

Abstract
Mucopolysaccharidosis type III (MPS III), or Sanfilippo disease, is a neurodegenerative lysosomal storage disease (LSD) caused by defective lysosomal degradation of heparan sulfate (HS). No effective disease-modifying therapy is yet available. In contrast to some other neuronopathic LSDs, bone marrow-derived hematopoietic stem cell transplantation (HSCT) fails to prevent neurological deterioration in MPS III patients. We report on the 5-year outcome of early transplantation, i.e., before onset of clinical neurological disease, in combination with the use of umbilical cord blood-derived hematopoietic stem cells (UCBT), in two MPS III patients. Both patients had a normal developmental quotient at the time of UCBT. One patient had a combination of mutations predicting a classical severe phenotype (MPS IIIA), and one patient (MPS IIIB) had mutations predicting a very attenuated phenotype. Transplantation was uncomplicated with full engraftment of donor cells in both.
Both patients showed progressive neurological deterioration with regression of cognitive skills and behavioral disturbances during 5 years after successful UCBT, comparable to the natural history of patients with the same combination of mutations. The concentration of HS in CSF in the patient with the attenuated phenotype of MPS IIIB 2 years after UCBT was very high and in the range of untreated MPS III patients.
We conclude that the course of cognitive development, behavioral problems, and absence of biochemical correction in CSF demonstrate the absence of relevant effect of UCBT in MPS III patients, even when performed before clinical onset of CNS disease.
Introduction
Mucopolysaccharidosis type III (MPS III), or Sanfilippo disease, is a rare autosomal recessive lysosomal storage disease, comprising of four subtypes, types A–D, each caused by a specific enzyme deficiency and all involved in the degradation of the glycosaminoglycan (GAG) heparan sulfate (HS). MPS IIIA (OMIM #252900), caused by a deficiency of heparan-N-sulfamidase (NS, EC 3.10.1.1), and MPS IIIB (OMIM #252920), caused by a deficiency of a-N-acetylglucosaminidase (NAGLU, EC: 3.2.1.50), are the most common subtypes (Valstar et al. 2008). Clinically, all subtypes are characterized by the same signs and symptoms. The most prominent symptom in MPS III is progressive neurocognitive impairment after an initial symptom-free interval of 1–4 years, ultimately leading to severe dementia and early death (Valstar et al. 2008). Behavioral difficulties are an early symptom and consist of hyperactivity, impulsivity, obstinacy, anxious behaviors, and autistic-like behaviors. Other symptoms include sleeping problems; frequent diarrhea; ear, nose, and throat infections; and conductive hearing loss. Death usually follows in the second or third decade of life. However, in recent years, a remarkably broad phenotypic spectrum, with attenuated patients living well into adulthood and expressing a developmental delay that may remain stable for many years, has been recognized (Valstar et al. 2010a). To date, there is no disease-modifying treatment available for any of the subtypes of Sanfilippo disease.
Hematopoietic stem cell transplantation (HSCT) is the treatment of choice for patients with the severe, neuronopathic, Hurler phenotype of MPS I (MPS I-H) when performed early, i.e., before the age of 2.5 years (de Ru et al. 2011), as HSCT may prevent or halt cognitive decline (Peters et al. 1996, 1998). HSCT for MPS I-H and other neuronopathic lysosomal storage disorders, including metachromatic leukodystrophy and Krabbe disease, is based on the engraftment of non-affected hematopoietic stem cells in various tissues including the CNS (Boelens et al. 2010). Presumably, donor-derived monocytes can cross the blood–brain barrier and differentiate into microglia cells. These microglia cells may secrete lysosomal enzymes into the extracellular space leading to cross-correction of the enzyme deficiency in neuronal cells (Krivit et al. 1995). Early transplantation, before the clinical onset of irreversible central nervous system (CNS) disease, is key for successful UCBT in neuronopathic lysosomal storage disorders. However, in MPS III patients, HSCT with bone marrow as the stem cell source (bone marrow transplantation, BMT) failed to prevent neurological deterioration (Klein et al. 1995). This might be caused by the relatively late timing of transplantation as diagnosis before clinical symptoms of CNS disease is exceptional due to the fact that first symptoms are almost invariably due to CNS disease. Early HSCT, using bone marrow as donor source, in MPS III has been reported in two neurologically unaffected siblings with MPS IIIB, both <2 years of age (Vellodi et al. 1992) and in one asymptomatic MPS IIIA patient, aged <1 year (Sivakumur and Wraith 1999), all diagnosed because of an affected older sibling. Unfortunately, neurological deterioration was not prevented by the early transplantation. This lack of efficacy might be due to the stem cell source, as in all three transplants a (carrier) parent was used as donor. The lower enzyme activity in the heterozygous donor cells may have impacted on the efficacy of enzymatic cross-correction, as has been demonstrated for MPS I (Wynn et al. 2009). Indeed, a more recent study suggested a moderate positive effect of UCBT on CNS disease in MPS III patients, with a more prominent effect in patients that were transplanted at a younger age (Prasad et al. 2008).
We report on the outcome in two MPS III patients (one with MPS IIIA and one with MPS IIIB) who had a successful UCBT at an age of 2.5 and 3 years, respectively, before the onset of neurological symptoms.
Case Histories
The experimental nature of the UCBT in MPS III was discussed with the parents of both children, and informed consent was obtained. The experimental interventions of UCBT in MPS III were approved by the local IRB.
Patient A
This male patient was born after an uncomplicated pregnancy and delivery as the first child of non-consanguineous parents. He had frequent ENT infections and, at the age of 2 years and 3 months, he was evaluated because of a cardiac murmur detected during a routine physical examination. He was found to have an enlarged liver, and cardiac ultrasound revealed significant mitral valve insufficiency, a ventricular septal defect and as a result dilatation of the left ventricle. A month later he was admitted to the Pediatric Intensive Care Unit (PICU) because of a cardiovascular collapse due to cardiac failure aggravated by supraventricular tachycardia. Because of the presence of mild coarse facial features in combination with hepatomegaly and cardiac defects, a metabolic screening for inborn metabolic diseases was performed, with a focus on lysosomal storage diseases. This led to the enzymatic diagnosis of MPS IIIA. Mutation analysis showed compound heterozygosity for the p.R245H (c.734G>A) and p.Q380R (c.1139A>G) mutations. Both mutations are associated with the classical severe phenotype (Weber et al. 1997; Valstar et al. 2010b). Testing at the age of 25 months revealed a normal neurocognitive development (GQ 106 on the Griffith scale). At the age of 30 months, a UCBT was carried out.
Patient B
This male patient was born as the second child of non-consanguineous parents after an uncomplicated pregnancy and delivery. He had frequent upper airway infections and an adenotonsillectomy with placement of grommets done at the age of 13 months. At the age of 19 months, hepatomegaly was found on routine physical examination. Metabolic screening led to the enzymatic diagnosis MPS IIIB. Mutation analysis showed compound heterozygosity for the p.R297X (c.889C>T) and the p.S612G (c.1834A>G) mutations. This combination of mutations has been reported previously in two brothers with an attenuated phenotype of MPS IIIB (Valstar et al. 2010a). Neurocognitive testing at the age of 21 months demonstrated a normal cognitive development (DQ 94, BSID-II-NL). At the age of 25 months, a UCBT was carried out.
Materials and Methods
Umbilical Cord Blood Transplantation (UCBT)
The conditioning regime in both patients consisted of busulfan (day −9 until day −7; cumulative dose 360 mg/m2), cyclophosphamide (day −5 until day −2; cumulative dose 200 mg/kg), and thymoglobulin (day −4 until day −2; cumulative dose 10 mg/kg). Ciclosporin (blood level 0.20–0.25 mg/L, started day −1, phased out from day 28) and prednisone (1 mg/kg/day; started day 0, phased out from day 28) were used as graft-versus-host disease prophylaxis. Donor specifications are as follows: patient A cord blood 6/6, DR mismatch, ABO incompatibility; patient B cord blood 5/6, DR mismatch, ABO incompatibility.
Donor Cell Engraftment
Full donor chimerism (on whole blood) was defined as the presence of >95% donor-derived hematopoietic cells. Neutrophil engraftment was defined as the first day of achieving an absolute neutrophil count of ≥0.5 × 109/L for 3 consecutive days.
Glycosaminoglycans in Urine and Cerebrospinal Fluid (CSF)
Urine
Total GAGs in urine were measured by the DMB test which involves binding of GAGs to the dye dimethylene blue and subsequent spectrophotometric analysis of the GAG-DMB complex (de Jong et al. 1992).
CSF
Heparan sulfate levels in liquor were analyzed essentially as described by de Ru and coworkers (de Ru et al. 2013). In short, 25 μL of CSF was incubated with 5 mIU each of heparinase I, II, and III in 100 mM NH4Ac (pH 7.0), 10 mM Ca(Ac)2, and 2 mM DTT at 30°C for 2 h for digestion of heparan sulfate into disaccharides (final volume of 150 mL). Next, 125 ng of internal standard (4UA-2S-GlcNCOEt-6S, HD009, Iduron) in 15 μL of 150 mM EDTA (pH 7.0) was added. Samples were boiled for 5 min and centrifuged at 20,000 × g for 5 min and the supernatant was centrifuged at 14,000 × g for 15 min at 25°C over an Amicon Ultra 30 K centrifugal filter (Millipore). The disaccharides in the filtrate were quantified on a Waters Quattro Premier XE (tandem) mass spectrometer (Waters Corporation, Milford, MA, USA) coupled to an ACQUITY UPLC system. All samples were digested and analyzed in triplicate. The concentrations of D0A0, D0S0, D0A6+D2A0, and D0S6+D2S0 (following the nomenclature of (Lawrence et al. 2008)) were calculated using a calibration curve for each of the disaccharides. The HS level in liquor was calculated as the sum of these disaccharides.
Neurocognitive Testing
Developmental testing was repeatedly performed in both patients within the scope of care. Different tests were used to assess neurocognitive development, appropriate for the developmental age of the child: the second edition of the Bayley Scales of Infant Development (BSID-II), which is the most recent version of the Bayley Scales that is available in Dutch (Van der Meulen et al. 2002), the Griffiths Mental Development Scales (Griffiths 1970), the Snijders-Oomen Nonverbal Intelligence Test 2.5–7 (SON-R) (Moore et al. 1998), the Vineland Adaptive Behavior Scales survey version (VABS) (De Bildt and Kraijer 2003), and the shorter Vineland Screener (VABS-S) (Scholte et al. 2008).
The results of the different tests were expressed as a Developmental Quotient (DQ). DQ was calculated as (age equivalent (AE) in months/age in months)*100. This DQ is comparable to IQ, with a mean score of 100 in the case of normal development.
Cardiac Surgery in Patient A
Successful mitral valve repair was performed in patient A at the age of 3 years.
Results
UCBT
The transplantation procedure was well tolerated in both patients, and no serious complications or side effects occurred.
Donor Cell Engraftment
The chimerism posttransplantation of patient A was 100% on day 15 and at 11 months and 5 years after transplantation. The chimerism in patient B was 96% on day 12 and 100% 4 months and 2.5 years after transplantation.
Neutrophil counts of > 0.5 × 109/L for 3 consecutive days were achieved on day +13 in patient A and on day +25 in patient B.
Enzyme activities in lymphocytes were measured in patient A and patient B at 5 years and 5 months and 2.5 years after transplantation, respectively, and were all normal.
GAG Levels
The urinary GAG excretion was increased in both patients prior to transplantation and was found to be normal at 5 months and 5 years after HSCT in patient A and at 5 years after HSCT in patient B.
Heparan sulfate in CSF was measured only in patient B, 2 years after HSCT, and was found to be increased (2,035 ng/mL; range in MPS III patients: 809–2,261 (n = 6); range in healthy controls: 36–181 ng/mL (n = 16)).
Cognitive Development
The developmental quotient shortly before HSCT was normal in both patients (Table 1). The course of DQ after transplantation is presented in Table 1 and Fig. 1.
Table 1.
Cognitive development in patients A and B. AE: age equivalent; DQ: developmental quotient
| Age (months) | Test (scale) | AE (months) | DQ |
|---|---|---|---|
| Patient A | |||
| 29 | Griffith scales | – | 106 |
| 45 | BSID-II | 17 | 38 |
| 56 | BSID-II | 15 | 27 |
| 68 | VABS | 23 | 34 |
| 92 | VABS | 19 | 21 |
| Patient B | |||
| 21 | BSID-II | 20 | 95 |
| 33 | BSID-II | 23 | 70 |
| 39 | SON-R | 29 | 74 |
| 45 | VABS | 31 | 69 |
| 58 | SON-R | 39 | 67 |
| 83 | SON-R | 49 | 59 |
| VABS | 48 | 58 | |
Fig. 1.
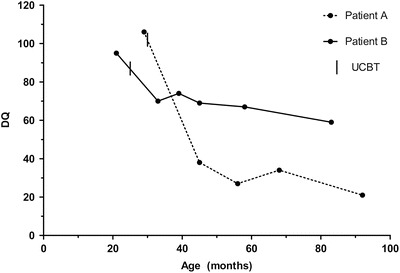
Developmental quotient in patients A and B relative to the UCBT. DQ: developmental quotient
Behavior
No behavioral problems were noted in both patients before UCBT. Both patients developed hyperactivity, temper tantrums, and a short attention span in the years following transplantation. Both patients have used risperidone for control of the behavioral problems.
Clinical Course
Patient A developed severe mitral valve insufficiency with left ventricular failure, for which a mitral valve repair was performed.
Patient B developed no somatic symptoms of interest.
Discussion
We show that early, i.e., before the clinical onset of CNS disease, and successful umbilical cord stem cell transplantation (UCBT) in two patients with MPS III did not significantly influence the course of the neurological disease. It is not clear why HSCT failed to correct the neurological disease in MPS IIIA and IIIB patients. Indeed, this lack of efficacy is in strong contrast with the results observed in patients with the severe phenotype of MPS I (MPS I-H). A potential explanation is that the donor-derived microglia cells secrete insufficient amounts of enzymes for cross-correction of the neuronal tissue in MPS III (Shapiro et al. 1995; Sivakumur and Wraith 1999). A recent study in MPS IIIA mice indeed showed that ex vivo lentiviral-mediated HSCT, probably resulting in high expression of SGSH, normalized heparan sulfate storage in brain and significantly improved neuroinflammation and behavior, while wild-type HSCT did not (Langford-Smith et al. 2012).
Our observations demonstrate that it is essential to take into account the predicted severity of the phenotype when assessing efficacy of a therapeutic intervention in MPS III patients. While the clinical course of the disease in patient A (MPS IIIA) essentially followed the course observed in patients with the classical phenotype of Sanfilippo disease (Buhrman et al. 2014), the disease in patient B (MPS IIIB) showed a much more attenuated course and his cognitive development still progressed at the age of 5 years. This may erroneously lead to a conclusion of clinical efficacy of the UCBT in this patient. However, the mutation combination in this patient has been reported previously in two brothers (Valstar et al. 2010a). One of them died at the age of 41 due to pneumonia. The other patient is still alive at the age of 52 years. He could still walk and read small sentences at the age of 48 years. The disease course in patient B closely resembles the clinical history of these brothers, and we can therefore conclude that the UCBT in this patient was ineffective. Mutations and mutation combinations conveying an attenuated phenotype have been reported frequently in MPS IIIB (Weber et al. 1999; Valstar et al. 2010a) and, though more rare, also occur in MPS IIIA (Meyer et al. 2008; Valstar et al. 2010b).
The complete normalization of the urinary GAG excretion in both patients suggests a full biochemical correction of the somatic enzymatic deficiency by the UCBT. Our observation that the HS concentration in CSF was still very high 2 years after transplantation in patient B shows that even full engraftment after UCBT does not lead to biochemical correction of the disease in the CSF which may explain the lack of clinical efficacy. The lack of clinical efficacy in both patients is further supported by the fact that both developed the typical behavioral problems associated with MPS III in the years following the UCBT.
Our study has some limitations. The patients were not studied in a single prospective study, and we did not use a standardized complete protocol for neurocognitive testing, and testing was done at different sites. In addition, no complete standardized assessment of behavior and sleep difficulties was done. This might have resulted in a less precise assessment of the potential benefits of the transplantation. However, the course of cognitive development in combination with the severe behavioral difficulties in both patients in combination with the high concentration of HS in CSF in patient B demonstrates that efficacy of UCBT in Sanfilippo disease is at its best very limited. Finally, we cannot exclude that a transplantation earlier in life might lead to clinical benefit in patients with MPS III. However, a very early diagnosis in MPS III is very rare and in general only possible in case of an older sibling with MPS III.
Several studies on alternative treatment strategies for MPS III are ongoing, including intrathecal enzyme replacement therapy in MPS IIIA (clinicaltrials.gov, identifier: NCT01299727), gene therapy in MPS IIIA (clinicaltrials.gov, identifier: NCT01474343), and high dose genistein in MPS IIIA, B, and C (public.ukcrn.org.uk; UKCRN ID 16209). Given the failure of early UCBT in MPS III and the unmet clinical need in this severe neurodegenerative disease, these studies are of the highest importance.
Compliance with Ethics Guidelines
Conflicts of Interest
Lindsey Welling, Jaap Jan Boelens, Jan Pieter Marchal, Peter van Hasselt Ans T. van der Ploeg, and Frits A. Wijburg declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from the parents of the patients included in the study. Additional informed consent was obtained for all patients for which identifying information is included in this article.
Contributions of the Individual Authors
Lindsey Welling: reporting
Jaap Jan Boelens: treating physician, coordinating the performed examinations
Jan Pieter Marchal: interpreting neuropsychological test results, reporting
Peter van Hasselt: treating physician, reporting
Ans T. van der Ploeg: reporting
Frits A. Wijburg: reporting
Footnotes
Competing interests: None declared
Contributor Information
Frits A. Wijburg, Email: f.a.wijburg@amc.uva.nl
Collaborators: Johannes Zschocke, Matthias Baumgartner, K Michael Gibson, Marc Patterson, and Shamima Rahman
References
- De Bildt AA, Kraijer DW. Vineland-Z (Handleiding) Leiden: PITS BV; 2003. [Google Scholar]
- Boelens JJ, Prasad VK, Tolar J, et al. Current international perspectives on hematopoietic stem cell transplantation for inherited metabolic disorders. Pediatr Clin North Am. 2010;57:123–145. doi: 10.1016/j.pcl.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Buhrman D, Thakkar K, Poe M, Escolar ML. Natural history of Sanfilippo syndrome type A. J Inherit Metab Dis. 2014;37:431–437. doi: 10.1007/s10545-013-9661-8. [DOI] [PubMed] [Google Scholar]
- Griffiths R. The abilities of young children. London: Child Development Research Center; 1970. [Google Scholar]
- De Jong JG, Wevers RA, Liebrand-van Sambeek R. Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidoses based on dimethylmethylene blue. Clin Chem. 1992;38:803–807. [PubMed] [Google Scholar]
- Klein K, Krivit W, Whitley C et al. (1995) Poor cognitive outcome of eleven children with Sanfilippo syndrome after bone marrow transplantation and successful engraftment. Bone Marrow Trans 15(SUPPL):S176–S181
- Krivit W, Ho Sung J, Shapiro EG, Lockman LA. Microglia: the effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transplant. 1995;4:385–392. doi: 10.1016/0963-6897(95)00021-O. [DOI] [PubMed] [Google Scholar]
- Langford-Smith A, Wilkinson FL, Langford-Smith KJ, et al. Hematopoietic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol Ther. 2012;20:1610–1621. doi: 10.1038/mt.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence R, Lu H, Rosenberg R, et al. Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat Methods. 2008;5:291–292. doi: 10.1038/nmeth0408-291. [DOI] [PubMed] [Google Scholar]
- Van der Meulen B, Ruiter S, Lutje Spelberg H, Smrkovskỳ M. BSID-II-NL, Dutch manual. Lisse: Swets; 2002. [Google Scholar]
- Meyer A, Kossow K, Gal A, et al. The mutation p.Ser298Pro in the sulphamidase gene (SGSH) is associated with a slowly progressive clinical phenotype in mucopolysaccharidosis type IIIA (Sanfilippo a syndrome) Hum Mutat. 2008;29:770. doi: 10.1002/humu.20738. [DOI] [PubMed] [Google Scholar]
- Moore C, O’Keefe S, Lawhown D, Tellegen P. Concurrent validity of the Snijders-Oomen nonverbal intelligence test 2 1/2-7-revised with the Wechsler Preschool and primary scale of intelligence-revised. Psychol Rep. 1998;82:619–625. doi: 10.2466/pr0.1998.82.2.619. [DOI] [Google Scholar]
- Peters C, Balthazor M, Shapiro EG, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood. 1996;87:4894–4902. [PubMed] [Google Scholar]
- Peters C, Shapiro EG, Anderson J et al. (1998) Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. 2601–2608 [PubMed]
- Prasad VK, Mendizabal A, Parikh SH, et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: influence of cellular composition of the graft on transplantation outcomes. Blood. 2008;112:2979–2989. doi: 10.1182/blood-2008-03-140830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ru MH, Boelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis. 2011;6:55. doi: 10.1186/1750-1172-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ru MH, van der Tol L, van Vlies N, et al. Plasma and urinary levels of dermatan sulfate and heparan sulfate derived disaccharides after long-term enzyme replacement therapy (ERT) in MPS I: correlation with the timing of ERT and with total urinary excretion of glycosaminoglycans. J Inherit Metab Dis. 2013;36:247–255. doi: 10.1007/s10545-012-9538-2. [DOI] [PubMed] [Google Scholar]
- Scholte E, Van Duijn G, Dijkxhoorn Y, et al. Vineland screener 0–6 years: manual of the Dutch adaptation. Leiden: PITS; 2008. [Google Scholar]
- Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J Inherit Metab Dis. 1995;18:413–429. doi: 10.1007/BF00710053. [DOI] [PubMed] [Google Scholar]
- Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: a comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis. 1999;22:849–850. doi: 10.1023/A:1005526628598. [DOI] [PubMed] [Google Scholar]
- Valstar MJ, Bruggenwirth HT, Olmer R, et al. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis. 2010;33:759–767. doi: 10.1007/s10545-010-9199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valstar MJ, Neijs S, Bruggenwirth HT, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010;68:876–887. doi: 10.1002/ana.22092. [DOI] [PubMed] [Google Scholar]
- Valstar MJ, Ruijter GJG, van Diggelen OP, et al. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis. 2008;31:240–252. doi: 10.1007/s10545-008-0838-5. [DOI] [PubMed] [Google Scholar]
- Vellodi A, Young E, New M, et al. Bone marrow transplantation for Sanfilippo disease type B. J Inherit Metab Dis. 1992;15:911–918. doi: 10.1007/BF01800232. [DOI] [PubMed] [Google Scholar]
- Weber B, Guo XH, Kleijer WJ, et al. Sanfilippo type B syndrome (mucopolysaccharidosis III B): allelic heterogeneity corresponds to the wide spectrum of clinical phenotypes. Eur J Hum Genet. 1999;7:34–44. doi: 10.1038/sj.ejhg.5200242. [DOI] [PubMed] [Google Scholar]
- Weber B, Guo XH, Wraith JE, et al. Novel mutations in Sanfilippo A syndrome: implications for enzyme function. Hum Mol Genet. 1997;6:1573–1579. doi: 10.1093/hmg/6.9.1573. [DOI] [PubMed] [Google Scholar]
- Wynn RF, Wraith JE, Mercer J, et al. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J Pediatr. 2009;154:609–611. doi: 10.1016/j.jpeds.2008.11.005. [DOI] [PubMed] [Google Scholar]