
Figure 2. Role of autophagy in inclusion body myositis pathogenesis and potential therapeutic avenues.
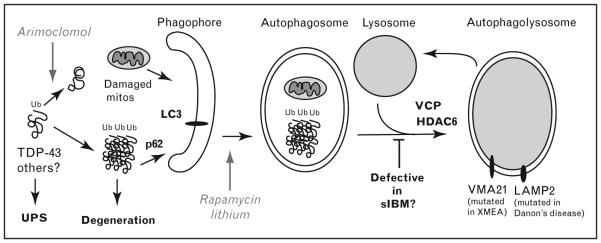
Myofiber degeneration in inclusion body myositis (IBM) is proposed to be due to toxic oligomers of misfolded proteins such as TDP-43. Arimoclomol is a drug being tried in IBM and neurodegenerative diseases due to its ability to induce expression of chaperones that help prevent accumulations of misfolded proteins. Normally, these ubiquitinated protein aggregates and damaged mitochondria (mitos) are degraded via the ubiquitin–proteasome system (UPS) or autophagy. Ubiquitinated aggregates may be recruited to the LC3 receptor on a newly forming double-membrane phagophore via the ubiquitin-binding protein p62/SQSTM1. Rapamycin and lithium both stimulate autophagy and help clear toxic aggregates such as TDP-43. VCP, mutated in familial IBM, functions in maturation of autophagosomes into an autophagolysosome. Overexpression of HDAC6 rescues the VCP phenotype, by stimulating autophagosome maturation. This step may be defective in sporadic inclusion body myositis (sIBM) as well, explaining the accumulation of rimmed vacuoles in addition to the numerous aggregated proteins. Upon fusion of autophagosomes with the lysosome, the ubiquinated aggregates and damaged mitochondria are degraded. A similar vacuolar pathology is seen in two rare inherited vacuolar myopathies in which myofiber degeneration is caused by defective lysosomal function termed XMEA (X-linked myopathy with ‘excessive’ autophagy, caused by loss of VMA21 function, required for lysosomal acidification) and Danon’s disease (caused by mutations in lysosome-associated membrane protein 2 (LAMP2) involved in lysosome fusion).