, St Germain DL.2009Life without thyroxine to 3,5,3′-triiodothyronine conversion: studies in mice devoid of the 5′-deiodinases. Endocrinology 150:2957–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC.2008Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev 29:898–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagase H, Nagasawa Y, Tachida Y, Sakakibara S, Okutsu J, Suematsu N, Arita S, Shimada K.2013Deiodinase 2 upregulation demonstrated in osteoarthritis patients cartilage causes cartilage destruction in tissue-specific transgenic rats. Osteoarthritis Cartilage 21:514–523 [DOI] [PubMed] [Google Scholar]
- 15.Waung JA, Bassett JH, Williams GR.2012Thyroid hormone metabolism in skeletal development and adult bone maintenance. Trends Endocrinol Metab 23:155–162 [DOI] [PubMed] [Google Scholar]
- 16.Dreier R.2010Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res Ther 12:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin AC, Seeto BL, Bartoszko JM, Khoury MA, Whetstone H, Ho L, Hsu C, Ali SA, Alman BA.2009Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat Med 15:1421–1425 [DOI] [PubMed] [Google Scholar]
- 18.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, Flannery CR, Peluso D, Kanki K, Yang Z, Majumdar MK, Morris EA.2005Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434:644–648 [DOI] [PubMed] [Google Scholar]
- 19.Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, Little CB, Last K, Farmer PJ, Campbell IK, Fourie AM, Fosang AJ.2005ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 434:648–652 [DOI] [PubMed] [Google Scholar]
- 20.Billinghurst RC, Dahlberg L, Ionescu M, Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O, Tschesche H, Chen J, Van Wart H, Poole AR.1997Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest 99:1534–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen-An P, Andreassen KV, Henriksen K, Karsdal MA, Bay-Jensen AC.2013Investigation of chondrocyte hypertrophy and cartilage calcification in a full-depth articular cartilage explants model. Rheumatol Int 33:401–411 [DOI] [PubMed] [Google Scholar]
- 22.Himeno M, Enomoto H, Liu W, Ishizeki K, Nomura S, Kitamura Y, Komori T.2002Impaired vascular invasion of Cbfa1-deficient cartilage engrafted in the spleen. J Bone Miner Res 17:1297–1305 [DOI] [PubMed] [Google Scholar]
- 23.Makihira S, Yan W, Murakami H, Furukawa M, Kawai T, Nikawa H, Yoshida E, Hamada T, Okada Y, Kato Y.2003Thyroid hormone enhances aggrecanase-2/ADAM-TS5 expression and proteoglycan degradation in growth plate cartilage. Endocrinology 144:2480–2488 [DOI] [PubMed] [Google Scholar]
- 24.Pereira RC, Jorgetti V, Canalis E.1999Triiodothyronine induces collagenase-3 and gelatinase B expression in murine osteoblasts. Am J Physiol 277:E496–504 [DOI] [PubMed] [Google Scholar]
- 25.Robson H, Siebler T, Stevens DA, Shalet SM, Williams GR.2000Thyroid hormone acts directly on growth plate chondrocytes to promote hypertrophic differentiation and inhibit clonal expansion and cell proliferation. Endocrinology 141:3887–3897 [DOI] [PubMed] [Google Scholar]
- 26.Kerkhof HJ, Lories RJ, Meulenbelt I, Jonsdottir I, Valdes AM, Arp P, Ingvarsson T, Jhamai M, Jonsson H, Stolk L, Thorleifsson G, Zhai G, Zhang F, Zhu Y, van der Breggen R, Carr A, Doherty M, Doherty S, Felson DT, Gonzalez A, Halldorsson BV, Hart DJ, Hauksson VB, Hofman A, Ioannidis JP, Kloppenburg M, Lane NE, Loughlin J, Luyten FP, Nevitt MC, Parimi N, Pols HA, Rivadeneira F, Slagboom EP, Styrkarsdottir U, Tsezou A, van de Putte T, Zmuda J, Spector TD, Stefansson K, Uitterlinden AG, van Meurs JB.2010A genome-wide association study identifies an osteoarthritis susceptibility locus on chromosome 7q22. Arthritis Rheum 62:499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evangelou E, Kerkhof HJ, Styrkarsdottir U, Ntzani EE, Bos SD, Esko T, Evans DS, Metrustry S, Panoutsopoulou K, Ramos YF, Thorleifsson G, Tsilidis KK, Arden N, Aslam N, Bellamy N, Birrell F, Blanco FJ, Carr A, Chapman K, Day-Williams AG, Deloukas P, Doherty M, Engstrom G, Helgadottir HT, Hofman A, Ingvarsson T, Jonsson H, Keis A, Keurentjes JC, Kloppenburg M, Lind PA, McCaskie A, Martin NG, Milani L, Montgomery GW, Nelissen RG, Nevitt MC, Nilsson PM, Ollier WE, Parimi N, Rai A, Ralston SH, Reed MR, Riancho JA, Rivadeneira F, Rodriguez-Fontenla C, Southam L, Thorsteinsdottir U, Tsezou A, Wallis GA, Wilkinson JM, Gonzalez A, Lane NE, Lohmander LS, Loughlin J, Metspalu A, Uitterlinden AG, Jonsdottir I, Stefansson K, Slagboom PE, Zeggini E, Meulenbelt I, Ioannidis JP, Spector TD, van Meurs JB, Valdes AM.2014A meta-analysis of genome-wide association studies identifies novel variants associated with osteoarthritis of the hip. Ann Rheum Dis 73:2130–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bos SD, Bovee JV, Duijnisveld BJ, Raine EV, van Dalen WJ, Ramos YF, van der Breggen R, Nelissen RG, Slagboom PE, Loughlin J, Meulenbelt I.2012Increased type II deiodinase protein in OA-affected cartilage and allelic imbalance of OA risk polymorphism rs225014 at DIO2 in human OA joint tissues. Ann Rheum Dis 71:1254–1258 [DOI] [PubMed] [Google Scholar]
- 29.Bomer N, den Hollander W, Ramos YF, Bos SD, van der Breggen R, Lakenberg N, Pepers BA, van Eeden AE, Darvishan A, Tobi EW, Duijnisveld BJ, van den Akker EB, Heijmans BT, van Roon-Mom WM, Verbeek FJ, van Osch GJ, Nelissen RG, Slagboom PE, Meulenbelt I.2014Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann Rheum Dis [Epub ahead of print]; DOI: 10.1136/annrheumdis-2013-204739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waarsing JH, Kloppenburg M, Slagboom PE, Kroon HM, Houwing-Duistermaat JJ, Weinans H, Meulenbelt I.2011Osteoarthritis susceptibility genes influence the association between hip morphology and osteoarthritis. Arthritis Rheum 63:1349–1354 [DOI] [PubMed] [Google Scholar]
- 31.Panicker V, Cluett C, Shields B, Murray A, Parnell KS, Perry JR, Weedon MN, Singleton A, Hernandez D, Evans J, Durant C, Ferrucci L, Melzer D, Saravanan P, Visser TJ, Ceresini G, Hattersley AT, Vaidya B, Dayan CM, Frayling TM.2008A common variation in deiodinase 1 gene DIO1 is associated with the relative levels of free thyroxine and triiodothyronine. J Clin Endocrinol Metab 93:3075–3081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider MJ, Fiering SN, Thai B, Wu SY, St Germain E, Parlow AF, St Germain DL, Galton VA.2006Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology 147:580–589 [DOI] [PubMed] [Google Scholar]
- 33.Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA.2001Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol 15:2137–2148 [DOI] [PubMed] [Google Scholar]
- 34.Bassett JH, Boyde A, Howell PG, Bassett RH, Galliford TM, Archanco M, Evans H, Lawson MA, Croucher P, St Germain DL, Galton VA, Williams GR.2010Optimal bone strength and mineralization requires the type 2 iodothyronine deiodinase in osteoblasts. Proc Natl Acad Sci U S A 107:7604–7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glasson SS, Chambers MG, Van Den Berg WB, Little CB.2010The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18:S17–23 [DOI] [PubMed] [Google Scholar]
- 36.Sondergaard BC, Catala-Lehnen P, Huebner AK, Bay-Jensen AC, Schinke T, Henriksen K, Schilling S, Haberland M, Nielsen RH, Amling M, Karsdal MA.2012Mice over-expressing salmon calcitonin have strongly attenuated osteoarthritic histopathological changes after destabilization of the medial meniscus. Osteoarthritis Cartilage 20:136–143 [DOI] [PubMed] [Google Scholar]
- 37.Hoegh-Andersen P, Tanko LB, Andersen TL, Lundberg CV, Mo JA, Heegaard AM, Delaisse JM, Christgau S.2004Ovariectomized rats as a model of postmenopausal osteoarthritis: validation and application. Arthritis Res Ther 6:R169–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waung JA, Maynard SA, Gopal S, Gogakos A, Logan JG, Williams GR, Bassett JH.2014Quantitative X-ray microradiography for high-throughput phenotyping of osteoarthritis in mice. Osteoarthritis Cartilage 22:1396–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bassett JH, van der Spek A, Gogakos A, Williams GR.2012Quantitative X-ray imaging of rodent bone by Faxitron. Methods Mol Biol 816:499–506 [DOI] [PubMed] [Google Scholar]
- 40.Burr DB, Schaffler MB.1997The involvement of subchondral mineralized tissues in osteoarthrosis: quantitative microscopic evidence. Microsc Res Tech 37:343–357 [DOI] [PubMed] [Google Scholar]
- 41.van der Kraan PM, van den Berg WB.2012Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage 20:223–232 [DOI] [PubMed] [Google Scholar]
- 42.Hayami T, Zhuo Y, Wesolowski GA, Pickarski M, Duong le T.2012Inhibition of cathepsin K reduces cartilage degeneration in the anterior cruciate ligament transection rabbit and murine models of osteoarthritis. Bone 50:1250–1259 [DOI] [PubMed] [Google Scholar]
- 43.Orth P, Cucchiarini M, Wagenpfeil S, Menger MD, Madry H.2014PTH [1–34]-induced alterations of the subchondral bone provoke early osteoarthritis. Osteoarthritis Cartilage 22:813–821 [DOI] [PubMed] [Google Scholar]
- 44.Dentice M, Bandyopadhyay A, Gereben B, Callebaut I, Christoffolete MA, Kim BW, Nissim S, Mornon JP, Zavacki AM, Zeold A, Capelo LP, Curcio-Morelli C, Ribeiro R, Harney JW, Tabin CJ, Bianco AC.2005The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol 7:698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carvalho-Bianco SD, Kim BW, Zhang JX, Harney JW, Ribeiro RS, Gereben B, Bianco AC, Mende U, Larsen PR.2004Chronic cardiac-specific thyrotoxicosis increases myocardial beta-adrenergic responsiveness. Mol Endocrinol 18:1840–1849 [DOI] [PubMed] [Google Scholar]
- 46.Cheng AW, Bolognesi M, Kraus VB.2012DIO2 modifies inflammatory responses in chondrocytes. Osteoarthritis Cartilage 20:440–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roman-Blas JA, Jimenez SA.2006NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage 14:839–848 [DOI] [PubMed] [Google Scholar]
- 48.Zeold A, Doleschall M, Haffner MC, Capelo LP, Menyhert J, Liposits Z, da Silva WS, Bianco AC, Kacskovics I, Fekete C, Gereben B.2006Characterization of the nuclear factor-kappa B responsiveness of the human dio2 gene. Endocrinology 147:4419–4429 [DOI] [PubMed] [Google Scholar]
- 49.Canani LH, Capp C, Dora JM, Meyer EL, Wagner MS, Harney JW, Larsen PR, Gross JL, Bianco AC, Maia AL.2005The type 2 deiodinase A/G (Thr92Ala) polymorphism is associated with decreased enzyme velocity and increased insulin resistance in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 90:3472–3478 [DOI] [PubMed] [Google Scholar]
- 50.Acosta BM, Bianco AC.2010New insights into thyroid hormone replacement therapy. F1000 Med Rep 2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larsen PR.2009Type 2 iodothyronine deiodinase in human skeletal muscle: new insights into its physiological role and regulation. J Clin Endocrinol Metab 94:1893–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, Rosier RN, O'Keefe RJ, Zuscik M, Chen D.2009Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res 24:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu M, Chen M, Zuscik M, Wu Q, Wang YJ, Rosier RN, O'Keefe RJ, Chen D.2008Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum 58:2053–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen J, Li J, Wang B, Jin H, Wang M, Zhang Y, Yang Y, Im HJ, O'Keefe R, Chen D.2013Deletion of the transforming growth factor beta receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum 65:3107–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC, Askin FB, Frassica FJ, Chang W, Yao J, Carrino JA, Cosgarea A, Artemov D, Chen Q, Zhao Z, Zhou X, Riley L, Sponseller P, Wan M, Lu WW, Cao X.2013Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med 19:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glasson SS, Blanchet TJ, Morris EA.2007The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15:1061–1069 [DOI] [PubMed] [Google Scholar]
- 57.Fonseca TL, Correa-Medina M, Campos MP, Wittmann G, Werneck-de-Castro JP, Arrojo e Drigo R, Mora-Garzon M, Ueta CB, Caicedo A, Fekete C, Gereben B, Lechan RM, Bianco AC.2013Coordination of hypothalamic and pituitary T3 production regulates TSH expression. J Clin Invest 123:1492–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B.2009Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis 47:805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.