

Abstract
Pathogenic mycobacteria are difficult to culture, requiring specialized media and a long incubation time, and have complex and exceedingly robust cell walls. Mycobacterium avium subsp. paratuberculosis (MAP), the causative agent of Johne's disease, a chronic wasting disease of ruminants, is a typical example. Culture of MAP from the feces and intestinal tissues is a commonly used test for confirmation of infection. Liquid medium offers greater sensitivity than solid medium for detection of MAP; however, support for the BD Bactec 460 system commonly used for this purpose has been discontinued. We previously developed a new liquid culture medium, M7H9C, to replace it, with confirmation of growth reliant on PCR. Here, we report an efficient DNA isolation and quantitative PCR methodology for the specific detection and confirmation of MAP growth in liquid culture media containing egg yolk. The analytical sensitivity was at least 104-fold higher than a commonly used method involving ethanol precipitation of DNA and conventional PCR; this may be partly due to the addition of a bead-beating step to manually disrupt the cell wall of the mycobacteria. The limit of detection, determined using pure cultures of two different MAP strains, was 100 to 1,000 MAP organisms/ml. The diagnostic accuracy was confirmed using a panel of cattle fecal (n = 54) and sheep fecal and tissue (n = 90) culture samples. This technique is directly relevant for diagnostic laboratories that perform MAP cultures but may also be applicable to the detection of other species, including M. avium and M. tuberculosis.
INTRODUCTION
Paratuberculosis, or Johne's disease, affects many economically important livestock species, including cattle, sheep, goats, and deer (1). It is caused by Mycobacterium avium subsp. paratuberculosis (MAP) and is characterized by chronic enteritis, wasting, and death. Control programs attempting to limit the impact and spread of the disease in many countries are based on the detection of infected animals by using fecal culture, which is one of the most specific antemortem tests for the diagnosis of paratuberculosis infection (2). The gold standard indications for disease confirmation are a pathological lesion consistent with paratuberculosis and a positive culture for MAP from the affected tissues (3).
For many mycobacterial species, culture requires long incubation times, from weeks to months, and highly specialized media. For MAP, liquid culture media, such as Bactec 12B medium, have been found to be more sensitive than solid culture media, such as Herrold's egg yolk medium (HEYM) supplemented with mycobactin J (4–6). MAP in cultures with positive growth is confirmed by the demonstration of mycobactin dependency or directly by molecular techniques (7). The advantages of liquid culture media systems include shorter culture incubation times (5, 7), and, importantly, some liquid culture media are capable of supporting the growth of all strains of MAP, including the fastidious sheep strain (8). Monitoring of bacterial growth in liquid culture systems has been achieved by ion sensors reading the production of 14CO2 (Bactec 460) for radiometric Bactec 12B medium, while other systems utilize oxygen consumption (VersaTREK system) or pressure changes (mycobacterial growth indicator tube [MGIT] system) as growth indicators.
Due to the discontinuation of Bactec 12B by Becton Dickinson, an alternate liquid culture medium capable of supporting the growth of all MAP strains from feces and intestinal tissues was required in the Australian diagnostic laboratory setting. The M7H9C liquid culture medium was developed and shown to have comparable performance to Bactec 12B medium for the growth of sheep and cattle strains of MAP (4). A drawback is the need to perform DNA extractions and PCR on all samples, as the M7H9C medium currently does not incorporate a growth indicator. The original validation of M7H9C medium performance used a simple DNA isolation method involving ethanol precipitation (7). This method effectively removes PCR inhibitors present in the culture medium, such as egg yolk constituents. Prior to this, subcultures were performed into non-egg yolk–containing Bactec medium to overcome the issue of PCR inhibition (5). However, the ethanol precipitation method is labor intensive and has low throughput, and the batch size is limited by processing time and microcentrifuge capacity. A mechanical lysis step such as bead beating has been found to enhance the recovery of DNA from mycobacteria in various matrices (9–12). Recently, a magnetic-bead-based DNA extraction technique was successfully applied to egg yolk-containing broth cultures to overcome issues of PCR inhibition (13).
The aims of this study were to develop and validate a sensitive, specific, and efficient method for detection of the growth of MAP in liquid broth cultures containing egg yolk due to the limitations of the DNA isolation method (ethanol precipitation) used currently for diagnostic cultures of MAP and the need to process large numbers of samples. An advantage of the new method is that the IS900 quantitative PCR (qPCR) assay applied for the detection of MAP DNA (with primers MP 10-1 and MP 11-1) has high analytical specificity for MAP across a panel of >50 mycobacterial species, including mycobacterial species with IS900-like sequences (11, 14). The diagnostic accuracy of the new method was determined, and the method gave results comparable to those with the ethanol precipitation DNA isolation method but with the advantages of increased throughput and greater analytical sensitivity.
MATERIALS AND METHODS
Culture media preparation.
M7H9C liquid culture medium for MAP containing Middlebrook 7H9 broth base (Becton Dickinson [BD]), PANTA antibiotic preparation (BD), mycobactin J (Allied Monitor), Middlebrook ADC enrichment (containing bovine albumin fraction V, dextrose, and catalase; BD), and egg yolk was prepared and cultures were performed as previously described (4). Modified Bactec 12B medium (Becton Dickinson) containing egg yolk, PANTA Plus, and mycobactin J was prepared as described previously (7).
Bacterial strains.
The reference S (sheep) and C (cattle) strains of MAP were Telford 9.2 and CM00/416, respectively. Telford 9.2 is a pure culture at passage level 5 (including its primary isolation from sheep feces) and is IS900 restriction fragment length polymorphism (RFLP) type S1, IS1311 type S, pathogenic in sheep, and distinct from MAP K10 based on whole-genome microarray comparison (15, 16). CM00/416 is a nonclonal culture which is at passage level 5 (including its primary isolation from cattle tissues) and is IS1311 type C, pathogenic for cattle (unpublished data), and apparently identical to MAP strain K10 based on whole-genome microarray comparison (16). These were reconstituted from lyophilized stock and grown in radiometric Bactec 12B culture as previously described and then subcultured onto modified 7H10 slopes (7, 17).
Analytical sensitivity using serial dilutions of MAP in culture media.
MAP organisms were harvested from slopes by gentle scraping of the surface of each slope using a sterile plastic inoculating loop and resuspended in phosphate-buffered saline (PBS) with 0.1%, wt/vol Tween 20. A single-cell suspension was prepared by vortexing for 5 min and then passing the suspension through a 26-gauge needle and filtering through a sterile 8-μm filter. A 1:100 dilution of the suspension was examined (×400 magnification) to confirm that the majority of cells were single. If clumping was evident, the 26-gauge needle and filter steps were repeated until a single-cell suspension was achieved. Enumeration of MAP was performed using a Helber counting chamber (1:10 or 1:100 dilution). To determine analytical sensitivity, serial 10-fold dilutions of known quantities of MAP were performed in M7H9C medium (108 to 101 MAP/ml) prior to DNA isolation using the ethanol precipitation or the magnetic bead DNA isolation method described below. This was followed by IS900 qPCR detection (see below). During the optimization experiments, only the S strain of MAP was used to examine analytical sensitivity, but for later studies, both S and C strains of MAP were included.
In a separate experiment, MAP was cultured for 12 weeks in either Bactec 12B or M7H9C medium. The study design was based on serial 10-fold dilutions of MAP S and C strains cultured in triplicate. Growth was monitored in Bactec 12B medium by 14CO2 detection and confirmed by IS900 qPCR at one time point or by IS900 qPCR at 0, 4, 8, and 12 weeks in M7H9C medium (Fig. 1). Viable counts of MAP in the suspension were estimated using the most probable number (MPN) method previously described (18).
FIG 1.

Flow chart of the experiment comparing the analytical sensitivity of the magnetic bead method to detect growth in serially diluted suspensions of MAP between Bactec culture medium and M7H9C nonradioactive liquid medium. Tenfold dilutions were performed of pure cultures of S or C strain MAP, from 1/10 to 1/109. GI, growth index.
Clinical samples.
A panel of Bactec 12B liquid culture samples was selected by convenience sampling in order to compare results from the different DNA extraction methods, as shown in Table 1. There were a total of 54 cattle and 70 sheep fecal culture samples and 20 sheep tissue culture samples. Culture-positive and culture-negative samples were included in similar numbers for each species and sample type in a retrospective study. The original culture status was based on the results of culture in Bactec 12B medium, with growth confirmed by IS900 PCR, as described previously (7). Sample collection, storage, and subsampling of cultures were performed as previously described (19).
TABLE 1.
Samples included in the feasibility study to compare the ethanol precipitation and optimized magnetic bead DNA isolation methods for MAP in liquid culture
| Species of origin | Sample type | Strain of MAP present | No. of samples and original culture result |
No of samples detected bya: |
||
|---|---|---|---|---|---|---|
| Positive (wk to GI 999b) | Negative | EtOH precip. | Mag bead | |||
| Cattle | Feces | C | 30 | 2c | 3c | |
| Cattle | Feces | C | 4 (4) | 4 | 4 | |
| 10 (5) | 10 | 10 | ||||
| 5 (6) | 5 | 5 | ||||
| 3 (7) | 3 | 3 | ||||
| 2 (8) | 2 | 2 | ||||
| Sheep | Feces | S | 30 | All negative | All negative | |
| Sheep | Tissues | S | 10 | All negative | All negative | |
| Sheep | Feces | S | 10 (4) | 10 | 10 | |
| 10 (5) | 10 | 10 | ||||
| 10 (6) | 10 | 10 | ||||
| 10 (7) | 10 | 10 | ||||
| Sheep | Tissues | S | 10 | 10 | 10 | |
EtOH precip., ethanol precipitation and IS900 qPCR method; Mag bead, optimized magnetic bead DNA isolation and IS900 qPCR method.
Weeks of growth in Bactec 12B liquid culture medium to reach a growth index (GI) of 999.
These samples had a GI of 999, but the original PCR results were negative. Two were positive on secondary testing with both methods, and an additional sample was detected by magnetic bead isolation/qPCR. The remainder of the samples in this group were negative using both methods.
DNA isolation from liquid culture media. (i) Ethanol precipitation method.
DNA was extracted using the commonly used ethanol precipitation method, as previously described (7). Briefly, 500 μl absolute ethanol was added to 200 μl of culture medium, left to stand for 2 min, and then vortexed briefly and centrifuged at 28 × g for 2 min to allow the partially flocculated egg yolk to accumulate at the base and sides of the tube in order to remove inhibitory egg components. The supernatant was transferred to a clean 1.5-ml centrifuge tube and centrifuged at 18,000 × g for 5 min. The resulting pellet was washed twice in 200 μl of sterile PBS, resuspended in 50 μl of sterile distilled water, and incubated at 100°C for 20 min; 5 μl was added to each PCR.
(ii) Magnetic bead DNA isolation.
DNA was isolated from liquid cultures using bead beating followed by a semiautomated magnetic bead isolation technique. During optimization, zirconia/silica beads (BioSpec Products, Inc., Daintree Scientific) and ceramic bead tubes (lysing matrix D; MP Biomedicals) were compared to control samples processed without a bead-beating step. The use of buffer RLT or buffer AL (Qiagen) as a lysis buffer in this step was also compared. The optimized method involved a bead-beating step, in which 200 μl of the liquid culture medium was added to 200 μl buffer RLT and then transferred to a 2-ml conical base screw-cap tube containing 0.3 g of zirconia/silica beads and disrupted using a Fast Prep-24 bead beater (MP Biomedicals) at 6.5 m/s for 60 s twice. The tubes were centrifuged at 16,000 × g for 2 min, and 100 μl of the homogenate was added to the deep 96-well (S block) plate. DNA was isolated using the BioSprint 96 one-for-all vet kit (Qiagen) and a MagMAX-96 automated magnetic processor, following the protocol for tissue homogenates using buffer RLT, according to the manufacturer's instructions.
IS900 PCR to detect MAP.
Conventional IS900 PCR was performed as previously described (7), using primers P90 (5′-GAAGGGTGTTCGGGGCCGTCGCTTAGG) and P91 (5′-GGCGTTGAGGTCGATCGCCCACGTGAC). IS900 qPCR was performed as previously described (14), using the specific primers reported by Kawaji et al. (11). Briefly, reaction mixtures contained 5 μl template DNA, 250 nM forward and reverse primers (MP10-1 forward 5′-ATGCGCCACGACTTGCAGCCT-3′; MP11-1 reverse 5′-GGCACGGCTCTTGTTGTAGTCG-3′), and SensiMix SYBR low-ROX qPCR master mix (Bioline). Cycling conditions were initial denaturation at 95°C for 8 to 10 min, 40 cycles of denaturation at 95°C for 30 s, and annealing/extension at 68°C for 60 s with fluorescence acquisition at the end of the annealing/extension step, followed by a melt curve analysis from 65°C to 95°C. A 5-step standard curve of MAP genomic DNA was included in every qPCR experiment (10 to 0.001 pg/reaction) as a reference standard and to normalize results between experiments.
Statistical analysis.
McNemar's chi-square test for paired observations was used to compare the results of the conventional PCR to those of the qPCR and to compare method performance on clinical samples (20). The comparison of qPCR DNA quantity to conventional PCR band intensity was performed using analysis of variance (ANOVA) with the Kruskal-Wallis posttest for nonparametric data (GraphPad Prism v4.0). Results for the analytical sensitivity and time course studies were analyzed with a restricted maximum-likelihood (REML) linear mixed model, using the log10-transformed MAP IS900 qPCR DNA quantity for different treatments as the outcome variable (GenStat 12th edition; VSN International Ltd.). Differences between treatments, time points, and/or groups were tested using the least significant difference derived from the REML model.
RESULTS
Quantitative PCR method for the confirmation of MAP in cultures.
Initially, the existing conventional IS900 PCR (primers P90 and P91) was compared to a highly specific IS900 qPCR method. The qPCR method was designed for the direct detection of MAP in feces, using primers that differentiate MAP IS900 from IS900-like sequences previously reported in the literature (11). DNA was extracted using the existing methodology (ethanol precipitation) for a panel of 412 fecal samples cultured in Bactec 12B medium, and a two-by-two contingency table of conventional PCR versus qPCR results was produced (Table 2). There was no significant difference between the results from the conventional IS900 PCR and the IS900 qPCR assays when DNA was obtained from cultures using the ethanol precipitation method (chi-square value, 2.286; P = 0.131). An increased MAP DNA quantity detectable by qPCR was associated with the band intensity originally detected in the conventional PCR (Fig. 2).
TABLE 2.
Two-by-two contingency table of frequencies for detection of MAP using different IS900 PCR techniques
| IS900 qPCR | Conventional IS900 PCR (no. of samples) |
||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 308 | 6 | 314 |
| Negative | 1 | 97 | 98 |
| Total | 309 | 103 | 412 |
FIG 2.
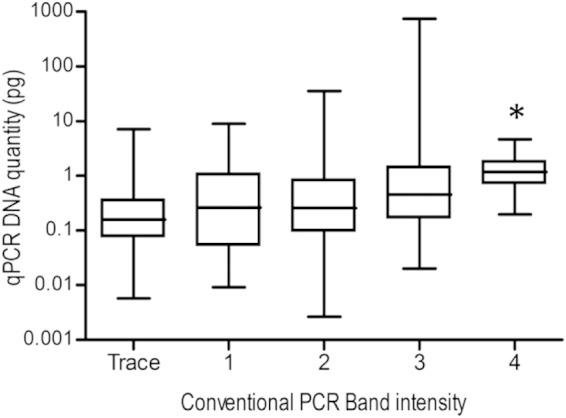
Box-and-whiskers plot of the comparison between the IS900 conventional PCR band intensity and IS900 qPCR DNA quantity. The two MAP IS900 PCR methods were performed in parallel on DNA extracted from a panel of culture samples using the ethanol precipitation method. Band intensity in conventional PCR was graded from trace (barely visible band) to 4 (strong band) of the appropriate molecular size in an agarose gel. *, Significantly higher DNA quantity (P < 0.05) compared to band intensities of trace, 1, and 2.
Optimization and analytical sensitivity.
A pilot study was conducted to determine whether a bead-beating/magnetic bead DNA isolation method was likely to detect MAP in liquid culture (Fig. 3A). This showed that zirconia/silica beads in the bead-beating step appeared to be superior to ceramic beads, and samples processed using both types of beads were far superior to those processed without a bead-beating step. Two buffers supplied with the BioSprint 96 one-for-all vet magnetic bead nucleic acid isolation kit were tested, and buffer RLT appeared to give superior sensitivity (Fig. 3A). This was confirmed in a separate experiment comparing the two buffers, using four replicate extractions for each MAP concentration, which showed that buffer RLT performed significantly better (P < 0.001) than buffer AL (Fig. 3B). The final protocol used for all subsequent experiments combined a bead-beating step using zirconia/silica beads and a magnetic bead isolation step using buffer RLT, referred to as the optimized magnetic bead DNA isolation method.
FIG 3.
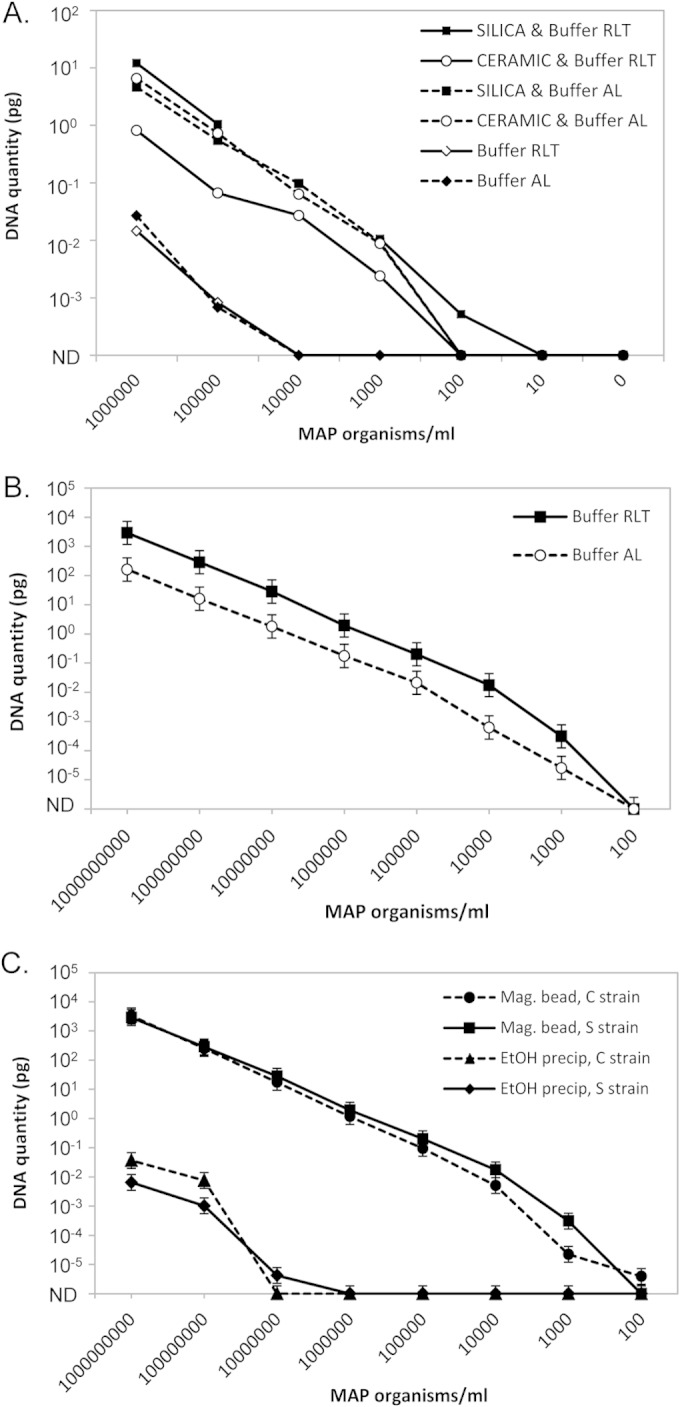
Detection of MAP in liquid culture media by qPCR. (A) Pilot study to test the feasibility of using magnetic bead isolation of MAP from liquid culture media. M7H9C medium samples were spiked with known quantities of sheep strain MAP (106 to 101 MAP/ml culture medium) and a control with no MAP (0); n = 1/treatment and MAP concentration. (B) Comparison of buffer RLT to buffer AL. M7H9C medium samples were spiked with known quantities of sheep strain MAP (108 to 101 MAP/ml culture medium); n = 4/treatment and MAP concentration. Predicted means ± SEM from the REML linear mixed model. Buffer RLT was significantly different from buffer AL (P < 0.001). (C) Comparison of the standard ethanol precipitation method to the optimized magnetic bead DNA isolation method performed on samples spiked with known quantities of either cattle or sheep strain MAP (108 to 101 MAP/ml culture medium); n = 4/treatment and MAP concentration. Predicted means ± SEM from the REML linear mixed model. The optimized magnetic bead method was significantly different from the ethanol precipitation method (P < 0.001). ND, not detected.
The analytical sensitivity was assessed on replicate samples of M7H9C culture medium (n = 4/treatment and dilution) spiked with known quantities of MAP. The optimized magnetic bead DNA isolation method was significantly more sensitive than the ethanol precipitation method (P < 0.001), as shown in Fig. 3C. No significant difference in the detection of the two MAP strains was found. The limit of detection was 104- to 105-fold lower for the optimized magnetic bead method than for the ethanol precipitation method (100 to 1,000 compared to 10,000,000 MAP organisms/ml, respectively).
Analytical sensitivity comparison of Bactec 12B and M7H9C culture media using the optimized magnetic bead DNA isolation method.
The analytical sensitivities of Bactec 12B and M7H9C media were previously shown to be comparable when using either the S or C strain of MAP (19). For M7H9C cultures, growth is normally detected by PCR at a single time point (12 weeks). Here, we assessed sampling at various times postinoculation (0, 4, 8, and 12 weeks) to examine (i) changes over time with various initial concentrations of MAP and (ii) whether multiple sampling times are needed to detect MAP growth or a single test at the endpoint of culture (12 weeks) is sufficient. The experimental design is shown in Fig. 1.
Representative results of the MAP DNA quantity detected by optimized magnetic bead DNA isolation and IS900 qPCR at 0, 4, 8, and 12 weeks for S or C strain in M7H9C medium are shown in Fig. 4. The DNA quantity in these cultures increased over time and appeared to plateau between 8 and 12 weeks of culture at a MAP DNA quantity of ∼104 pg, equivalent to CT values between 9 and 12. Across all cultures tested, the mean MAP DNA quantity at 12 weeks in M7H9C cultures was significantly higher than at 8 weeks (14,406 pg versus 9,982 pg; P < 0.0001), indicating that replication may still be occurring in some cultures during this time. The MAP DNA quantities detected at 12 weeks in Bactec 12B and M7H9C cultures were very similar, with mean values of 13,239 pg and 14,406 pg, respectively (nonsignificant difference, P = 0.3). This suggested similar levels of MAP proliferation in the two media.
FIG 4.
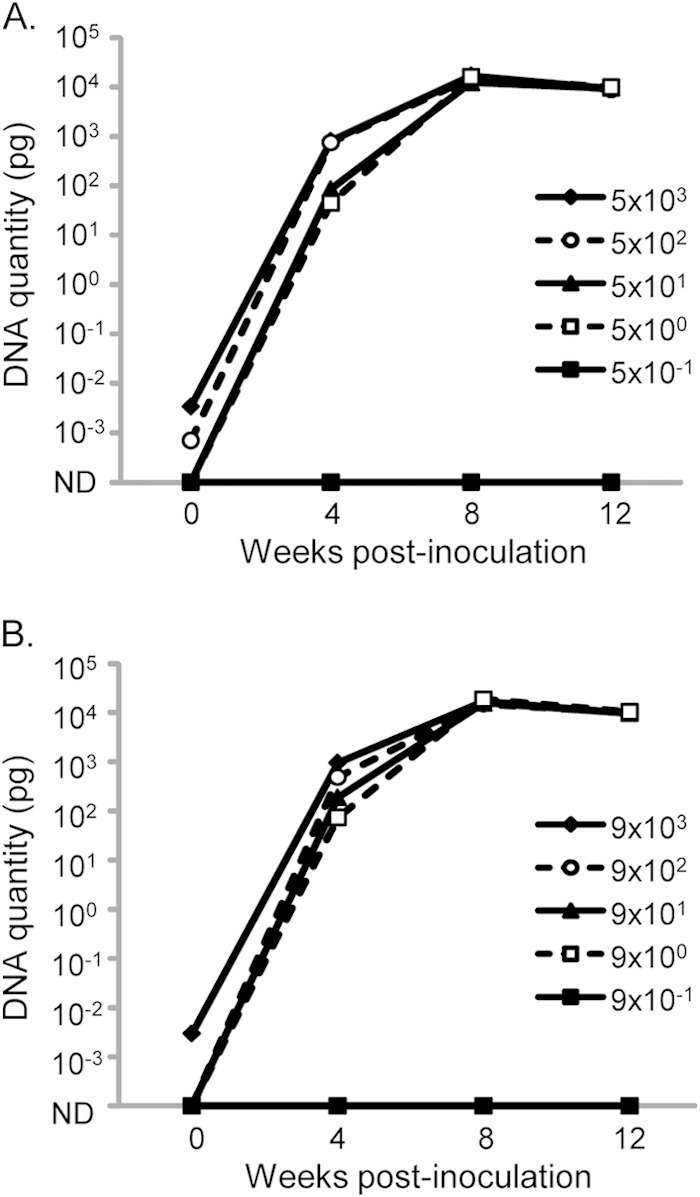
Isolation and detection of MAP DNA from M7H9C cultures at 0, 4, 8, and 12 weeks postinoculation. Triplicate cultures of 10-fold serially diluted suspensions of MAP S strain (A) and C strain (B) were evaluated. The mean DNA quantity detected in IS900 qPCR is shown for estimated starting concentrations of viable MAP between 104 and 10−1, based on an MPN estimate. ND, not detected.
In cultures with high starting concentrations of MAP (>1,000 MAP in the inoculum), there were low levels of MAP DNA (CT values, >35 in qPCR, equivalent to <0.01 pg MAP genomic DNA) detectable in the culture media at week 0 when using the optimized magnetic bead DNA isolation method and qPCR detection. This level of detection is consistent with the analytical sensitivity of the method. When followed to 12 weeks, these cultures had very high MAP DNA quantities, with CT values in the range of 9 to 12, which was indicative of MAP growth that was easily distinguishable from the week 0 results.
Validation on clinical samples.
Cattle fecal and sheep fecal and tissue samples were included in a validation study to compare the optimized magnetic bead DNA isolation method with the ethanol precipitation method (Table 1). Samples chosen were previously grown in radiometric liquid medium (Bactec 12B); in this way, growth indices were used to give a measure of mycobacterial growth independent of the PCR detection. All of the samples were tested previously and categorized as negative (n = 70) or positive (n = 74) based on the original culture results (using the standard ethanol precipitation/IS900 conventional PCR method). The range of positive growth indices ranged from 4 to 8 weeks of culture (Table 1). Individual culture samples were divided into two equal aliquots and processed in parallel using the two DNA isolation methods, with detection of MAP DNA using the IS900 qPCR on both extracts.
There was a high level of agreement in the positive and negative results obtained using the two methodologies (Table 3) (McNemar's chi-square test, P = 1.0). Two cattle samples with late growth indices (12 weeks) that were negative in the original PCR had MAP DNA detectable using the ethanol precipitation and the optimized magnetic bead DNA isolation methods coupled with qPCR, and an additional sample was positive with only the magnetic bead isolation method (Table 1).
TABLE 3.
Two-by-two contingency table of frequencies for detection of MAP in the feasibility study, comparing the ethanol precipitation/conventional PCR method and the optimized magnetic bead DNA isolation/qPCR method
| Mag bead/IS900 qPCR | EtOH/conv. IS900 PCR (no. of samples) |
||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 76 | 1 | 77 |
| Negative | 0 | 67 | 67 |
| Total | 76 | 68 | 144 |
A significant difference was seen between the two DNA isolation methods in relation to the quantity of MAP DNA detected by qPCR for individual samples. Representative results are shown in Fig. 5 for a subset of the samples with peak growth indices between 4 and 6 weeks of culture. Amplification of IS900 MAP DNA for the samples processed using the optimized magnetic bead DNA isolation method occurred very early in the PCR cycling, before the highest MAP DNA standard used in the qPCR (standard 1, 10 pg MAP genomic DNA), whereas the samples processed using the ethanol precipitation method showed amplification late in the qPCR. The range of DNA quantities detected in the samples is shown in Table 4, with the optimized magnetic bead method consistently giving significantly higher DNA quantities (P < 0.001).
FIG 5.

Representative amplification plots from IS900 qPCR experiments on sheep culture-positive samples from the validation set. DNA was isolated using the ethanol precipitation method (A) or high-throughput magnetic bead isolation method (B). Amplification curves for the five qPCR standard concentrations included in each experiment are shown for each graph. Std 1, amplification curve for qPCR standard 1 containing 10 pg MAP genomic DNA; Std 5, amplification curve for qPCR standard 5 containing 0.001 pg MAP genomic DNA.
TABLE 4.
Detection of MAP DNA in cultures by IS900 qPCR, comparing the ethanol precipitation and optimized magnetic bead DNA isolation methods
| Bactec GI (wk to 999) | No. of samples | Quantity of MAP DNAa (pg) detected by: |
|||
|---|---|---|---|---|---|
| Ethanol precipitation method |
Magnetic bead method |
||||
| Mean | Range | Mean | Range | ||
| 4 | 14 | 0.06 | 0.005–0.286 | 1,516 | 570–3,415 |
| 5 | 20 | 0.11 | 0.016–0.451 | 1,998 | 779–4,636 |
| 6 | 15 | 0.04 | 0.001–0.161 | 1,390 | 394–2,086 |
| 7 | 13 | 0.02 | 0.003–0.054 | 2,204 | 937–5,328 |
| 8 | 2 | 0.02 | 0.015–0.018 | 1,099 | 975–1,222 |
Determined from the standard curve included in each qPCR experiment.
DISCUSSION
Here, we describe the optimization and validation of a detection method involving bead beating, magnetic bead DNA isolation, and qPCR for the confirmation of MAP growth in liquid culture media. The method is many times more sensitive than the previously used detection method. The increased sensitivity is likely to be attributable to the inclusion of a bead-beating step to disrupt the cell membrane and to the advantages offered by using magnetic bead DNA isolation technology and a qPCR detection assay. Critical to this application, the optimized magnetic bead method removes inhibitors that can affect the detection of MAP in liquid culture by PCR.
Liquid culturing of MAP has undergone successive improvements in methodology. Liquid culturing offered advantages over solid media in reducing the culture time and in the ability to grow type S or I strains of MAP (7, 17). Due to PCR inhibition by media containing egg yolk, subcultures into non-egg yolk–containing media were required (5) until a simple DNA isolation method involving ethanol precipitation was developed (7). There were disadvantages for processing large numbers of samples with this method, as well as a significant dependence of the results on the technical capability of the operator. This study describes an improvement in DNA isolation from liquid culture media and in PCR detection of MAP DNA compared to the previous methodology. The advantages of this method are that it is rapid, efficient, and semiautomated, requiring fewer operator handling steps, which is important as detection of growth in the M7H9C liquid medium is dependent on PCR assay of every individual culture. The method may also be applicable to other liquid culture systems based on Middlebrook 7H9 medium with egg yolk, such as the BBL MGIT system (BD) and VersaTREK mycobacteria detection system (Thermo Scientific), although this would require validation.
The analytical sensitivity of the new method was far higher than that of the ethanol precipitation method. This is in part due to the magnetic bead DNA isolation technique; however, the incorporation of a bead-beating step, previously reported to be important for release of DNA from within mycobacteria (9), significantly improved the sensitivity. During the validation on clinical samples, additional positive samples (n = 3) from cultures with evidence of growth was identified, indicating that this sample may have remained undetected due to the limits of the sensitivity of the previous methodology. In the experiment on pure cultures of MAP C and S strains, the majority (95%) of positive cultures were PCR positive by 4 weeks of culture, although this may not reflect the growth rate of MAP organisms present in other matrices, such as feces. The MAP DNA quantity detected in these cultures tended to plateau between 8 and 12 weeks, which suggests that MAP proliferation had peaked and that the DNA was stable during the culture period. The high sensitivity of the new method may open the potential to detect DNA from dead organisms present in the original sample. This was investigated in samples at week 0 of culture, and it was determined that low levels (CT values of >35) of DNA were detectable at high MAP concentrations, equivalent to ∼103 to 104 MAP organisms/gram of the original fecal sample, not taking into consideration the effect of decontamination. When followed, these cultures subsequently had very high MAP DNA quantities, indicative of growth and thus the presence of viable MAP organisms in culture. However, further consideration may need to be given to this issue with regard to the testing of week 0 cultures for comparison or to the establishment of a cut point for positive culture results.
Many studies have assessed molecular versus culture methods of detection of bacteria in biological and environmental samples (14, 21, 22); however, few studies compared alternative methods for confirmation of growth in culture. Many of the methods that are in use for mycobacterial cultures are historical, such as phenotypical differences in colony morphology, biochemical characteristics, and acid-fast staining of smears. Molecular probes or low-throughput DNA isolation followed by single/multiplex PCR is generally used only for a subset of samples that require confirmation. For PCR-based methods, the type of organism and the media components can significantly affect the success of DNA isolation and the presence of PCR inhibitors in the extract. Some simple cleanup procedures may not be applicable for all cultures, as identified in MAP cultures containing egg yolk (7, 13). One method for the differentiation of nontuberculous mycobacteria from M. tuberculosis in clinical cultures involves protein extraction followed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (23). This enabled identification at the species level for a high proportion (94%) of samples. However, this type of analysis is dependent on specialized equipment and expertise. Thus, an efficient technique of nucleic acid isolation from cultures that removes PCR inhibitors and can be aligned with downstream PCR methods is of value. It can be used for confirmation and characterization of strain type and other phenotypic properties (e.g., antibiotic resistance), in the mycobacterial field particularly but also more broadly.
Magnetic bead methods have been used for the isolation of bacterial and viral nucleic acids from various substrates (13, 14, 24–27). Okwumabua et al. (13) provide proof that a magnetic bead DNA isolation method can be used to isolate MAP DNA in broth media. Pozzato et al. (26) combined bead beating and a magnetic bead isolation method using a liquid culture medium similar to M7H9C; however, this study included serial dilutions of one infected and one artificially spiked cattle fecal sample, without validation of the method. In the present study, the bead-beating/magnetic bead isolation method was optimized and coupled with an IS900 qPCR assay of known high analytical specificity, followed by validation on samples cultured from both sheep and cattle to confirm the robust performance of this method for clinical specimens.
The high-throughput method validated in this study offers a significant advance for those using nonradiometric liquid cultures for the detection of MAP and may be applicable for use with other media used for MAP cultivation, as many of these contain egg yolk that is inhibitory of PCR. As the MAP cell wall is extremely difficult to disrupt, this method is likely to be suitable for nucleic acid isolation from cultures of other mycobacterial species with similar properties, when coupled with the appropriate PCR detection assay, or more broadly to bacterial species with robust cell walls.
ACKNOWLEDGMENTS
This work was supported by Meat and Livestock Australia and by Cattle Council of Australia, Sheepmeat Council of Australia, and WoolProducers Australia through Animal Health Australia.
We thank Ann-Michele Whittington and Adelyn Bolithon for technical support and Craig Kristo, Nobel Toribio, and James Dalton for animal husbandry support.
REFERENCES
- 1.Behr MA, Collins DM. 2010. Paratuberculosis: organism, disease, control. CABI, Wallingford, United Kingdom. [Google Scholar]
- 2.Nielsen SS, Toft N. 2008. Ante mortem diagnosis of paratuberculosis: a review of accuracies of ELISA, interferon-gamma assay and faecal culture techniques. Vet Microbiol 129:217–235. doi: 10.1016/j.vetmic.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 3.OIE World Organization for Animal Health. 2014. Paratuberculosis (Johne's disease). In Terrestrial manual 2014. http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.01.11_PARATB.pdf.
- 4.Whittington RJ, Whittington A-M, Waldron A, Begg DJ, de Silva K, Purdie AC, Plain KM. 2013. Development and validation of a liquid culture medium (M7H9C) for routine culture of Mycobacterium avium subsp. paratuberculosis to replace modified Bactec 12B medium. J Clin Microbiol 51:3993–4000. doi: 10.1128/JCM.01373-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cousins DV, Evans RJ, Francis BR. 1995. Use of BACTEC radiometric culture method and polymerase chain reaction for the rapid screening of faeces and tissues for Mycobacterium paratuberculosis. Aust Vet J 72:458–462. doi: 10.1111/j.1751-0813.1995.tb03489.x. [DOI] [PubMed] [Google Scholar]
- 6.Eamens GJ, Whittington RJ, Marsh IB, Turner MJ, Saunders V, Kemsley PD, Rayward D. 2000. Comparative sensitivity of various faecal culture methods and ELISA in dairy cattle herds with endemic Johne's disease. Vet Microbiol 77:357–367. doi: 10.1016/S0378-1135(00)00321-7. [DOI] [PubMed] [Google Scholar]
- 7.Whittington RJ, Marsh I, Turner MJ, McAllister S, Choy E, Eamens GJ, Marshall DJ, Ottaway S. 1998. Rapid detection of Mycobacterium paratuberculosis in clinical samples from ruminants and in spiked environmental samples by modified Bactec 12B radiometric culture and direct confirmation by IS900 PCR. J Clin Microbiol 36:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whittington RJ. 2010. Cultivation of Mycobacterium avium subsp. paratuberculosis., p 244–266. In Behr MA, Collins DM (ed), Paratuberculosis: organis, disease, control. CABI, Wallingford, United Kingdom. [Google Scholar]
- 9.Hermon-Taylor J, Bull TJ, Sheridan JM, Cheng J, Stellakis ML, Sumar N. 2000. Causation of Crohn's disease by Mycobacterium avium subspecies paratuberculosis. Can J Gastroenterol 14:521–539. [DOI] [PubMed] [Google Scholar]
- 10.Odumeru J, Gao A, Chen S, Raymond M, Mutharia L. 2001. Use of the bead beater for preparation of Mycobacterium paratuberculosis template DNA in milk. Can J Vet Res 65:201–205. [PMC free article] [PubMed] [Google Scholar]
- 11.Kawaji S, Taylor DL, Mori Y, Whittington RJ. 2007. Detection of Mycobacterium avium subsp. paratuberculosis in ovine faeces by direct quantitative PCR has similar or greater sensitivity compared to radiometric culture. Vet Microbiol 125:36–48. doi: 10.1016/j.vetmic.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Madiraju MVVS, Qin MH, Rajagopalan M. 2000. Development of simple and efficient protocol for isolation of plasmids from mycobacteria using zirconia beads. Lett Appl Microbiol 30:38–41. doi: 10.1046/j.1472-765x.2000.00619.x. [DOI] [PubMed] [Google Scholar]
- 13.Okwumabua O, Shull E, O'Connor M, Moua TV, Danz T, Strelow K. 2010. Comparison of three methods for extraction of Mycobacterium avium subspecies paratuberculosis DNA for polymerase chain reaction from broth-based culture systems. J Vet Diagn Invest 22:67–69. doi: 10.1177/104063871002200111. [DOI] [PubMed] [Google Scholar]
- 14.Plain KM, Marsh IB, Waldron AM, Galea F, Whittington AM, Saunders VF, Begg DJ, de Silva K, Purdie AC, Whittington RJ. 2014. High-throughput direct fecal PCR assay for detection of Mycobacterium avium subsp. paratuberculosis in sheep and cattle. J Clin Microbiol 52:745–757. doi: 10.1128/JCM.03233-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Begg D, Whittington R. 2010. Paratuberculosis in sheep, p 157–168. In Behr MA, Collins DM (ed), Paratuberculosis: organism, diseas, control. CABI, Wallingford, United Kingdom. [Google Scholar]
- 16.Marsh IB, Bannantine JP, Paustian ML, Tizard ML, Kapur V, Whittington RJ. 2006. Genomic comparison of Mycobacterium avium subsp. paratuberculosis sheep and cattle strains by microarray hybridization. J Bacteriol 188:2290–2293. doi: 10.1128/JB.188.6.2290-2293.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whittington RJ, Marsh I, McAllister S, Turner MJ, Marshall DJ, Fraser CA. 1999. Evaluation of modified Bactec 12B radiometric medium and solid media for culture of Mycobacterium avium subsp. paratuberculosis from sheep. J Clin Microbiol 37:1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddacliff LA, Nicholls PJ, Vadali A, Whittington RJ. 2003. Use of growth indices from radiometric culture for quantification of sheep strains of Mycobacterium avium subsp. paratuberculosis. Appl Environ Microbiol 69:3510–3516. doi: 10.1128/AEM.69.6.3510-3516.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whittington RJ, Whittington AM, Waldron A, Begg DJ, de Silva K, Purdie AC, Plain KM. 2013. Development and validation of a liquid medium (M7H9C) for routine culture of Mycobacterium avium subsp. paratuberculosis to replace modified Bactec 12B medium. J Clin Microbiol 51:3993–4000. doi: 10.1128/JCM.01373-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motulsky H. 1995. Intuitive biostatistics. Oxford Universtiy Press, Oxford, United Kingdom. [Google Scholar]
- 21.Adams AP, Bolin SR, Fine AE, Bolin CA, Kaneene JB. 2013. Comparison of PCR versus culture for detection of Mycobacterium bovis after experimental inoculation of various matrices held under environmental conditions for extended periods. Appl Environ Microbiol 79:6501–6506. doi: 10.1128/AEM.02032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Therese KL, Gayathri R, Dhanurekha L, Sridhar R, Meenakshi N, Madhavan HN. 2013. Diagnostic appraisal of simultaneous application of two nested PCRs targeting MPB64 gene and IS6110 region for rapid detection of M. tuberculosis genome in culture proven clinical specimens. Indian J Med Microbiol 31:366–369. doi: 10.4103/0255-0857.118887. [DOI] [PubMed] [Google Scholar]
- 23.Balada-Llasat JM, Kamboj K, Pancholi P. 2013. Identification of mycobacteria from solid and liquid media by matrix-assisted laser desorption ionization-time of flight mass spectrometry in the clinical laboratory. J Clin Microbiol 51:2875–2879. doi: 10.1128/JCM.00819-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hick P, Whittington RJ. 2010. Optimisation and validation of a real-time reverse transcriptase-polymerase chain reaction assay for detection of betanodavirus. J Virol Methods 163:368–377. doi: 10.1016/j.jviromet.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 25.Pan S, Gu B, Wang H, Yan Z, Wang P, Pei H, Xie W, Chen D, Liu G. 2013. Comparison of four DNA extraction methods for detecting Mycobacterium tuberculosis by real-time PCR and its clinical application in pulmonary tuberculosis. J Thorac Dis 5:251–257. doi: 10.3978/j.issn.2072-1439.2013.05.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pozzato N, Gwozdz J, Gastaldelli M, Capello K, Ben CD, Stefani E. 2011. Evaluation of a rapid and inexpensive liquid culture system for the detection of Mycobacterium avium subsp. paratuberculosis in bovine faeces. J Microbiol Methods 84:413–417. doi: 10.1016/j.mimet.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 27.Yang G, Erdman DE, Kodani M, Kools J, Bowen MD, Fields BS. 2011. Comparison of commercial systems for extraction of nucleic acids from DNA/RNA respiratory pathogens. J Virol Methods 171:195–199. doi: 10.1016/j.jviromet.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]