

Abstract
The 1,5-benzodiazepine clobazam is indicated for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in patients 2 years of age or older in the United States, and for treatment of anxiety and various forms of epilepsy elsewhere. Clobazam has been reported to exhibit different in vivo adverse effects and addiction liability profile than the classic 1,4-benzodiazepines. In this study, it was investigated whether the in vitro pharmacological properties of clobazam and its major active metabolite N-desmethylclobazam could explain some of these clinical differences. The functional properties of the two 1,5-benzodiazepines were characterized at the human γ-aminobutyric acid type A receptor (GABAAR) subtypes α1β2γ2S, α2β2γ2S, α3β2γ2S, α5β2γ2S and α6β2δ expressed in Xenopus laevis oocytes by use of two-electrode voltage-clamp electrophysiology and compared to those exhibited by the 1,4-benzodiazepine clonazepam. All three compounds potentiated GABA EC20-evoked responses through the α1,2,3,5β2γ2S GABAARs in a reversible and concentration-dependent manner, with each displaying similar EC50 values at the four subtypes. Furthermore, the degrees of potentiation of the GABA EC20 currents through the four receptors mediated by saturating modulator concentrations did not differ substantially for any of the three benzodiazepines. The three compounds were substantially less potent (200-3900 fold) as positive allosteric modulators at the α6β2δ GABAAR than at the α1,2,3,5β2γ2S receptors. Interestingly, however, clobazam and especially N-desmethylclobazam were highly efficacious potentiators of α6β2δ receptor signaling. Although this activity component is unlikely to contribute to the in vivo effects of clobazam/N-desmethylclobazam, the 1,5-benzodiazepine could constitute an interesting lead for novel modulators targeting this low-affinity binding site in GABAARs. In conclusion, the non-selective modulation exerted by clobazam, N-desmethylclobazam and clonazepam at the α1β2γ2S, α2β2γ2S, α3β2γ2S and α5β2γ2S GABAARs indicate that the observed clinical differences between clobazam and 1,4-benzodiazepines are likely to arise from factors other than their respective pharmacological properties at the GABAARs as investigated here.
Introduction
As the main inhibitory neurotransmitter in the central nervous system (CNS), γ-aminobutyric acid (GABA) is directly involved in, or contributes to, an exhaustive number of physiological processes and pathophysiological states. GABA exerts its effects through two receptor classes, the GABAA and GABAB receptors [1, 2]. The GABAA receptors (GABAARs) are membrane-bound, chloride-permeable ligand-gated ion channels belonging to the Cys-loop receptor superfamily, which also includes receptors for acetylcholine, serotonin, and glycine [2–5]. The GABAAR complex is composed of five subunits, and the existence of a total of 19 human GABAA subunits (α 1–6, β1–3, γ 1–3, δ, ε, π, θ and ρ1–3) gives rise to an array of physiologically relevant receptor subtypes [6]. It is estimated that approximately 80% of all GABAARs are α βγ receptors predominantly composed of α 1/2/3/5, β2/3, and γ 2 subunits in an anticlockwise α β α γ β arrangement (viewed from the extracellular space) [6–9]. However, numerous other physiologically important receptor subtypes exist, including the α 4βδ and α 6βδ receptors that through their predominant expression as extra- and perisynaptic receptors are key mediators of the GABAergic tonic inhibition [10, 11].
The signaling through the GABAAR is susceptible to modulation by numerous ligands acting through different allosteric sites in the receptor complex, and many of these ligands are used to treat human pathologies [12–16]. Delineation of the molecular modes of action of these modulators at the receptors has provided considerable insight into the signal transduction mechanism of the GABAAR as well as the molecular compositions of its allosteric sites. The allosteric modulation of the α 1,2,3,5βγ GABAARs exerted by benzodiazepines is predominantly mediated through a high-affinity binding site located in the extracellular α (+)/γ (–) subunit interface of the receptor [14, 17]. However, benzodiazepines have also been proposed to target a low-affinity binding site located in the transmembrane domains of both αβ and αβγ GABAARs [18]. Thus, the benzodiazepines have been proposed to possess a nM activity component arising exclusively from α 1,2,3,5βγ GABAARs and a μM activity component that potentially could involve all GABAARs [18]. Furthermore, several recent studies have proposed the existence of a low-affinity binding site for some benzodiazepines and other benzodiazepine-site ligands such as CGS 9895, LAU 177 and Ro 15–4513 in the extracellular α (+)/β(–) subunit interface of the GABAAR, a site homologous to the extracellular high-affinity α(+)/γ(–) binding site for benzodiazepines in the αβγ GABAAR [19–23].
The selectivity profile of a particular benzodiazepine at the different α1,2,3,5βγ GABAAR subtypes is believed to correlate to its clinical efficacy and adverse effects. Insights gained from studies of knock-in mice expressing benzodiazepine-insensitive subtypes have provided the rationale for the development of positive allosteric modulators (PAMs) of α1-containing subtypes as hypnotics, of PAMs of α 2- or α3-containing subtypes as anxiolytics and analgesics, and of negative allosteric modulators (NAMs) of α 5-containing receptors as cognitive enhancers [12, 13, 15, 16, 24]. Moreover, other studies suggest that the anticonvulsive and anxiolytic effects of benzodiazepines may be mediated via α 2-containing GABAARs, whereas modulation of these subtypes has little or no sedative effects [25–29]. However, experiments in rodents indicate that the anticonvulsant effects require modulation of more than one specific α-subunit-containing GABAAR and that modulation of different subtypes may act synergistically [29].
To date, most benzodiazepines reported capable of modulating GABAAR signaling are 1,4-benzodiazepines. One of the few exceptions is the 1,5-benzodiazepine clobazam (Fig. 1). Clobazam (Onfi) has recently been approved in the United States and is indicated for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in patients aged 2 years and older, and outside the United States the drug is routinely administered for anxiety disorders and epilepsy [30–32]. Interestingly, clobazam has been reported to exhibit different in vivo adverse effects and addiction liability profile than the 1,4-benzodiazepines, including clonazepam (Klonopin, Fig. 1), an antiepileptic drug approved for the treatment of Lennox-Gastaut syndrome (petit mal variant), and akinetic and myoclonic seizures. For example, studies suggest that clobazam induces fewer psychomotor disturbances than clonazepam when dosed at clinically effective concentrations in healthy volunteers [33, 34]. Moreover, preclinical studies suggest that clobazam in contrast to clonazepam and other 1,4-benzodiazepines may exert more specific anticonvulsant/antiepileptic over sedative effects [35, 36]. The major active metabolite of clobazam, N-desmethylclobazam (Fig. 1), has been shown to have a longer plasma half-life than the parent compound (79 h vs. 36 h), resulting in greater metabolite plasma concentrations following long-term clobazam dosing in humans [37]. Hence, the metabolite is likely to contribute significantly to the clinical efficacy of clobazam, and interestingly N-desmethylclobazam has been reported to produce fewer adverse effects than clobazam [38, 39].
Fig 1. Chemical structures of clobazam, N-desmethylclobazam and clonazepam.

Considering that clobazam has been administered for various indications in the clinic for decades, the current insight into the pharmacological characteristics of clobazam and N-desmethylclobazam at GABAARs is surprisingly limited [40]. In a recent study, we delineated the binding characteristics of clobazam, N-desmethylclobam and the 1,4-benzodiazepine clonazepam at native GABAARs in rat brain membranes and at human α 1,2,3,5β2γ2SGABAARs expressed in HEK293 cells in a [3H]flumazenil competition binding assay [41]. In the present study, we have characterized the functional properties of clobazam and N-desmethylclobazam at human α 1β2γ2S, α 2β2γ2S, α 3β2γ2S, α 5β2γ2S and α 6β2δ GABAARs expressed in Xenopus laevis oocytes using two-electrode voltage clamp (TEVC) electrophysiology and compared these functionalities to that exhibited by clonazepam.
Materials and Methods
Materials
GABA, ZnCl2 and chemicals for buffers were obtained from Sigma-Aldrich (Denmark), and DS2 was obtained from Tocris Cookson (Bristol, UK). Clobazam (synthesized at H. Lundbeck A/S, Denmark), N-desmethylclobazam (from Johnson Matthey Pharma Services, MA, USA), clonazepam (from Lipomed AG, Switzerland), diazepam, and zolpidem (both from Sigma-Aldrich, Denmark) were dissolved in DMSO and diluted in Ringer buffer on the given experimental day. The cDNAs encoding human α1, α2, α3, α5, α6, β2, γ2S and δ GABAAR subunits were kind gifts from Dr. P J Whiting and Merck, Sharp & Dohme (Harlow, Essex, UK), and they were subcloned into mammalian expression vector pcDNA3.1 (Invitrogen, Denmark) as described previously [42, 43].
Preparation of cRNA and injection in Xenopus laevis oocytes
The cDNAs encoding the human GABAAR subunits were linearized with DraIII (α1, α2, α3, α5 and α6), SmaI (β2 and γ2S) or StuI (δ) and used as templates for in vitro cRNA synthesis using the T7 mMESSAGE mMACHINE High Yield Capped RNA Transcription Kit (Life Technologies Corporation, Carlsbad, CA, USA). Except where otherwise indicated, Xenopus oocytes were injected with 36.8 nL cRNA solution encoding for α1β2γ2S, α2β2γ2S, α3β2γ2S and α5β2γ2S GABAARs in a 1:1:1 α:β2:γ2S ratio (2.7 ng/μL of each subunit), with 46 nL cRNA solution encoding for the α6β2δ GABAAR in a 10:1:10 α6:β2:δ ratio (1:0.1:1 μg/μL), with 18 nL cRNA solution encoding for the α1β2 GABAAR in a 1:1 α1:β2 ratio (0.6:0.6 μg/μL), or with 46 nL cRNA encoding for α6β2 (α6:β2 ratio: 1:0.1 μg/μL). Following injection, the oocytes were incubated at 18°C in modified Barth’s solution [88 mM NaCl, 1 mM KCl, 15 mM HEPES (pH 7.5), 2.4 mM NaHCO3, 0.41 mM CaCl2, 0.82 mM MgSO4, 0.3 mM Ca(NO3)2, 100 U/mL penicillin and 100 μg/mL streptomycin]. Electrophysiological recordings were performed 3 to 6 days after injection.
Electrophysiological recordings
Electrophysiological recordings were performed using the TEVC technique on Xenopus oocytes expressing various GABAAR combinations using a protocol adapted from previous studies [44, 45]. Oocytes were placed in a recording chamber and gravity perfused with Ringer buffer [115 mM NaCl, 2.5 mM KCl, 10 mM HEPES (pH 7.5), 1.8 mM CaCl2, 0.1 mM MgCl2]. Cells were impaled with agar-plugged 0.5–1 MΩ electrodes containing 3 M KCl and voltage clamped at −70 mV by a Gene Clamp 500B amplifier (Axon Instruments, Union City, CA, USA) and recorded with pClamp 10 (Windows version, Molecular Devices, LLC, Sunnyvale, CA, USA). The oocytes were continuously perfused with Ringer buffer, and the test compounds were applied in the perfusate. Experiments were performed at room temperature and each data point represents the mean ± S.E.M. value of recordings performed on at least two oocytes from at least two different batches of oocytes. The recorded baseline-to-peak current amplitudes were analyzed using Clampfit 10.1 (Axon Instruments, Union City, CA, USA). Analogously to the procedures used in a recent study [43], the incorporation of the γ2S subunit into the GABAARs assembled at the cell surface of α1,2,3,5β2γ2S-expressing oocytes was confirmed on a routinely basis with 100 μM ZnCl2 [46]. The presence of δ in cell surface-expressed receptors in α6β2δ-injected oocytes was confirmed on a routinely basis using the δ-GABAAR selective PAM DS2 (1 μM) [47]. Furthermore, taking advantage of the differential sensitivity of αβ and αβδ receptors to Zn2+ as antagonist [45, 48–50], 1 μM ZnCl2 was used to verify that a homogenous population of ternary α6β2δ receptor complexes was expressed in these oocytes.
The GABA EC20 (the GABA concentration eliciting 20% of the maximum effect) for each receptor subtype was determined on two oocytes on each day of the experiment. A maximum concentration of GABA was applied until the peak of the response was observed, usually within 30 seconds. When two consecutive applications of the maximum GABA concentration were observed to elicit responses of similar effect (± 5%), 3–4 different concentrations of GABA were applied to the perfusate until the peak of the response was observed. Two to five minutes of wash time between each application were allowed to prevent receptor desensitization. Data for the GABA concentrations was normalized to the maximal response elicited by GABA on each oocyte, and the concentration-response curves were fitted in Prism GraphPad 5.0a (GraphPad Software, Inc. La Jolla, CA, USA) by nonlinear regression using the equation for sigmoidal dosage-response with variable slope (Equation 1): (1) Y = Bottom + [(Top—Bottom)/(1 + 10(logEC50-X)Hillslope)].
Bottom = response at the bottom plateau; EC50 = concentration giving rise to 50% of the maximum response; Top = response at the top plateau; X = logarithm of the concentration; Y = response.
The GABA EC20 response was calculated using Equation 2 (F = 20), and this concentration was subsequently used on a given experimental day. (2) log EC50 = (1/HillSlope) [log (F/(100-F)].
The functional characteristics of clobazam, N-desmethylclobazam, clonazepam, diazepam, and zolpidem on the GABAARs were determined by co-application of different concentrations of the compounds with GABA EC20. The test compounds were pre-applied 30 seconds prior to the co-application with GABA EC20. At the end of an experiment, a maximum concentration of GABA was applied in the perfusate to determine the maximum response elicited by GABA through the receptor, which served as the internal standard and as a control of any potential drift in the system during the recordings. Two to five minutes of wash time between each application were permitted to overcome receptor desensitization. Data for the benzodiazepines was normalized to the responses elicited by GABA EC20 at the receptor (the EC20 response was defined as 100%). Concentration-response curves for the benzodiazepines were fitted using Equation 1.
Data analysis
Using GraphPad Prism 4 (GraphPad Software, Inc., La Jolla, CA, USA), the pEC50 and Imax values were evaluated for statistical differences across the receptor subtypes per compound using one-way ANOVA with Tukey’s Multiple-Comparison Post-hoc Test, where P<0.05 was considered significant.
Results
Functional characterization of GABA and determination of GABA EC20 values at human α1β2γ2S, α2β2γ2S, α3β2γ2S, α5β2γ2S and α6β2δ GABAARs expressed in Xenopus oocytes. Prior to the functional characterization of the benzodiazepines at the five GABAAR subtypes, the pharmacological properties of GABA at the receptors were determined. The concentration-response relationships displayed by GABA at the receptors are given in Fig. 2, and the pharmacological data are summarized in Table 1. The EC50 and Hill slope values determined for the agonist were in good agreement with those observed in previous studies of these five receptors expressed in Xenopus oocytes [46, 51].
Fig 2. Functional properties of GABA at six human GABAARs expressed in Xenopus oocytes.
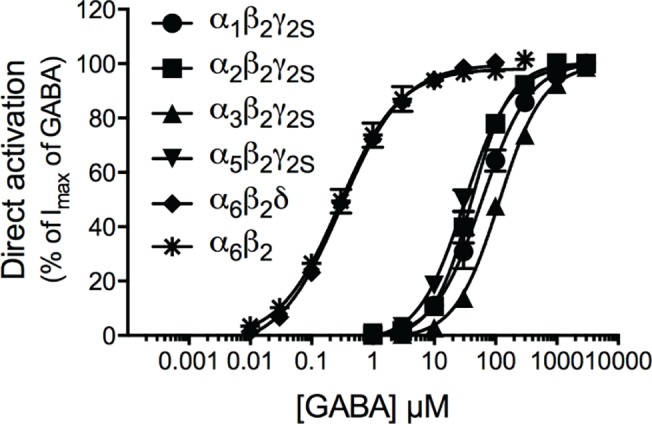
Concentration-response curves of GABA at the α1β2γ2S (circle), α2β2γ2S (square), α3β2γ2S (triangle), α5β2γ2S (inverted triangle), α6β2δ (diamond) and α6β2 (asterisk) GABAARs (means ± S.E.M.; N = 4–7).
Table 1. Functional properties of GABA at the human α1β2γ2S, α2β2γ2S, α3β2γ2S, α5β2γ2S, α6β2δ and α6β2GABAARs expressed in Xenopus oocytes.
The EC50 values are given in μM with pEC50 ± S.E.M. values in brackets, and the Hill slopes (nH ± S.E.M.) and the numbers of experiments (N) are also given.
| Receptor | EC50 [pEC50 ± S.E.M.] | nH ± S.E.M. | N |
|---|---|---|---|
| α1β2γ2S | 57 [4.25 ± 0.10] | 1.19 ± 0.05 | 6 |
| α2β2γ2S | 40 [4.40 ± 0.07] | 1.44 ± 0.07 | 4 |
| α3β2γ2S | 120 [3.94 ± 0.03] | 1.22 ± 0.05 | 7 |
| α5β2γ2S | 31 [4.50 ± 0.06] | 1.19 ± 0.08 | 6 |
| α6β2δ | 0.30 [6.52 ± 0.04] | 0.89 ± 0.02 | 6 |
| α6β2 | 0.29 [6.54 ± 0.07] | 0.89 ± 0.09 | 7 |
For the functional characterization of the benzodiazepines at the receptors, the GABA EC20 values were determined on the days of the experiments. The actual GABA concentrations constituting the EC20 values for the respective receptor subtypes varied within 2- to 4-fold from day to day (α1β2γ2S: 20–30 μM; α2β2γ2S: 25–45 μM; α3β2γ2S: 25–60 μM; α5β2γ2S: 15–45 μM; α6β2δ0.05–0.20 μM). Thus, this procedure enabled us to use very accurate GABA EC20 concentrations for these studies. In fact, retrospective evaluation of the specific GABA concentrations used for characterization of the benzodiazepines in these subsequent studies revealed that these varied very little from the calculated EC20 (as percentage of GABA Imax, means ± S.E.M., N): α1β2γ2S (20.2 ± 0.74, N = 15); α2β2γ2S (19.8 ± 0.89, N = 17); α3β2γ2S (21.0 ± 1.07, N = 11); α5β2γ2S (18.6 ± 1.30, N = 13); and α6β2δ (15.7 ± 1.47, N = 16).
Functional properties of clobazam, N-desmethylclobazam, and clonazepam at human α1β2γ2S, α2β2γ2S, α3β2γ2S, α5β2γ2S and α6β2δ GABAARs expressed in Xenopus oocytes. The functional properties of clobazam, N-desmethylclobazam, and clonazepam when preincubated and co-applied with EC20 GABA at the four α1,2,3,5β2γ2S GABAARs expressed in Xenopus oocytes are given in Table 2. Representative traces for each of the three compounds at the α5β2γ2Ssubtype are given in Fig. 3A, and the concentration-response relationships obtained for the three compounds at the four receptors are outlined in Fig. 3B.
Table 2. Functional properties of clobazam, N-desmethylclobazam, and clonazepam at the human α1β2γ2S, α2β2γ2S, α3β2γ2S, α5β2γ2S, α6β2δ and α6β2GABAARs expressed in Xenopus oocytes.
| Receptor | EC50 [pEC50 ± S.E.M.] | Imax ± S.E.M. (%) | N |
|---|---|---|---|
| Clobazam | |||
| α1β2γ2S | 132 [6.88 ± 0.05] | 256 ± 12 | 5 |
| α2β2γ2S | 138 [6.86 ± 0.04] | 261 ± 6.7 | 6 |
| α3β2γ2S | 240 [6.62 ± 0.10] | 269 ± 0.8 | 4 |
| α5β2γ2S | 174 [6.76 ± 0.03] | 216 ± 7.0 | 6 |
| α6β2δ | 55,000 [4.26 ± 0.04] | 528 ± 46 | 6 |
| N-desmethylclobazam | |||
| α1β2γ2S | 151 [6.82 ± 0.07] | 203 ± 8.5 | 6 |
| α2β2γ2S | 138 [6.86 ± 0.18] | 270 ± 20 | 6 |
| α3β2γ2S | 282 [6.55 ± 0.11] | 270 ± 27 | 5 |
| α5β2γ2S | 98 (123 and 79) b | 233 (248 and 217) b | 2 |
| α6β2δ | ∼300,000 [∼3.5] a | 2420 ± 210 a | 5 |
| α6β2 | ∼300,000 [∼3.5] a | 2350 ± 200 a | 4 |
| Clonazepam | |||
| α1β2γ2S | 15 [7.81 ± 0.12] | 209 ± 14 | 4 |
| α2β2γ2S | 7.4 [8.13 ± 0.04] | 253 ± 17 | 5 |
| α3β2γ2S | 16 (20 and 13) b | 316 (334 and 297) b | 2 |
| α5β2γ2S | 21 [7.68 ± 0.17] | 255 ± 25 | 5 |
| α6β2δ | 29,000 [4.53 ± 0.10] | 283 ± 38 | 5 |
a The concentration-response curves for N-desmethylclobazam at the α6β2δ and α6β2GABAARs were not saturated at the maximal concentration used (1 mM). Thus, for this receptors the EC50 and pEC50 values for N-desmethylclobazam are estimated from the data, and the currents evoked by 1 mM N-desmethylclobazam (in % of the GABA EC20 response) is given instead of Imax.
b The properties for N-desmethylclobazam at α5β2γ2S and for clonazepam at α3β2γ2S are based on two independent experiments (n = 2), and thus the mean EC50 and Imax values for the modulators are given with specific EC50 and Imax values determined in the two experiments in parantheses.
Fig 3. Functional properties of clobazam, N-desmethylclobazam and clonazepam at four human GABAARs expressed in Xenopus oocytes.

(A) Representative traces for various concentrations of clobazam (top), N-desmethylclobazam (middle) and clonazepam (bottom) co-applied with GABA EC20 to oocytes expressing the α5β2γ2S GABAAR. The black bars represent applications of GABA EC20 and of 3 mM GABA that elicits maximal current through the receptor. The grey bars represent applications of various concentrations of clobazam, N-desmethylclobazam or clonazepam (a 30 s pre-incubation with the compound followed by co-application of the compound and GABA EC20). (B) Concentration-response relationships for clobazam (top), N-desmethylclobazam (middle) and clonazepam (bottom) at α1β2γ2S, α2β2γ2S, α3β2γ2S and α5β2γ2S GABAARs in the presence of GABA EC20 (means ± S.E.M.; N = 2–6).
EC50 values are given in nM with pEC50 ± S.E.M. values in brackets, Imax values are given as % of the response evoked by GABA EC20 at the receptor, and the numbers of experiments (N) are also given.
Clobazam, N-desmethylclobazam, and clonazepam all potentiated GABA EC20-mediated responses through the α1β2γ2S, α2β2γ2S, α3β2γ2S and α5β2γ2S GABAARs in a reversible and concentration-dependent manner (Fig. 3A and 3B). Clobazam and N-desmethylclobazam displayed EC50 values within the 100–300 nM range at all four receptors, whereas clonazepam was 9-, 19-, 15-, and 8-fold more potent than clobazam and 10-, 19-, 18-, and 5-fold more potent than N-desmethylclobazam as a PAM at α1β2γ2S, α2β2γ2S, α3β2γ2S and α5β2γ2S, respectively (Table 2). In terms of modulatory efficacy, saturating concentrations of clobazam and N-desmethylclobazam potentiated the GABA EC20-evoked currents through the receptors 203–270%, corresponding to 40–54% of the maximum responses evoked by GABA through the respective receptors. Clonazepam potentiated the GABA EC20-evoked responses through the receptors of 209–316%, corresponding to 42–63% of the maximum GABA responses (Fig. 3B, Table 2).
Statistical evaluation of the differences in pEC50 and Imax values exhibited by clobazam, N-desmethylclobazam, and clonazepam at the four α1,2,3,5β2γ2S GABAAR subtypes was performed (Table 3). Significant differences (P<0.05) were identified for the pEC50 values for clobazam between α1β2γ2S and α3β2γ2S subtypes as well as between the α2β2γ2S and α3β2γ2S receptors. The Imax value of clobazam at α5β2γ2S was significantly smaller than those at α1β2γ2S (P<0.05), α2β2γ2S (P<0.01), and α3β2γ2S (P<0.01). The pEC50 value of N-desmethylclobazam at α3β2γ2S was significantly smaller than those at α2β2γ2S (P<0.01) and α5β2γ2S (P<0.05), and its Imax value at α1β2γ2S was significantly smaller than those obtained at α2β2γ2S and α3β2γ2S receptors (P<0.05). As for the functional properties of clonazepam at the four receptors, only a significant difference was identified for its Imax values at α3β2γ2S and α1β2γ2S (P<0.05).
Table 3. Statistical analysis of the functional properties of clobazam, N-desmethylclobazam, and clonazepam at the human α1β2γ2S, α2β2γ2S, α3β2γ2S and α5β2γ2S GABAARs.
| pEC50 Values | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clobazam | α1β2γ2S | α2β2γ2S | α3β2γ2S | α5β2γ2S | N-desmethylclobazam | α1β2γ2S | α2β2γ2S | α3β2γ2S | α5β2γ2S | Clonazepam | α1β2γ2S | α2β2γ2S | α3β2γ2S | α5β2γ2S |
| α1β2γ2S | — | NS | P<0.05 | NS | α1β2γ2S | — | NS | NS | NS | α1β2γ2S | — | NS | NS | NS |
| α2β2γ2S | — | — | P<0.05 | NS | α2β2γ2S | — | — | P<0.01 | NS | α2β2γ2S | — | — | NS | NS |
| α3β2γ2S | — | — | — | NS | α3β2γ2S | — | — | — | P<0.05 | α3β2γ2S | — | — | — | NS |
| α5β2γ2S | — | — | — | — | α5β2γ2S | — | — | — | — | α5β2γ2S | — | — | — | — |
| Imax Values | ||||||||||||||
| Clobazam | α1β2γ2S | α2β2γ2S | α3β2γ2S | α5β2γ2S | N-desmethylclobazam | α1β2γ2S | α2β2γ2S | α3β2γ2S | α5β2γ2S | Clonazepam | α1β2γ2S | α2β2γ2S | α3β2γ2S | α5β2γ2S |
| α1β2γ2S | — | NS | NS | P<0.05 | α1β2γ2S | — | P<0.05 | P<0.05 | NS | α1β2γ2S | — | NS | P<0.05 | NS |
| α2β2γ2S | — | — | NS | P<0.01 | α2β2γ2S | — | — | NS | NS | α2β2γ2S | — | — | NS | NS |
| α3β2γ2S | — | — | — | P<0.01 | α3β2γ2S | — | — | — | NS | α3β2γ2S | — | — | — | NS |
| α5β2γ2S | — | — | — | — | α5β2γ2S | — | — | — | — | α5β2γ2S | — | — | — | — |
P-values from one-way ANOVA testing if mean values (pEC50 and Imax) experimentally determined for the compounds with Tukey’s Multiple Comparison Post-hoc Test. NS; not significant.
Investigations into the benzodiazepine-mediated modulation of the α1,2,3,5β2γ2S GABAARs
As will be outlined in the Discussion section, the modulatory efficacies exhibited by benzodiazepines at αβγ GABAARs in recombinant expression systems in previous studies have varied considerably. To address this aspect, we compared the maximum responses mediated by saturating concentrations (3 μM) of clobazam, N-desmethylclobazam, and clonazepam at the GABAARs with the maximal responses mediated by a saturating concentration of diazepam, a 1,4-benzodiazepine often referred to as a “full benzodiazepine agonist”. The maximum responses evoked by clobazam and N-desmethylclobazam when co-applied with GABA EC20 at α1β2γ2S and α2β2γ2S GABAARs, respectively, were slightly but significantly smaller than those mediated by diazepam at the respective receptors (p<0.1; ordinary one-way ANOVA) (Table 2, Figs. 3B and 4A). In contrast, the maximum responses mediated by clobazam at α2,3,5β2γ2S, by N-desmethylclobazam at α1,3,5β2γ2S and by clonazepam at all the four receptors did not differ significantly from those exhibited by diazepam (Table 2, Figs. 3B and 4A). We also compared the maximum degree of potentiation mediated by clobazam at the α2β2γ2S GABAAR with the maximal responses induced by a saturating concentration of zolpidem. Albeit not a benzodiazepine, zolpidem acts as a PAM through the high-affinity benzodiazepine site in α1,2,3,5βγ GABAARs, and the compound has previously been reported to be a “full benzodiazepine-site agonist” at the α2β2γ2 subtype [52]. In this experiment cRNA for α2β2γ2S was injected in a ratio of 1:1:5 (α2:β2:γ2S) to facilitate the expression of a homogenous population of γ2S-containing complexes. The modulatory efficacies displayed by clobazam and zolpidem at the α2β2γ2S receptors in these experiments did not differ significantly (Fig. 4B). Moreover, the respective degrees of potentiation mediated by clobazam and zolpidem at the receptors expressed in these oocytes did not differ significantly from those at oocytes injected with an α2:β2:γ2S cRNA ratio of 1:1:1 (Fig. 4B and data not shown).
Fig 4. Comparison of the functional efficacies of clobazam, N-desmethylclobazam, and clonazepam at α1,2,3,5β2γ2S GABAARs with those of diazepam and zolpidem.
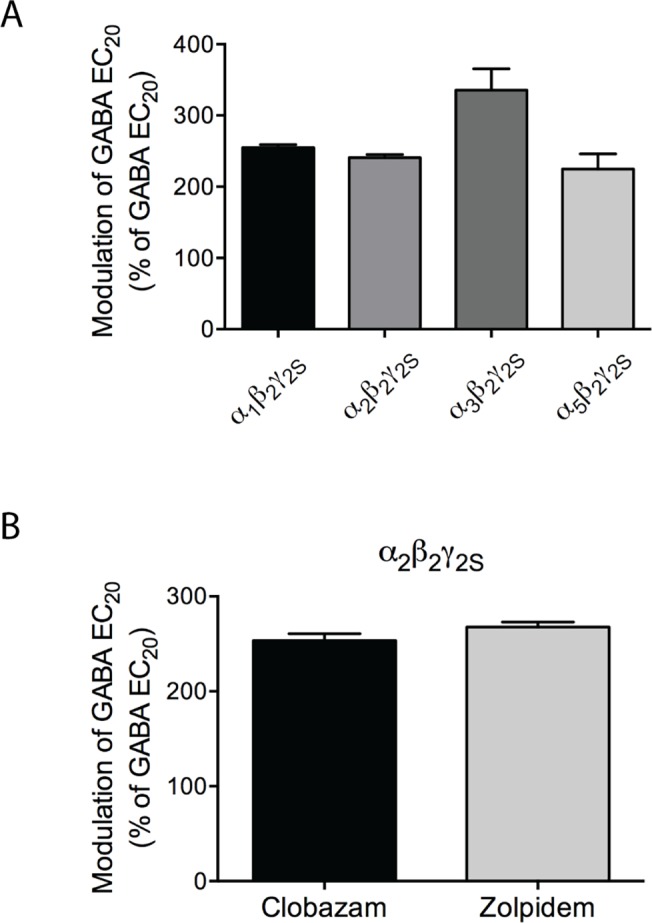
(A) Potentiation of the response elicited by GABA EC20 by 3 μM diazepam in Xenopus oocytes injected with cRNAs encoding for α1β2γ2S, α2β2γ2S, α3β2γ2S and α5β2γ2S GABAARs in a subunit ratio of 1:1:1 (means ± S.E.M.; N = 2–4) (B) Potentiation of the response elicited by EC20 GABA by 3 μM clobazam and 3 μM zolpidem in Xenopus oocytes injected with cRNAs encoding the α2β2γ2S GABAAR injected in a subunit ratio of 1:1:5 (means ± S.E.M.; N = 2).
In another series of experiments, we took advantage of the well-documented ability of zinc to discriminate between αβand αβγ GABAARs [46, 53, 54] to investigate whether oocytes injected with α1,2,3,5β2γ2S cRNAs in a 1:1:1 subunit ratio express homogeneous populations of ternary receptor complexes. As can be seen from Fig. 5, the GABA EC80-evoked response through the α1β2 GABAAR was almost completely eliminated by 100 μM Zn2+, whereas the presence of this concentration of the metal ion had negligible effect on the currents elicited in α1β2γ2S-expressing oocytes. This strongly suggests that the functional properties of clobazam, N-desmethylclobazam and clonazepam at the α1,2,3,5β2γ2S GABAARs have been determined at homogenous populations of γ2S-containing receptors.
Fig 5. Zinc-mediated inhibition of human α1β2 and α1β2γ2S GABAAR signalling in Xenopus oocytes.
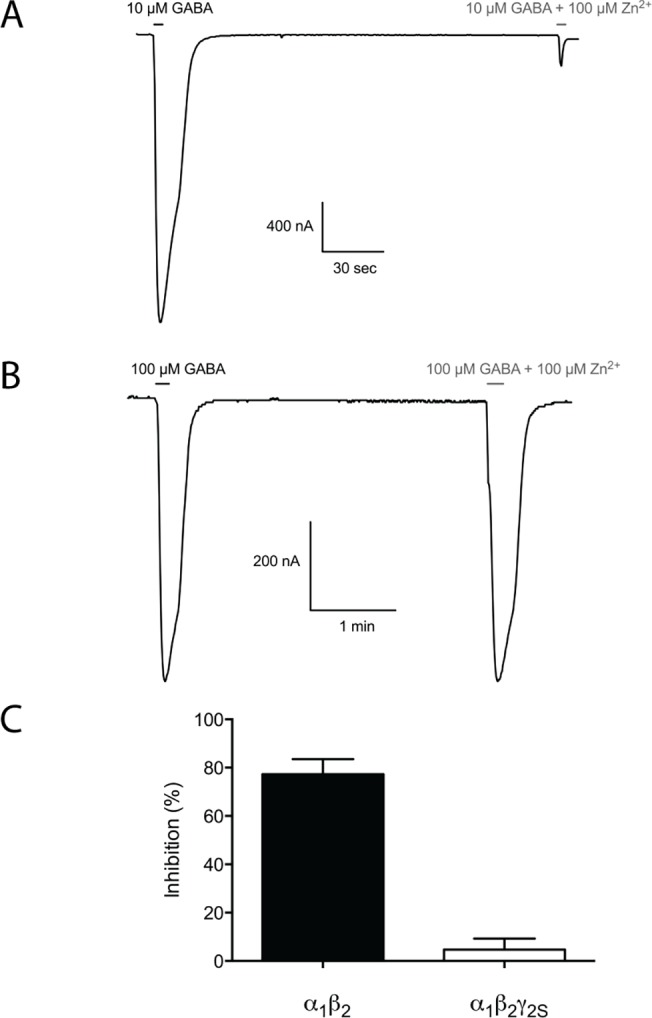
(A) Representative trace of the inhibition mediated 100 μM Zn2+ of the currents elicited by 10 μM GABA (EC80) through the α1β2 GABAAR. (B) Representative trace of the inhibition mediated 100 μM Zn2+ of the currents induced by 100 μM GABA (EC80) through the α1β2γ2S GABAAR. (C) The degree of inhibition mediated by 100 μM Zn2+ of GABA EC80-evoked currents in oocytes expressing α1β2 mean ± S.E.M.; 77 ± 6.3%; N = 7and α1β2γ2S (mean ± S.E.M.; 4.7 ± 4.6%; N = 6) GABAARs.
Functional properties of clobazam, N-desmethylclobazam, and clonazepam at the human α6β2δ GABAAR expressed in Xenopus oocytes
To investigate whether the functional properties of clobazam and its metabolite at αβδ GABAARs potentially differ from those of clonazepam, the three compounds were tested at a representative of these receptors, the human α6β2δ subtype (Table 2). Representative traces recorded for clobazam, N-desmethylclobazam and clonazepam when pre-incubated and co-applied with GABA EC20 at the receptor are presented in Fig. 6A, and concentration-response relationships determined for the compounds at the receptor are given in Fig. 6B.
Fig 6. . Functional properties of clobazam, N-desmethylclobazam and clonazepam at the human α6β2δ and α6β2 GABAARs expressed in Xenopus oocytes.

(A) Representative traces for various concentrations of clobazam (top), N-desmethylclobazam (middle) and clonazepam (bottom) co-applied with GABA EC20 to oocytes expressing the α6β2δ GABAAR. The black bars represent applications of GABA EC20 and of 300 M GABA that elicits maximal current through the receptor. The grey bars represent applications of various concentrations of clobazam, N-desmethylclobazam or clonazepam (a 30 s pre-incubation with the compound followed by co-application of the compound and GABA EC20). (B) Concentration-response relationships for clobazam (top), N-desmethylclobazam (middle) and clonazepam (bottom) at the α6β2δ GABAAR and for N-desmethylclobazam at the α6β2 GABAAR (middle) in the presence of GABA EC20 (means ± S.E.M.; N = 4–6).
All three modulators potentiated GABA EC20-mediated signaling in α6β2δ-oocytes in a concentration-dependent and reversible manner, exhibiting EC50 values in the mid-to-high micromolar range at the receptor (Table 2). Thus, the modulatory potencies of clobazam, N-desmethylclobazam and clonazepam at this receptor were 200–400, 1100–3100 and 1400–3900 fold lower than those exhibited by the respective benzodiazepines at the α1,2,3,5β2γ2S GABAARs, respectively (Table 2). Strikingly, however, the modulatory efficacies of the two 1,5-benzodiazepines at this receptor were very different from those at the four α1,2,3,5β2γ2S receptors. Whereas the degree of potentiation of the GABA EC20-evoked response through α6β2δ mediated by clonazepam was comparable to those at the α1,2,3,5β2γ2S GABAARs, clobazam and in particular N-desmethylclobazam were much more efficacious PAMs at the receptor (Table 2, Fig. 6). Albeit the concentration-response relationship for N-desmethylclobazam at the α6β2δ receptor was not saturated within the concentration range tested, 1 mM of the drug was found to potentiate the GABA EC20-evoked response through the receptor by ∼24-fold (Fig. 6A and 6B). Interestingly, a distinct inhibition phase in the concentration-response relationship was observed in some of these recordings for clobazam and clonazepam, with 1 mM of the modulator resulting in a lower degree of potentiation than 300 μM (Fig. 6A). Moreover, rebound currents were observed at these high modulator concentrations (Fig. 6A).
To elucidate the molecular basis for the high-efficacious modulation exerted by N-desmethylclobazam at the α6β2δ GABAAR, we characterized the functional characteristics of the compound at the binary α6β2 receptor. N-desmethylclobazam also potentiated the GABA EC20-mediated signaling through this receptor in a concentration-dependent manner, exhibiting modulatory potency and efficacy not significantly different from those displayed at α6β2δ (Table 2, Fig. 6B). However, it should be stressed that the EC50 and Imax values given for N-desmethylclobazam at α6β2δ and α6β2 are estimates, since neither of the concentration-response curves for the modulator at these two receptors reached saturation.
Discussion
In view of the clinical use of clobazam for the treatment of various diseases over the last decades, surprising little is known about the in vitro pharmacology of the drug. In the present study, we have performed an elaborate functional characterization of clobazam, its major active metabolite N-desmethylclobazam and the 1,4-benzodiazepine clonazepam at the human α1β2γ2S, α2β2γ2S, α3β2γ2S, α5β2γ2S and α6β2δ GABAARs expressed in Xenopus oocytes. A detailed functional characterization of clonazepam at recombinant GABAARs has to our knowledge not been published previously, and thus this study provides substantial insights into the molecular pharmacology of all three modulators.
Each of the three benzodiazepines exhibited similar EC50 values as a PAM at the four α1,2,3,5β2γ2S GABAARs, and the maximum responses evoked by saturating concentrations of each of the modulators upon co-application with GABA EC20 at these four subtypes were also very similar (Table 2). Although statistical analysis identified differences between some of the potencies and efficacies displayed by the respective compounds at the four receptors (Table 3), these differences are not considered pertinent from a biological perspective. It is important to stress that the α1,2,3,5β2γ2S receptors included in this study only constitute a selection of all αβγ receptors targeted by benzodiazepines in clinically relevant concentrations. However, the fact that benzodiazepines have been reported to be less potent and less efficacious modulators of αβγ1 and αβγ3 receptors than of the corresponding αβγ2 receptors [55–57] combined with the very restricted expression of γ1 and γ3 in the CNS [58, 59] strongly suggests that the contributions of these receptor assemblies to the overall clinical effects of benzodiazepines are negligible. More importantly, the identity of the β subunit in the αβγ2 GABAAR complex is not believed to influence benzodiazepine pharmacology substantially [56, 57], just as there to our knowledge are no reports of benzodiazepines exhibiting significantly different pharmacological properties at γ2S- and γ2L-containing GABAARs. Thus, although we can not exclude the possibility that the functional properties of clobazam, N-desmethylclobazam and/or clonazepam at α1,2,3,5βγ2 complexes comprising β1β3 and/or γ2L subunits could differ from those observed at the receptors in this study, we propose that the three benzodiazepines are likely to act as non-selective PAMs at all α1,2,3,5βγ2 receptors.
In our recent study of the binding properties of the three benzodiazepines, clobazam and N-desmethylclobazam displayed slightly but significantly higher binding affinities at the α2β2γ2S GABAAR compared to the α1β2γ2S subtype (2.5- and 4.3-fold, respectively), whereas clonazepam exhibited significantly higher binding affinities to α1,2,5β2γ2S subtypes than to the α3β2γ2S receptor (2.8- to 3.4-fold) [41]. The fact that these binding subtype-preferences are not mirrored in the functional profiles of the modulators is not particular surprising, given the different methodologies used to assess binding affinities and functional potencies of modulators. In support of this, another interesting observation that can be extracted from the two studies is that the 200–1000 fold higher binding affinities displayed by clonazepam compared to clobazam and N-desmethylclobazam at the four α1,2,3,5β2γ2S receptors in the [3H]flumazenil binding assay translate into considerably less pronounced differences (5–19 fold) between the EC50 values of the 1,4-benzodiazepine and the two 1,5-benzodiazepines at the receptors in the oocyte recordings (Table 2) [41]. These observations may reflect differences in the degrees of receptor desensitization in the two assays.
A comprehensive literature search has only identified two previous studies of the functional properties of clobazam and N-desmethylclobazam at recombinant GABAARs [55, 60]. Clobazam has been reported to exert negligible potentiation of GABA EC5-EC10-evoked responses through α1β2γ1 receptors in Xenopus oocytes, suggesting that the presence of this γ subunit in the GABAAR complex alters the functionality of the 1,5-benzodiazepine substantially [55]. Of particular interest for this study, Fisher has performed a direct comparison of the modulation mediated by clonazepam, clobazam, and N-desmethylclobazam on the current elicited by 3 μM GABA (EC10–EC20) through the rat α3β3γ2L GABAAR expressed in HEK-293T cells by patch clamp electrophysiology [60]. The EC50 values determined for clonazepam, clobazam, and N-desmethylclobazam at the rat α3β3γ2L receptor in the Fisher study were 90 nM, 490 nM and 550 nM, respectively, which are in good agreement with the potencies exhibited by three benzodiazepines at the human α3β2γ2S receptor in this study (Table 2) [60]. However, Fisher found clobazam and diazepam to be substantially more efficacious potentiators of rat α3β3γ2L currents (Imax values of 487% and 508% of the responses evoked by GABA EC10–EC20, respectively) than N-desmethylclobazam and clonazepam (Imax values of 270% and 263%, respectively) [60]. Whereas the modulatory efficacies of N-desmethylclobazam and clonazepam at α3β3γ2L are in good agreement with those at the α3β2γ2S receptor in the present study, the higher efficacies exhibited by clobazam and diazepam in the Fisher study clearly contrast the comparable Imax values determined for the four PAMs at the α3β2γ2S receptor in this study (Table 2, Figs. 3 and 4). Several factors might explain this apparent discrepancy, including the different receptors studied (rat α3β3γ2L vs. human α3β2γ2S), the different expression systems (HEK-293T cells vs. Xenopus oocytes), and the different recording techniques (patch clamp vs. TEVC electrophysiology). We propose that the precise determination of the GABA EC20 used for the characterization of the benzodiazepines in the present study can have facilitated a more precise determination of the degree of maximum potentiation of the GABA-evoked responses than the GABA EC10–EC20 concentrations employed in the Fisher study. On the other hand, the faster application rate of the compounds in patch clamp recordings using HEK-293T cells may have resulted in less concomitant desensitization of the receptors during application than in the Xenopus oocyte recordings system, which is characterized by slower exchange rates. Thus, we cannot exclude the possibility that concurrent desensitization of the receptors upon co-application of GABA EC20 with high benzodiazepine concentrations could constitute a ceiling effect with respect to the degree of maximum response elicited by the benzodiazepine-bound receptor, and that this effect potentially can have masked putative differential efficacies of the benzodiazepines at the receptors. However, as will be outlined below, the divergent efficacies reported for benzodiazepines at GABAARs expressed in oocytes in the literature strongly suggest that establishing a “true efficacy” for any given benzodiazepine is not trivial.
In the absence of other previous studies of the functional characteristics of clobazam, N-desmethylclobazam and/or clonazepam at GABAARs expressed in Xenopus oocytes, we investigated whether the determined modulatory efficacies for these compounds could be considered reliable by comparing them to the efficacies mediated by two reference benzodiazepine-site modulators. A saturating concentration of the prototypic benzodiazepine diazepam was observed to induce comparable or slightly higher responses than those mediated by saturating concentrations of clobazam, N-desmethylclobazam and clonazepam at the four α1,2,3,5β2γ2S subtypes (Figs. 3B and 4A). Furthermore, the maximum responses induced by zolpidem at α1β2γ2S and α2β2γ2S receptors were comparable to those mediated by clobazam (Fig. 4B and data not shown). Unfortunately, the efficacies determined for diazepam and zolpidem at GABAARs expressed in oocytes in previous studies have varied considerably. Although often referred to as a “full benzodiazepine agonist,” diazepam has exhibited very different efficacies as a PAM of GABA EC10- to EC20-evoked currents through α1β2γ2 receptors in previous studies, including degrees of potentiation in the same 2.3- to 3.4-fold range as exhibited by the modulator at the α1,2,3,5β2γ2S receptors in this study [61–63]. Likewise, whereas zolpidem has been reported to potentiate the GABA EC15- to EC20-evoked responses through α1β2γ2 and α2β2γ2 receptors 4- to 6-fold in some studies [52, 64, 65], its maximum modulation of GABA-evoked currents through the receptors in other studies have been very similar to the 2.7-fold potentiation observed at α2β2γ2S in this study [61, 62, 66]. In conclusion, the results in this study indicate that clobazam and N-desmethylclobazam are equally efficacious or almost as efficacious as the 1,4-benzodiazepines clonazepam and diazepam at the human α1,2,3,5β2γ2S receptors. However, considering the variation in the efficacies reported for standard benzodiazepines in the literature, the absolute degrees of potentiation exerted by the two 1,5-benzodiazepines at the receptors in other recording set-ups could potentially differ from those observed in this study.
The α6β2δ receptor was included in this study as a representative of the δ-containing GABAARs, and clobazam, N-desmethylclobazam and clonazepam all displayed mid-to-high micromolar potencies as PAMs of this receptor (Fig. 6, Table 2). Apart from the obvious absence of an α(+)/γ(–) subunit interface in the α6β2δ complex, the distinct functional characteristics exhibited by the three benzodiazepines at this receptor support the notion of them acting through another allosteric site than the classical high-affinity benzodiazepine binding site, and the comparable potencies displayed by N-desmethylclobazam as a PAM at the α6β2 and α6β2δ receptors strongly suggest that this site is comprised within the αβ regions of the αβδ complex (Fig. 6B, Table 2). Some of the characteristics displayed by the benzodiazepines at the α6β2δ GABAAR could be argued to be indicative of a binding site in the transmembrane domain of the receptor. The tendencies towards bell-shaped concentration-response curves observed for clobazam and clonazepam as well as the rebound currents observed at high concentrations of the modulators are certainly reminiscent of the characteristics previously reported for PAMs/ago-PAMs acting through transmembrane domains of GABAARs [55, 67–72]. Moreover, whereas PAMs targeting extracellular non-cannonical subunit interfaces in GABAARs and other Cys-loop receptors historically predominantly have been found to increase agonist potency without affecting the maximal agonist response through the receptors significantly [20, 73, 74], several PAMs acting through the transmembrane domains of the receptors have been shown capable of increasing agonist-evoked maximal peak currents, analogously to the high-efficacious potentiation of α6β2δ signaling mediated by N-desmethylclobazam [67, 74–76]. However, this phenotypic difference between PAMs acting through extracellular and transmembrane regions of Cys-loop receptors does not appear to be black-and-white, as illustrated by the pronounced enhancement of GABA efficacy at the α1β3δ GABAAR mediated by LAU 177 through a site in the extracellular α1 (+)/β3 (–) interface [22] and by the augmentation of agonist efficacy exerted by the anthelmintic drug morantel through the extracellular β2 (+)/α3 (–) interface of the α3β2 nicotinic acetylcholine receptor [77]. Thus, not having explored the modes of action of clobazam, N-desmethylclobazam and clonazepam at the α6β2δ GABAAR in detail, we will refrain from speculations about whether the modulators target a low-affinity binding site located in the transmembrane domain, in the extracellular α(+)/β(–) subunit interface, or in another region of the receptor complex.
The nature and efficacies of the modulation exerted by clobazam and N-desmethylclobazam at other αβ and αβδ receptors could potentially differ from those observed for the compounds at α6β2 and α6β2δ. However, the two 1,5-benzodiazepines are likely to be weak modulators at all αβand αβδ GABAARs, thus mediating their effects at these receptors at substantially higher concentrations than those required to modulate αβγ receptors. Although the μM activity component of benzodiazepines previously has been proposed to contribute to the CNS depression observed at high in vivo concentrations [18], we find it highly improbable that this component contributes significantly to the clinical efficacy of clobazam/N-desmethylclobazam. On the other hand, the highly efficacious potentiation of α6β2δ GABAAR signaling exerted by N-desmethylclobazam is quite interesting from a molecular perspective, and we propose that the 1,5-benzodiazepine could constitute an interesting lead scaffold for the development of novel allosteric modulators targeting this elusive low-affinity benzodiazepine binding site in the GABAARs.
Conclusion
The present study represents the first elaborate in vitro functional characterization of clobazam, N-desmethylclobazam and clonazepam at recombinant human GABAARs. While both 1,5-benzodiazepines are potent PAMs of human α1,2,3,5β2γ2S receptors, they appear to be non-selective both in terms of potency and efficacy, a characteristic they share with many classical 1,4-benzodiazepines including clonazepam (Table 2) [15, 16]. Obviously we can not completely exclude the possibility that the functionalities of clobazam and/or N-desmethylclobazam at one or a few of the GABAAR subtypes not included in this study could differ substantially from those of clonazepam and other 1,4-benzodiazepines, and that this difference could contribute to the distinct properties observed for clobazam compared with the classical 1,4-benzodiazepines in the clinic. However, judging from the results in this study we propose that these clinical differences are more likely to be rooted in other factors than the in vitro pharmacological properties of the modulators, such as their respective pharmacokinetic characteristics.
Acknowledgments
Dr. Paul J. Whiting and Merck, Sharp & Dohme are thanked for their generous gifts of the GABAAR subunit cDNAs. Anders A. Jensen thanks the Novo Nordisk Foundation for financial support. Editorial support during manuscript preparation was provided by Apurva Davé, PhD, of Prescott Medical Communications Group (Chicago, IL), and Michael A. Nissen, ELS, Lundbeck LLC (Deerfield, IL).
Data Availability
All relevant data are within the paper.
Funding Statement
HH and AAJ received an unrestricted grant from Lundbeck (US) to conduct this work. AAJ thanks the Novo Nordisk Foundation for financial support. Lundbeck (Denmark) provided support in the form of salaries for authors HSJ and BE, but neither Lundbeck (US) or Lundbeck (Denmark) have had any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABAB receptors. Physiol Rev 2004;84: 835–867. [DOI] [PubMed] [Google Scholar]
- 2. Olsen RW, Sieghart W. GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 2009;56: 141–148. 10.1016/j.neuropharm.2008.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lynch JW. Native glycine receptor subtypes and their physiological roles. Neuropharmacology 2009;56: 303–309. 10.1016/j.neuropharm.2008.07.034 [DOI] [PubMed] [Google Scholar]
- 4. Barnes NM, Hales TG, Lummis SC, Peters JA. The 5-HT3 receptor—the relationship between structure and function. Neuropharmacology 2009;56: 273–284. 10.1016/j.neuropharm.2008.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jensen AA, Frølund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem 2005;48: 4705–4745. [DOI] [PubMed] [Google Scholar]
- 6. Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacological reviews 2008;60: 243–260. 10.1124/pr.108.00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whiting PJ. GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov Today 2003;8: 445–450. [DOI] [PubMed] [Google Scholar]
- 8. Baumann SW, Baur R, Sigel E. Forced subunit assembly in α1β2γ2 GABAA receptors. Insight into the absolute arrangement. J Biol Chem 2002;277: 46020–46025. [DOI] [PubMed] [Google Scholar]
- 9. Baur R, Minier F, Sigel E. A GABAA receptor of defined subunit composition and positioning: concatenation of five subunits. FEBS Lett 2006;580: 1616–1620. [DOI] [PubMed] [Google Scholar]
- 10. Brickley SG, Mody I. Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 2012;73: 23–34. 10.1016/j.neuron.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW. Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci 2009;29: 12757–12763. 10.1523/JNEUROSCI.3340-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Atack JR. GABAA receptor subtype-selective modulators. I. α2/α3-selective agonists as non-sedating anxiolytics. Curr Top Med Chem 2011;11: 1176–1202. [DOI] [PubMed] [Google Scholar]
- 13. Atack JR. GABAA receptor subtype-selective modulators. II. α5-selective inverse agonists for cognition enhancement. Curr Top Med Chem 2011;11: 1203–1214. [DOI] [PubMed] [Google Scholar]
- 14.Sieghart W. Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv Pharmacol in press. [DOI] [PubMed]
- 15. Ebert B, Wafford KA, Deacon S. Treating insomnia: Current and investigational pharmacological approaches. Pharmacol Ther 2006;112: 612–629. [DOI] [PubMed] [Google Scholar]
- 16. Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov 2011;10: 685–697. 10.1038/nrd3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sigel E, Lüscher BP. A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr Top Med Chem 2011;11: 241–246. [DOI] [PubMed] [Google Scholar]
- 18. Walters RJ, Hadley SH, Morris KD, Amin J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci 2000;3: 1274–1281. [DOI] [PubMed] [Google Scholar]
- 19. Baur R, Tan KR, Lüscher BP, Gonthier A, Goeldner M, Sigel E. Covalent modification of GABAA receptor isoforms by a diazepam analogue provides evidence for a novel benzodiazepine binding site that prevents modulation by these drugs. Journal of neurochemistry 2008;106: 2353–2363. 10.1111/j.1471-4159.2008.05574.x [DOI] [PubMed] [Google Scholar]
- 20. Ramerstorfer J, Furtmuller R, Sarto-Jackson I, Varagic Z, Sieghart W, Ernst M. The GABAA receptor α+β- interface: a novel target for subtype selective drugs. J Neurosci 2011;31: 870–877. 10.1523/JNEUROSCI.5012-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Varagic Z, Ramerstorfer J, Huang S, Rallapalli S, Sarto-Jackson I, Cook J, et al. Subtype selectivity of α+β- site ligands of GABAA receptors: identification of the first highly specific positive modulators at α6β2/3γ2 receptors. Br J Pharmacol 2013;169: 384–399. 10.1111/bph.12153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mirheydari P, Ramerstorfer J, Varagic Z, Scholze P, Wimmer L, Mihovilovic MM, et al. Unexpected properties of δ-containing GABAA receptors in response to ligands interacting with the α+ β- site. Neurochemical research 2014;39: 1057–1067. 10.1007/s11064-013-1156-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wallner M, Hanchar HJ, Olsen RW. Alcohol selectivity of β3-containing GABAA receptors: evidence for a unique extracellular alcohol/imidazobenzodiazepine Ro15–4513 binding site at the α+β- subunit interface in αβ3δ GABAA receptors. Neurochemical research 2014;39: 1118–1126. 10.1007/s11064-014-1243-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engin E, Liu J, Rudolph U. α2-containing GABAA receptors: a target for the development of novel treatment strategies for CNS disorders. Pharmacol Ther 2012;136: 142–152. 10.1016/j.pharmthera.2012.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanger DJ, Benavides J, Perrault G, Morel E, Cohen C, Joly D, et al. Recent developments in the behavioral pharmacology of benzodiazepine (omega) receptors: evidence for the functional significance of receptor subtypes. Neurosci Biobehav Rev 1994;18: 355–372. [DOI] [PubMed] [Google Scholar]
- 26. McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat Neurosci 2000;3: 587–592. [DOI] [PubMed] [Google Scholar]
- 27. Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR. Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates. Proceedings of the National Academy of Sciences of the United States of America 2005;102: 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acidA receptor subtypes. Nature 1999;401: 796–800. [DOI] [PubMed] [Google Scholar]
- 29. Fradley RL, Guscott MR, Bull S, Hallett DJ, Goodacre SC, Wafford KA, et al. Differential contribution of GABAA receptor subtypes to the anticonvulsant efficacy of benzodiazepine site ligands. J Psychopharmacol 2007;21: 384–391. [DOI] [PubMed] [Google Scholar]
- 30. Cramer JA, Sapin C, Francois C. Indirect comparison of clobazam and other therapies for Lennox-Gastaut syndrome. Acta Neurol Scand 2013;128: 91–99. 10.1111/ane.12086 [DOI] [PubMed] [Google Scholar]
- 31. Leahy JT, Chu-Shore CJ, Fisher JL. Clobazam as an adjunctive therapy in treating seizures associated with Lennox-Gastaut syndrome. Neuropsychiatr Dis Treat 2011;7: 673–681. 10.2147/NDT.S20173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ng YT, Collins SD. Clobazam. Neurotherapeutics 2007;4: 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Meyden CH, Bartel PR, Sommers DK, Blom M, Pretorius LC. Effects of clobazam and clonazepam on saccadic eye movements and other parameters of psychomotor performance. Eur J Clin Pharmacol 1989;37: 365–369. [DOI] [PubMed] [Google Scholar]
- 34. Wildin JD, Pleuvry BJ, Mawer GE, Onon T, Millington L. Respiratory and sedative effects of clobazam and clonazepam in volunteers. Br J Clin Pharmacol 1990;29: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steru L, Chermat R, Millet B, Mico JA, Simon P. Comparative study in mice of ten 1,4-benzodiazepines and of clobazam: anticonvulsant, anxiolytic, sedative, and myorelaxant effects. Epilepsia 1986;27 Suppl 1: S14–17. [DOI] [PubMed] [Google Scholar]
- 36. Miura Y, Amano S, Torii R, Ihara N. Clobazam shows a different antiepileptic action profile from clonazepam and zonisamide in Ihara epileptic rats. Epilepsy research 2002;49: 189–202. [DOI] [PubMed] [Google Scholar]
- 37.Tolbert D, Bekersky I. Pharmacokinetics of N-desmethylclobazam, the active and primary metabolite of Clobazam. 66th Annual Meeting of the American Epilepsy Society. San Diego, CA. Epilepsy Currents 2013;13: Suppl 1. Abstract #2.226.
- 38. Haigh JR, Pullar T, Gent JP, Dailley C, Feely M. N-desmethylclobazam: a possible alternative to clobazam in the treatment of refractory epilepsy? Br J Clin Pharmacol 1987;23: 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kinoshita M, Ikeda A, Begum T, Terada K, Shibasaki H. Efficacy of low-dose, add-on therapy of clobazam (CLB) is produced by its major metabolite, N-desmethyl-CLB. J Neurol Sci 2007;263: 44–48. [DOI] [PubMed] [Google Scholar]
- 40. Sankar R. GABAA receptor physiology and its relationship to the mechanism of action of the 1,5-benzodiazepine clobazam. CNS Drugs 2012;26: 229–244. 10.2165/11599020-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 41. Jensen HS, Nichol K, Lee D, Ebert B. Clobazam and its active metabolite N-desmethylclobazam display significantly greater affinities for α2- versus α1-GABAA-receptor complexes. PLoS One 2014;9: e88456 10.1371/journal.pone.0088456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jensen AA, Bergmann ML, Sander T, Balle T. Ginkgolide X is a potent antagonist of anionic Cys-loop receptors with a unique selectivity profile at glycine receptors. J Biol Chem 2010;285: 10141–10153. 10.1074/jbc.M109.079319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoestgaard-Jensen K, Dalby NO, Krall J, Hammer H, Krogsgaard-Larsen P, Frølund B, et al. Probing α4βδ GABAA receptor heterogeneity: differential regional effects of a functionally selective α4β1δ/α4β3δ receptor agonist on tonic and phasic inhibition in rat brain. J Neurosci 2014;34: 16256–16272 10.1523/JNEUROSCI.1495-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Storustovu S, Ebert B. Gaboxadol: in vitro interaction studies with benzodiazepines and ethanol suggest functional selectivity. European journal of pharmacology 2003;467: 49–56. [DOI] [PubMed] [Google Scholar]
- 45. Storustovu SI, Ebert B. Pharmacological characterization of agonists at δ-containing GABAA receptors: Functional selectivity for extrasynaptic receptors is dependent on the absence of γ2. J Pharmacol Exp Ther 2006;316: 1351–1359. [DOI] [PubMed] [Google Scholar]
- 46. Karim N, Wellendorph P, Absalom N, Johnston GA, Hanrahan JR, Chebib M. Potency of GABA at human recombinant GABAA receptors expressed in Xenopus oocytes: a mini review. Amino Acids 2013;44: 1139–1149. 10.1007/s00726-012-1456-y [DOI] [PubMed] [Google Scholar]
- 47. Wafford KA, van Niel MB, Mab QP, Horridge E, Herd MB, Peden DR, et al. Novel compounds selectively enhance δ subunit containing GABAA receptors and increase tonic currents in thalamus. Neuropharmacology 2009;56: 182–189. 10.1016/j.neuropharm.2008.08.004 [DOI] [PubMed] [Google Scholar]
- 48. Karim N, Wellendorph P, Absalom N, Bang LH, Jensen ML, Hansen MM, et al. Low nanomolar GABA effects at extrasynaptic α4β1/β3δ GABAA receptor subtypes indicate a different binding mode for GABA at these receptors. Biochem Pharmacol 2012;84: 549–557. 10.1016/j.bcp.2012.05.017 [DOI] [PubMed] [Google Scholar]
- 49. You H, Dunn SM. Identification of a domain in the δsubunit (S238-V264) of the α4β3δ GABAA receptor that confers high agonist sensitivity. Journal of neurochemistry 2007;103: 1092–1101. [DOI] [PubMed] [Google Scholar]
- 50. Krishek BJ, Moss SJ, Smart TG. Interaction of H+ and Zn2+ on recombinant and native rat neuronal GABAA receptors. The Journal of physiology 1998;507 (Pt 3): 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoestgaard-Jensen K, O’Connor RM, Dalby NO, Simonsen C, Finger BC, Golubeva A, et al. The orthosteric GABAA receptor ligand 5-(4-piperidyl)-3-isothiazolol (Thio-4-PIOL) displays distinctly different functional properties at synaptic and extrasynaptic receptors. Br J Pharmacol 2013;170: 919–932. 10.1111/bph.12340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petroski RE, Pomeroy JE, Das R, Bowman H, Yang W, Chen AP, et al. Indiplon is a high-affinity positive allosteric modulator with selectivity for α1 subunit-containing GABAA receptors. The Journal of pharmacology and experimental therapeutics 2006;317: 369–377. [DOI] [PubMed] [Google Scholar]
- 53. Smart TG, Moss SJ, Xie X, Huganir RL. GABAA receptors are differentially sensitive to zinc: dependence on subunit composition. Br J Pharmacol 1991;103: 1837–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hosie AM, Dunne EL, Harvey RJ, Smart TG. Zinc-mediated inhibition of GABAA receptors: discrete binding sites underlie subtype specificity. Nat Neurosci 2003;6: 362–369. [DOI] [PubMed] [Google Scholar]
- 55. Khom S, Baburin I, Timin EN, Hohaus A, Sieghart W, Hering S. Pharmacological properties of GABAA receptors containing γ1 subunits. Mol Pharmacol 2006;69: 640–649. [DOI] [PubMed] [Google Scholar]
- 56. Hevers W, Lüddens H. The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Molecular neurobiology 1998;18: 35–86. [DOI] [PubMed] [Google Scholar]
- 57. Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacol Rev 1995;47: 181–234. [PubMed] [Google Scholar]
- 58. Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 2000;101: 815–850. [DOI] [PubMed] [Google Scholar]
- 59. Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci 1992;12: 1040–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fisher JL. Interactions between modulators of the GABAA receptor: Stiripentol and benzodiazepines. European journal of pharmacology 2011;654: 160–165. 10.1016/j.ejphar.2010.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bowser DN, Wagner DA, Czajkowski C, Cromer BA, Parker MW, Wallace RH, et al. Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [γ2(R43Q)] found in human epilepsy. Proceedings of the National Academy of Sciences of the United States of America 2002;99: 15170–15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Buhr A, Baur R, Malherbe P, Sigel E. Point mutations of the α1β2γ2 gamma-aminobutyric acidA receptor affecting modulation of the channel by ligands of the benzodiazepine binding site. Mol Pharmacol 1996;49: 1080–1084. [PubMed] [Google Scholar]
- 63. Rabe H, Kronbach C, Rundfeldt C, Luddens H. The novel anxiolytic ELB139 displays selectivity to recombinant GABAA receptors different from diazepam. Neuropharmacology 2007;52: 796–801. [DOI] [PubMed] [Google Scholar]
- 64. Morlock EV, Czajkowski C. Different residues in the GABAA receptor benzodiazepine binding pocket mediate benzodiazepine efficacy and binding. Mol Pharmacol 2011;80: 14–22. 10.1124/mol.110.069542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rahman M, Lindblad C, Johansson IM, Backstrom T, Wang MD. Neurosteroid modulation of recombinant rat α5β2γ2L and α1β2γ2L GABAA receptors in Xenopus oocyte. European journal of pharmacology 2006;547: 37–44. [DOI] [PubMed] [Google Scholar]
- 66. Sanna E, Busonero F, Talani G, Carta M, Massa F, Peis M, et al. Comparison of the effects of zaleplon, zolpidem, and triazolam at various GABAA receptor subtypes. European journal of pharmacology 2002;451: 103–110. [DOI] [PubMed] [Google Scholar]
- 67. Feng HJ, Bianchi MT, Macdonald RL. Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABAA receptor currents. Mol Pharmacol 2004;66: 988–1003. [DOI] [PubMed] [Google Scholar]
- 68. Pistis M, Belelli D, Peters JA, Lambert JJ. The interaction of general anaesthetics with recombinant GABAA and glycine receptors expressed in Xenopus laevis oocytes: a comparative study. Br J Pharmacol 1997;122: 1707–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wooltorton JR, Moss SJ, Smart TG. Pharmacological and physiological characterization of murine homomeric β3 GABAA receptors. Eur J Neurosci 1997;9: 2225–2235. [DOI] [PubMed] [Google Scholar]
- 70. Feng HJ, Macdonald RL. Barbiturates require the N terminus and first transmembrane domain of the δ subunit for enhancement of α1β3δ GABAA receptor currents. J Biol Chem 2010;285: 23614–23621. 10.1074/jbc.M110.122564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Halliwell RF, Thomas P, Patten D, James CH, Martinez-Torres A, Miledi R, et al. Subunit-selective modulation of GABAA receptors by the non-steroidal anti-inflammatory agent, mefenamic acid. Eur J Neurosci 1999;11: 2897–2905. [DOI] [PubMed] [Google Scholar]
- 72. Hill-Venning C, Belelli D, Peters JA, Lambert JJ. Subunit-dependent interaction of the general anaesthetic etomidate with the γ-aminobutyric acid type A receptor. Br J Pharmacol 1997;120: 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sigel E, Steinmann ME. Structure, function, and modulation of GABAA receptors. J Biol Chem 2012;287: 40224–40231. 10.1074/jbc.R112.386664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Olsen JA, Kastrup JS, Peters D, Gajhede M, Balle T, Ahring PK. Two distinct allosteric binding sites at α4β2 nicotinic acetylcholine receptors revealed by NS206 and NS9283 give unique insights to binding activity-associated linkage at Cys-loop receptors. J Biol Chem 2013;288: 35997–36006. 10.1074/jbc.M113.498618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feng HJ, Macdonald RL. Multiple actions of propofol on αβγ and αβδ GABAA receptors. Mol Pharmacol 2004;66: 1517–1524. [DOI] [PubMed] [Google Scholar]
- 76. Young GT, Zwart R, Walker AS, Sher E, Millar NS. Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci USA 2008;105: 14686–14691. 10.1073/pnas.0804372105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu TY, Smith CM, Sine SM, Levandoski MM. Morantel allosterically enhances channel gating of neuronal nicotinic acetylcholine α3β2 receptors. Mol Pharmacol 2008;74: 466–475. 10.1124/mol.107.044388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.