
Fig. 1.
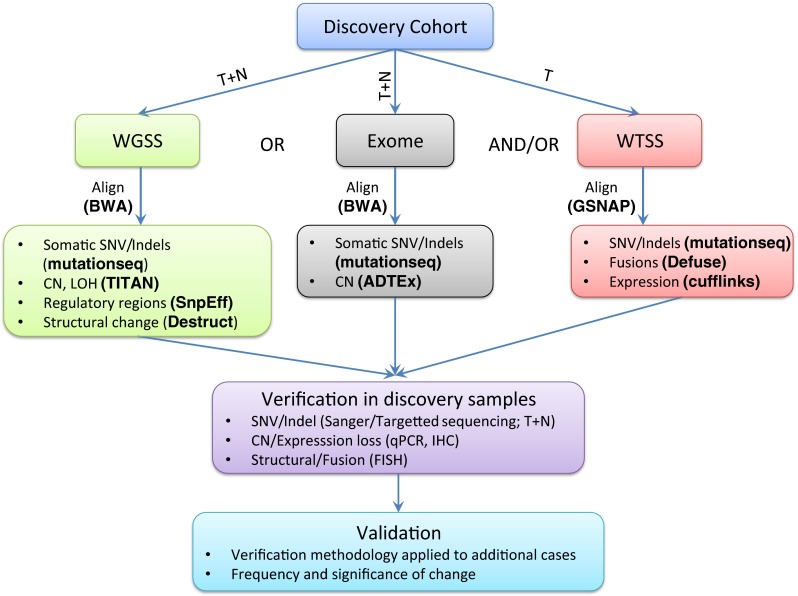
A flowchart of the typical approach to a NGS study to discover novel mutations. Representative bioinformatic programs are in parenthesis and in bold (further details can be found in [75–81]). For somatic mutations, tumor (T) and matched normal (N) samples, obtained from blood or adjacent normal tissue, are used in whole genome (WGSS) or exome sequencing to look for somatic mutations and copy number changes (CN). Transcriptome analysis (WTSS) of tumor samples will enable assessment of expressed mutations and fusions as well as expression patterns. Confirmation of the NGS findings using a different platform such as Sanger sequencing to eliminate false positives would be the next step. Finally, to understand the frequency of the findings in the disease of interest, analysis on a larger validation cohort of tumor samples should be completed. For hotspot mutations, sequencing; for inactivating mutations, sequencing or immunohistochemistry (IHC); and for fusions, fluorescent in situ hybridization (FISH) could be methods of choice for verification and validation