

Abstract
Hematopoietic stem cell transplantation (HSCT) is an effective approach for the treatment of severe combined immunodeficiency (SCID). However, SCID is not a homogeneous disease, and the treatment required for successful transplantation varies significantly between SCID subtypes and the degree of HLA mismatch between the best available donor and the patient. Recent studies are beginning to more clearly define this heterogeneity and how outcomes may vary. With a more detailed understanding of SCID, new approaches can be developed to maximize immune reconstitution, while minimizing acute and long-term toxicities associated with chemotherapy conditioning.
Introduction
For patients with severe combined immunodeficiency (SCID), there are several treatment options. Hematopoietic stem cell transplantation (HSCT) is potentially curative for all patients with SCID. Gene therapy (GT) may also be curative and currently is available on an experimental basis for eligible patients with IL2RG and adenosine deaminase deficiency (ADA) SCID (see Calero et al., review in this series). Finally, enzyme replacement therapy (ERT) is supportive therapy for patients with ADA-SCID and is often used while awaiting HSCT or GT. For the majority of patients meeting the diagnostic criteria for SCID,(1, 2) HSCT offers the most widely available approach to an effective cure and will be the focus of this chapter.
SCID is a rare disease with an incidence of 1:58000 (3) and most centers treating patients with SCID only see on average one patient per year. Thus, it is very difficult if not impossible for any single center to develop optimal treatment approaches for this disorder. Many of the recent advances in our understanding of the diagnosis of SCID and issues surrounding treatment for SCID are a result of collaborative working groups coordinated by the The Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplant (EBMT), (IEWP/EBMT) and PIDTC (Primary Immune Deficiency Treatment Consortium – a group of 44 centers in North America established in 2009 to study the definitive treatment of PIDs). These issues and their effects on immunologic and survival outcomes are discussed below. Specific considerations for SCID subtypes are outlined in Table 1.
Table 1.
SCID subtypes and relevant considerations for HSCT.
| T-B+NK- SCID-XL (IL2RG) JAK3 |
|
| T-B-NK- ADA |
|
| T-B+NK+ IL7Rα CD45 CD3 subunits |
|
| T-B-NK+ RAG1/2 Artemis DNA ligase IV Cernunnos-XLF DNA-PKcs |
|
| |
|
Timing of HSCT
In general, SCID patients who proceed to HSCT earlier in life have superior outcomes compared to those transplanted later.(5-7) The PIDTC recently performed a retrospective analysis of the largest cohort of SCID patients in North America published to date.(8) In this study, the likelihood of having an active infection at the time of transplant was significantly higher (52%) for patients transplanted at >3.5 months of age (old), compared to those transplanted at <3.5 months of age (young) (22%). Old infants with active infection had significantly (p<0.001) poorer 5 year survival (50%) versus young infants with/without infection (94%), old infants with no infection (90%), or infants whose infection cleared by the time of HSCT (82%).
The widespread use of newborn screening (NBS) by T-cell receptor excision circle (TREC) quantification has greatly shortened time to diagnosis, making treatment possible at a very early age.(3, 9, 10) (see Kwan et al., review in this series). In one prospective study, patients diagnosed by family history or NBS proceeded to HSCT at a much younger median age of 67 days (and 74% received HSCT at <3.5 months of age), compared to a median of 214 days (with only 17% receiving HSCT at <3.5 months of age) when diagnosed by clinical signs.(11). In cases where chemotherapy-based conditioning regimens are required, caution is recommended, as there is a likely risk of increased early and late toxicities (discussed below) in these very young patients treated with alkylating agents. However, available data indicate that, regardless of age, patients should be transplanted before the development of an infection if at all possible.
Conditioning for HSCT
In addition to recipient age, donor type and SCID type may influence the decision regarding conditioning. For example, matched sibling donors (MSDs), matched unrelated donors (URDs), and haploidentical maternal donors in which there is maternal chimerism in the recipient, all engraft readily without conditioning, although the likelihood of reconstituting B cell immunity decreases with each kind of donor as HLA mismatch increases.(12, 13) Additionally, SCID type may influence the decision regarding conditioning; some types are much more likely than others to recover B cell function without conditioning, as host B-cells in some SCID types can regain function with competent T-cell help. In a cohort of patients receiving unconditioned haploidentical or matched sibling donor transplants, patients with defects in IL7Rα, ADA, and CD3 were all more likely to recover B cell immunity compared to those with IL2RG or RAG1/2 defects.(14) Conversely, in IL2RG and JAK3-deficencies, phenotypically normal B cells are produced but are unable to function despite adequate T-cell help.(15) An advantage of using conditioning, particularly in uninfected patients, is the increased chance of achieving donor myeloid chimerism (62% vs 20%, p=0.007), and being off immunoglobulin replacement therapy in patients surviving beyond 2 years post HSCT (O.R. of 8.9; 84% vs 41%, p<0.001).(8) Overall, the highest rates of B cell function have consistently been observed in studies using myeloablative conditioning (MAC).(16-18)
While not absolutely necessary for T cell reconstitution, myeloid chimerism is also known to strongly predict recovery of thymic function as measured by both TREC levels and CD45RA+ circulating T cells, which reflect thymopoiesis and T-cell receptor (TCR) diversity.(19, 20) This was observed in a subset of PIDTC study patients analyzed for naïve CD4+CD45RA+ reconstitution; 100% of patients who received conditioning developed naive T cells >100/uL, but this was less common in those who did not receive conditioning (approximately 50%).(8) In addition, the use of a conditioning regimen was associated with an 8.8-fold increased chance of attaining a CD3 count >1000/mm3 at 2-5 years (89% vs 62%, p=0.007).(8) However, in some SCID subtypes (i.e. IL2RG and ADA defects) T cell recovery appears to be equivalent in MSD versus URD transplants even without the use of conditioning.(12)
In terms of which chemotherapy agents are most optimal for SCID patients who may require conditioning, there are several options. Busulfan-based regimens are most commonly used in both reduced-intensity (RIC) and myeloablative (MAC) transplants for SCID, although alternatives, i.e., melphalan-based regimens, have been used with comparable results.(8) Melphalan has been used successfully in a reduced intensity regimen, although metabolism of melphalan in infants is not well defined, and should be used with caution, if at all, in patients <6 months of age (P. Veys, personal communications, European group for Blood and Marrow Transplant guidelines).(21, 22) Treosulfan (not yet approved in the US), which is associated with less veno-occlusive disease and CNS toxicity than busulfan, has also been used in infants, although its metabolism in this age has not been well-studied.(22) Fludarabine is often used in combination with alkylating agents to provide T lymphocyte ablation, but it may not be sufficient to fully eradicate host NK cells in mismatched HSCT and should be used with some care in infants <6 months old.(23)
Serotherapy (anti-thymocyte globulin [ATG] or alemtuzumab [CAMPATH-1H]) is also frequently administered prior to stem cell infusion as part of conditioning, with or without chemotherapy. With its relatively long half-life, serotherapy provides host immunoablation (reducing the risk of rejection), as well as GVHD prophylaxis. Serotherapy with post- HSCT GVHD prophylaxis is essential in URD transplants for SCID patients to reduce the incidence and severity of GVHD.(12) It may be used in combination with ex vivo T cell depletion (TCD) prior to haploidentical HCT, but the dosing must be carefully considered to avoid prolonged T cell recovery.(24)
In summary, the use of conditioning remains controversial. It is clearly associated with poorer survival in patients with infection at the time of HSCT(8), and the safety in uninfected patients is not fully established. While it may result in enhanced T and B cell reconstitution, this may be at the cost of early toxicity and late effects, a question which has not been carefully studied.
Donor Selection
An approach at our center in UCSF for determining the most appropriate donor and conditioning regimen for patients with SCID is outlined in Figure 1. Regardless of SCID type, all siblings should be HLA-typed as soon as the diagnosis is made. Because of the high incidence of shared HLA haplotypes within some SCID populations, and the potential need for haploidentical donors, parents should also be typed.
Figure 1.
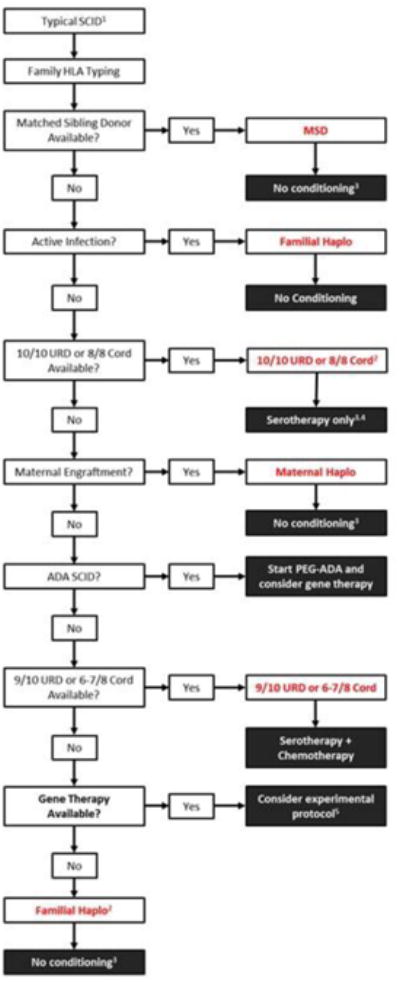
Donor selection and conditioning regimen in patients with typical SCID (UCSF approach).
1Algorithm excludes Omenn's syndrome and leaky SCID that would be classified as atypical (2). Also excludes patients with DNA sensitivity, as donor selection and conditioning for these patients are variable due to the high risk of rejection and potential for chemotherapy toxicity
2Based on availability, CMV status, donor age, or other variables. Patients with RAG SCID receiving a haploidentical transplant will generally require non-myeloablative chemotherapy and serotherapy.
3May consider chemotherapy-based conditioning for enhanced B cell and/or T cell reconstitution
4Patients with T-B-NK+ SCID receiving an URD transplant generally require a conditioning regimen with serotherapy
5Experimental gene therapy protocols are becoming more widely available and should be considered in cases where no appropriate donor can be identified
Matched Sibling Donors
An HLA-matched sibling is the donor of choice for all SCID types. In the 2014 PIDTC analysis, for patients receiving a MSD transplant, 5 year survival far exceeded that of any other donor type, at 97%.(8)
For patients with typical SCID (T cells <300/μL and T cell function <10% lower limit of normal),(2) most centers do not use a conditioning regimen for MSD transplants, regardless of SCID type, as the risk of rejection and severe GVHD are quite low. As shown in Table 2, excellent survival rates have been observed in studies where a conditioning regimen was not used, ranging from 89-95%.(7, 12, 25) Even if maternally engrafted cells are present, eradication of these cells prior to unconditioned MSD HSCT is not necessary.(12) Generally, for MSD, conditioning is only required for patients with reticular dysgenesis, leaky SCID (T cells <1000/μL, T cell function <30% lower limit of normal, and absence of maternal engraftment) or Omenn syndrome, which often requires some degree of myeloablation to correct the associated inflammatory and autoimmune manifestations, regardless of donor type. To date, there are no data comparing conditioned vs unconditioned HSCT for patients with SCID receiving a MSD.
Table 2.
Overall survival in studies of SCID patients by donor and conditioning regimen.
| First Author | Year | Ref | MRD (N) | Haplo (N) | URD (N) | UCB (N) | |||
|---|---|---|---|---|---|---|---|---|---|
| Conditioning | None | None | RIC/MAC | None | MAC | RIC | MAC | ||
| Bertrand | 1999 | (55) | 46% (50) | 54% (129) | |||||
| Dalal | 2000 | (56) | 67% (9) | ||||||
| Knutsen | 2000 | (57) | 88% (8) | ||||||
| Antoine | 2003 | (40) | 81% (104) | 63% (28) | |||||
| Rao | 2005 | (21) | 71% (7) | 83% (6) | |||||
| Bhattacharya | 2005 | (58) | 80% (10) | ||||||
| Grunebaum | 2006 | (17) | 92% (13) | 53% (40) | 81% (41) | ||||
| Dvorak | 2008 | (13) | 87% (15) | ||||||
| Gennery | 2010 | (30) | 84% (135) | 54% (415) | 66% (81) | ||||
| Buckley | 2011 | (5) | 100% (17) | 73% (149) | |||||
| Fernandes | 2012 | (28) | 62% (175) | 57% (74) | |||||
| Dvorak | 2014 | (12) | 92% (66) | 73% (37) | |||||
| Pai | 2014 | (8) | 97% (32) | 79% (87+16 IS only) | 66% (35) | 74% (19) | 58% (43) | ||
Immune reconstitution for patients receiving MSD transplants is largely determined by SCID subtype, as noted above. In the PIDTC analysis, where over 87% of MSD HSCT's were unconditioned, recovery of T cells (>1000/mm3 CD3) was achieved in 76% of patients, and B cell function was observed in 81% of patients.(8)
Acute GVHD, a reaction of donor immune cells against host tissues which classically occurs within 100 days of stem cell infusion, is not uncommon in transplants for SCID. Its incidence is modified primarily by donor-recipient HLA disparity as well as the use of GVHD prophylaxis. In MSD transplants, some centers omit GVHD prophylaxis entirely, while others administer a post-transplant calcineurin inhibitor with or without methotrexate, with the rationale that GVHD is of little benefit to a patient with SCID. Although the overall incidence of acute GVHD ranges from 23–50% in MSD HSCT for SCID, Grade III-IV aGVHD and chronic GVHD are uncommon even in two studies where many patients received no GVHD prophylaxis (3-6%).(8, 12)
Unrelated Donors
In the absence of a MSD, an URD search should be performed as soon as possible. Unrelated umbilical cord blood (UCB) sources should also be considered, and they are discussed below. Survival rates are typically lower with URD's compared to MSD's. This is likely due to a delay in proceeding to HSCT (by approximately 1-3 months) and subsequent immune reconstitution, an increased incidence of GVHD, and possibly due to increased use of chemotherapy-based conditioning. In considering an URD versus an available haploidentical donor, infection status at the time of transplant is an important determinant of survival. In infants of any age without an active infection, overall survival for URD HSCT is equivalent to that of haploidentical HSCT without conditioning (93% vs 91% respectively).(8) However, in the setting of an active infection at the time of transplant, overall survival for an URD HSCT is inferior compared to haploidentical HSCT without conditioning (53% versus 65%, p=0.006). In such cases, or when the likelihood of finding a matched URD is very low (rare ethnic background, etc.), haploidentical donors without conditioning should be considered as initial therapy.
Recipients of URD transplants may receive no conditioning, reduced intensity conditioning (RIC), or myeloablative conditioning (MAC), as shown in Table 2. Overall survival appears equivalent in patients receiving URD transplants with or without conditioning. In a multicenter survey of transplants for patients with SCID (mainly IL2RG and ADA) using URD's and UCB without RIC or MAC, 5-year overall survival for URD/UCB recipients was 71%, comparable to published reports for those receiving RIC or MAC.(12) In the PIDTC cohort, HSCT using adult URDs typically included conditioning, and overall survival was 74%.(8) In a direct comparison of RIC vs MAC, a small cohort of infants with SCID (and other immunodeficiencies) receiving URD HSCT's was analyzed: RIC was associated with 94% overall survival, significantly better than with MAC (53%, p=0.014).(21)
Overall, to determine the appropriate use of conditioning for an URD transplant, SCID type and infection risk should be considered. For patients without an intrinsic B cell defect, such as IL7Rα deficiency, there is little advantage to using conditioning; likewise, for patients with ADA deficiency or DNA repair defects, use of conditioning has been associated with poor outcomes and is not recommended.(26, 27) However, for other patients without active infection, a RIC regimen may be considered to promote B cell reconstitution, although the late effects of chemotherapy exposure in these babies have not been well-characterized.
For URD transplants without conditioning, acute GVHD occurs in up to 73% of patients if serotherapy is not used, and 32% may experience Grade III-IV aGVHD; this is reduced to 50% (8% with Grade III-IV) with serotherapy.(12) Standard GHVD prophylaxis with a calcineurin inhibitor and methotrexate (or mycophenolate mofetil or corticosteroids in the case of UCB) is recommended for these patients. The incidence of chronic GVHD (39%) was significantly higher in URD's compared to MSD recipients (5%; p<0.01), and was also partially abrogated by the use of serotherapy (47% vs 27%).
Umbilical Cord Blood Donors
An alternative stem cell source for a SCID patient without a conventional MSD or adult URD is UCB. In one retrospective comparison of UCB vs haploidentical related donors, no difference in 5-year overall survival was detected (57% vs. 62% respectively, p=0.68).(28) Although this study showed increased myeloid engraftment and IVIG independence and lower rates of second transplant (for non-engraftment) in the patients receiving UCB, this may have been related to the higher-intensity conditioning that these patients received. Of note, the degree of HLA mismatch in the UCB unit was a strong determinant of inferior overall survival. The PIDTC analysis also demonstrated that for patients <3.5 months old, UCB recipients had the lowest 5-year overall survival compared to MSD (58%, p=0.01).(8) Based on these and prior studies (29) demonstrating that outcomes using a 4/6 HLA matched UCB unit are inferior to those with a TCD haploidentical donor, this approach is only recommended when at least a 5/6 allele-matched unit is available.
Haploidentical Donors
If a MSD or a well-matched URD (adult or UCB) cannot be identified in a timely fashion, or if the patient has an active infection, a familial haploidentical transplant should be considered. As described above, survival using haploidentical donors without conditioning is comparable to that obtained with URD's, unless the patient has an active infection at the time of transplant, when haploidentical HCT is superior.(8) A European retrospective analysis demonstrated equivalent survival using a T cell depleted haploidentical relative (66%) compared to a URD (69%), although patients with active infection were not analyzed separately.(30)
An important parameter to consider in haploidentical transplants is the presence of transplacental maternal engraftment (TME). Maternally engrafted cells can be identified in approximately 40% (31) of SCID patients requiring transplant, and can interfere with engraftment of a paternal donor (32). Although these maternally-derived T cells are typically functionally incompetent, often expressing a limited TCR repertoire (33) with limited or no proliferative response to mitogen (34), they can be associated with pre-transplant GVHD.(32) If TME is present without GVHD, fetal-maternal tolerance is inferred and an unconditioned maternal T cell depleted transplant can be performed readily.(13) Engraftment is less likely if a non-maternal donor is used without conditioning in patients with circulating maternal T cells.(35) In addition, if TME is not present, at a minimum serotherapy should be considered to try to decrease the risk of rejection due to host NK cells and/or small numbers of host T cells.
For haploidentical transplants, use of any conditioning beyond serotherapy has a significant adverse effect on overall survival when used in the context of active infections (overall survival of 65% without conditioning vs 39% with conditioning, p=0.006); this difference was diminished for infants without infection (overall survival of 91% vs 81%, p =0.16).(8) Therefore, withholding conditioning is appropriate in most haploidentical HCTs for SCID, especially when performed for defects in IL2RG or JAK3 (18) or when TME is present. However, without conditioning, engraftment is less likely in cases where early T cells or NK cells are present and there is no TME. For defects involving RAG1/2 or radiosensitive SCID (19, 36) graft resistance may be higher, and these patients often require some immunosuppression to achieve T cell engraftment.(13) In an attempt to promote engraftment and immune reconstitution, high CD34 cell doses without busulfan or melphalan-containing conditioning was evaluated in a single-center study.(13) Out of 15 SCID patients who received at least 20×10ˆ6 CD34+ cells/kg, 11 engrafted without the need for 2nd transplant; 3-year overall survival was 87%, although the 4 patients who rejected received conditioning prior to a second transplant using an unrelated donor. All patients with IL2RG-deficiency or pre-HCT evidence of TME engrafted, while only 3 out of 7 patients with NK+ SCID without TME engrafted.
T-cell reconstitution in recipients of haploidentical transplants is variable. In the PIDTC study, 66% had CD3 recovery (>1000/mm3) at 2-5 years, compared to 76% of MSD recipients (p=0.01) and 76% of URD recipients (p=0.84).(8) Another study showed that the median time to T cell reconstitution (development of a PHA response >50% of control) was 4.9 months, and that to development of a PHA response 90% of control was 9.8 months.(13) This is consistent with prior studies showing that TCD is associated with a median time to regain functional T-cells of 5-9 months.(37, 38)
Recovery of B cell function following haplocompatible HCT is also heterogeneous and highly dependent upon SCID genotype.(6, 38) In an unconditioned cohort primarily consisting of haploidentical donors (and also including some MSDs), Buckley et al. observed long-term independence from immunoglobulin supplementation in 63% of patients. This is comparable to results obtained by Haddad et al in a study of only haploidentical HSCT's, where conditioning was variable: 69% of these patients recovered B cell function at last follow up. This compares favorably to the rate of B cell reconstitution in unconditioned URD's (including UCB) where only 30% of patients were IVIG-independent at last follow-up(12), though this latter result may be partly due to inclusion of only certain SCID genotypes.
The incidence of aGVHD in TCD haploidentical transplants, where no additional GVHD prophylaxis is given, ranges from 20% (grade II-IV) to 58% (all grade I-II).(8, 13) Chronic GVHD has been observed in up to 40% of these patients at 6 months post-transplant, but incidence at two years is relatively low (13-18%).(38)
Toxicity
Treatment-related mortality (TRM) occurred in 26% of patients analyzed in the PIDTC series, mostly in patients diagnosed and transplanted at >3.5 months of age, and 38% of the mortality was due to infection.(8) However, much of the infection-related mortality seen in HSCT for SCID may be due to infections that occurred prior to diagnosis and HSCT. With the advent of NBS for SCID, TRM due to infection should decrease significantly. Pulmonary toxicity was the other primary contributor to TRM in this study, accounting for 37% of mortality in the entire cohort, and 64% of mortality in patients receiving MAC with busulfan-based regimens.(8) A primary risk factor for pulmonary toxicity in SCID patients is pre-existing lung infection (CMV pneumonitis, PCP pneumonia). The pulmonary toxicity of busulfan, cyclophosphamide or fludarabine on young infants <6 months of age is not yet fully understood and requires further study.(39)
Long-term survival has increased over the last 5 decades among all SCID types.(30, 40) If good T-cell function is achieved within 1-2 years after HSCT, function is very likely to be maintained long-term (20, 41) although some reports suggest that T cell immunity may decline over time in some patients not receiving conditioning.(5) Because long-term survival is expected for most SCID patients, late effects need to be considered carefully. At least two studies have evaluated the late effects of conditioning in SCID patients overall (41) and in patients with RAG- or Artemis-deficient SCID.(42) In the latter study, patients with radiosensitive SCID due to Artemis-deficiency had significantly more problems beyond 2 years post-transplant with growth, tooth development and endocrinopathies when exposed to alkylating chemotherapy prior to HSCT. Organ toxicity (specifically to heart, lungs, and the central nervous system) should be monitored regularly for patients with SCID who have received chemotherapy. In patients transplanted for a variety of primary immunodeficiencies, true secondary malignancies occurred with an incidence of only 0.2% at 5-15 years.(43) However, late mortality (>2 years post-HSCT) due to infection, organ failure, and chronic GVHD still occurred in 7% of patients.(44)
The long-term cognitive function of SCID patients treated with HSCT is a subject of ongoing research. Cognitive effects are related to many factors, including the intrinsic genetic defect (e.g., ADA-SCID and some types of RS-SCID), psychosocial factors such as socioeconomic status, the duration and severity of illness / hospitalization, and possibly direct CNS toxicity from conditioning.(45, 46) Given the general observation that the incidence and severity of cognitive deficits increases when radiation is given to very young infants,(47) alkylating chemotherapy such as busulfan, which readily crosses the blood-brain-barrier, should be administered with caution. Close follow-up, including a full battery of age-appropriate neurocognitive testing, should be done biannually through childhood.
A diagnosis of SCID can also have significant psychosocial effects. Both the disease and the transplant process itself pose stressors to the patient and family. For example, since CMV may be transmitted through breastfeeding, mothers are advised to discontinue breastfeeding if they are CMV-seropositive, which can be very distressing for new mothers; early cessation of breastfeeding has been associated with post-partum depression.(48) Prolonged hospitalization after initial diagnosis is sometimes necessary, especially if the home environment poses a significant infectious risk to the infant. In addition, depending on the type of transplant, the duration of hospital admission for transplant for SCID patients can be months, adding significant stress to families. The social isolation required as part of HSCT for SCID may contribute to long-term deficits in both cognitive and motor development.(49)
Conclusions
HSCT is a life-saving treatment for patients with SCID, especially if therapy is instituted early, prior to onset of infections. Outcomes for all types of SCID have continued to steadily improve, and collaborative studies are underway that will help to further identify areas where improvement is still needed. While much progress has been made over the years to reduce side effects and improve survival, more studies are needed to address the questions that remain. For example, identifying the minimum dose of myeloablative conditioning that can achieve myeloid chimerism with reliable T and B-cell reconstitution is especially important for patients who are otherwise unlikely to reject even a mismatched donor graft. This question is especially salient in the era of NBS, as patients are more frequently being diagnosed at <1 month of age, and would clearly benefit from immediate transplant, were it not for the potential risks posed by alkylating chemotherapy. Newer agents, such as the stem cell targeting monoclonal antibodies against CD117 (c-kit) and CD45, hold the promise of allowing early HSCT without the risks of conventional chemotherapy, but need to be evaluated in prospective multicenter studies.(50, 51) In addition to refinement of conditioning approaches for newborns with SCID, the optimal GVHD prophylaxis regimen for both matched and mismatched donors requires further study, since GVHD is of little benefit to these patients, but GVHD prophylaxis can interfere with functional immune recovery. Future prospective studies of new conditioning regimens, GVHD prophylaxis strategies, and late effects will continue to refine HSCT as a therapeutic approach for patients with SCID.
Footnotes
Compliance with Ethics Guidelines: Conflicts of Interest: Justin T. Wahlstrom and Christopher C. Dvorak declare that they have no conflict of interest.
Mortan J.Cowan reports personal fees from Bluebird Bio, Inc. and served on the Scientific Advisory board for Exogen Bio, Inc.
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
Justin T. Wahlstrom, Email: wahlstromj@peds.ucsf.edu, 505 Parnassus Ave, Room M-659, San Francisco, CA 94143, Office: (415) 476-2188, Fax: Fax: (415) 502-4867.
Christopher C. Dvorak, Email: dvorakc@peds.ucsf.edu, 505 Parnassus Ave, Room M-659, San Francisco, CA 94143, Office: (415) 476-2188, Fax: Fax: (415) 502-4867.
Morton J. Cowan, Email: mcowan@peds.ucsf.edu, 505 Parnassus Ave, Room M-659, San Francisco, CA 94143, Office: (415) 476-2188, Fax: Fax: (415) 502-4867.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Geha RS, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. The Journal of allergy and clinical immunology. 2007 Oct;120:776. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2*.Shearer WT, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. The Journal of allergy and clinical immunology. 2014 Apr;133:1092. doi: 10.1016/j.jaci.2013.09.044. Provides the rationale and detailed analysis of criteria for making a diagnosis of SCID. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwan A, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA : the journal of the American Medical Association. 2014 Aug 20;312:729. doi: 10.1001/jama.2014.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith LM, et al. Primary Immune Deficiency Treatment Consortium (PIDTC) report. The Journal of allergy and clinical immunology. 2014 Feb;133:335. doi: 10.1016/j.jaci.2013.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckley RH. Transplantation of hematopoietic stem cells in human severe combined immunodeficiency: longterm outcomes. Immunologic research. 2011 Apr;49:25. doi: 10.1007/s12026-010-8191-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckley RH, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 1999 Feb 18;340:508. doi: 10.1056/NEJM199902183400703. [DOI] [PubMed] [Google Scholar]
- 7.Railey MD, Lokhnygina Y, Buckley RH. Long-term clinical outcome of patients with severe combined immunodeficiency who received related donor bone marrow transplants without pretransplant chemotherapy or post-transplant GVHD prophylaxis. The Journal of pediatrics. 2009 Dec;155:834. doi: 10.1016/j.jpeds.2009.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8**.Pai SY, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014 Jul 31;371:434. doi: 10.1056/NEJMoa1401177. This is the most recent retrospective study of a multi-institutional PIDTC cohort of over 200 SCID patients. The impact of SCID type, infection status, age at transplant, donor type, and conditioning regimen on immune reconstitution, GVHD, and survival are reported in detail. It showed that transplantation prior to infection is essential to maximize likelihood of survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwan A, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. The Journal of allergy and clinical immunology. 2013 Jul;132:140. doi: 10.1016/j.jaci.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. The Journal of allergy and clinical immunology. 2012 Mar;129:607. doi: 10.1016/j.jaci.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Dvorak CC, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. Journal of clinical immunology. 2013 Oct;33:1156. doi: 10.1007/s10875-013-9917-y. This report of the first 50 patients enrolled on the prospective multi-institutional PIDTC SCID study details the characteristics of SCID patients in the era of routine newborn screening and how this affects has affected the timing of HSCT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Dvorak CC, et al. Comparison of outcomes of hematopoietic stem cell transplantation without chemotherapy conditioning by using matched sibling and unrelated donors for treatment of severe combined immunodeficiency. The Journal of allergy and clinical immunology. 2014 Aug 7; doi: 10.1016/j.jaci.2014.06.021. This study of over 100 unconditioned SCID transplants reported the effects of SCID type, donor type, and serotherapy on outcomes including immune reconstitution, GVHD, and survival. It showed that unconditioned transplants for unrelated donors result in acceptable rates of GVHD and survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dvorak CC, et al. Megadose CD34(+) cell grafts improve recovery of T cell engraftment but not B cell immunity in patients with severe combined immunodeficiency disease undergoing haplocompatible nonmyeloablative transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2008 Oct;14:1125. doi: 10.1016/j.bbmt.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 14*.Buckley RH, et al. Post-transplantation B cell function in different molecular types of SCID. Journal of clinical immunology. 2013 Jan;33:96. doi: 10.1007/s10875-012-9797-6. This study provides a deeper analysis of how SCID type affects post-transplant immune reconstitution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Recher M, et al. IL-21 is the primary common gamma chain-binding cytokine required for human B-cell differentiation in vivo. Blood. 2011 Dec 22;118:6824. doi: 10.1182/blood-2011-06-362533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazzolari E, et al. Long-term immune reconstitution and clinical outcome after stem cell transplantation for severe T-cell immunodeficiency. The Journal of allergy and clinical immunology. 2007 Oct;120:892. doi: 10.1016/j.jaci.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Grunebaum E, et al. Bone marrow transplantation for severe combined immune deficiency. JAMA : the journal of the American Medical Association. 2006 Feb 1;295:508. doi: 10.1001/jama.295.5.508. [DOI] [PubMed] [Google Scholar]
- 18*.Haddad E, Leroy S, Buckley RH. B-cell reconstitution for SCID: should a conditioning regimen be used in SCID treatment? The Journal of allergy and clinical immunology. 2013 Apr;131:994. doi: 10.1016/j.jaci.2013.01.047. This review discusses the advantages and disadvantages of using conditioning in patients with SCID. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavazzana-Calvo M, et al. Long-term T-cell reconstitution after hematopoietic stem-cell transplantation in primary T-cell-immunodeficient patients is associated with myeloid chimerism and possibly the primary disease phenotype. Blood. 2007 May 15;109:4575. doi: 10.1182/blood-2006-07-029090. [DOI] [PubMed] [Google Scholar]
- 20.Sarzotti M, et al. T cell repertoire development in humans with SCID after nonablative allogeneic marrow transplantation. Journal of immunology. 2003 Mar 1;170:2711. doi: 10.4049/jimmunol.170.5.2711. [DOI] [PubMed] [Google Scholar]
- 21.Rao K, et al. Improved survival after unrelated donor bone marrow transplantation in children with primary immunodeficiency using a reduced-intensity conditioning regimen. Blood. 2005 Jan 15;105:879. doi: 10.1182/blood-2004-03-0960. [DOI] [PubMed] [Google Scholar]
- 22.Slatter MA, et al. Treosulfan-based conditioning regimens for hematopoietic stem cell transplantation in children with primary immunodeficiency: United Kingdom experience. Blood. 2011 Apr 21;117:4367. doi: 10.1182/blood-2010-10-312082. [DOI] [PubMed] [Google Scholar]
- 23.Robertson LE, et al. Natural killer cell activity in chronic lymphocytic leukemia patients treated with fludarabine. Cancer Chemother Pharmacol. 1996;37:445. doi: 10.1007/s002800050410. [DOI] [PubMed] [Google Scholar]
- 24.Dvorak CC, et al. A trial of alemtuzumab adjunctive therapy in allogeneic hematopoietic cell transplantation with minimal conditioning for severe combined immunodeficiency. Pediatric Transplantation. 2014 Sep;18:609. doi: 10.1111/petr.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. 2002 Feb 1;99:872. doi: 10.1182/blood.v99.3.872. [DOI] [PubMed] [Google Scholar]
- 26.Gaspar HB, et al. How I treat ADA deficiency. Blood. 2009 Oct 22;114:3524. doi: 10.1182/blood-2009-06-189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dvorak CC, Cowan MJ. Radiosensitive severe combined immunodeficiency disease. Immunology and allergy clinics of North America. 2010 Feb;30:125. doi: 10.1016/j.iac.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandes JF, et al. Transplantation in patients with SCID: mismatched related stem cells or unrelated cord blood? Blood. 2012 Mar 22;119:2949. doi: 10.1182/blood-2011-06-363572. [DOI] [PubMed] [Google Scholar]
- 29.Gluckman E, et al. Milestones in umbilical cord blood transplantation. Br J Haematol. 2011 Aug;154:441. doi: 10.1111/j.1365-2141.2011.08598.x. [DOI] [PubMed] [Google Scholar]
- 30.Gennery AR, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? The Journal of allergy and clinical immunology. 2010 Sep;126:602. doi: 10.1016/j.jaci.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 31.Muller SM, et al. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood. 2001 Sep 15;98:1847. doi: 10.1182/blood.v98.6.1847. [DOI] [PubMed] [Google Scholar]
- 32.Palmer BE, Mack DG, Martin AK, Maier LA, Fontenot AP. CD57 expression correlates with alveolitis severity in subjects with beryllium-induced disease. The Journal of allergy and clinical immunology. 2007 Jul;120:184. doi: 10.1016/j.jaci.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Scaradavou A, Carrier C, Mollen N, Stevens C, Rubinstein P. Detection of maternal DNA in placental/umbilical cord blood by locus-specific amplification of the noninherited maternal HLA gene. Blood. 1996 Aug 15;88:1494. [PubMed] [Google Scholar]
- 34.Knobloch C, Goldmann SF, Friedrich W. Limited T cell receptor diversity of transplacentally acquired maternal T cells in severe combined immunodeficiency. Journal of immunology. 1991 Jun 15;146:4157. [PubMed] [Google Scholar]
- 35.Palmer K, et al. Unusual clinical and immunologic manifestations of transplacentally acquired maternal T cells in severe combined immunodeficiency. The Journal of allergy and clinical immunology. 2007 Aug;120:423. doi: 10.1016/j.jaci.2007.02.047. [DOI] [PubMed] [Google Scholar]
- 36.Gaspar HB, et al. How I treat severe combined immunodeficiency. Blood. 2013 Nov 28;122:3749. doi: 10.1182/blood-2013-02-380105. [DOI] [PubMed] [Google Scholar]
- 37.Dror Y, et al. Immune reconstitution in severe combined immunodeficiency disease after lectin-treated, T-cell-depleted haplocompatible bone marrow transplantation. Blood. 1993 Apr 15;81:2021. [PubMed] [Google Scholar]
- 38.Haddad E, et al. Long-term immune reconstitution and outcome after HLA-nonidentical T-cell-depleted bone marrow transplantation for severe combined immunodeficiency: a European retrospective study of 116 patients. Blood. 1998 May 15;91:3646. [PubMed] [Google Scholar]
- 39.Long-Boyle JR, et al. High fludarabine exposure and relationship with treatment-related mortality after nonmyeloablative hematopoietic cell transplantation. Bone Marrow Transplantation. 2011 Jan;46:20. doi: 10.1038/bmt.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antoine C, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968-99. Lancet. 2003 Feb 15;361:553. doi: 10.1016/s0140-6736(03)12513-5. [DOI] [PubMed] [Google Scholar]
- 41.Neven B, et al. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood. 2009 Apr 23;113:4114. doi: 10.1182/blood-2008-09-177923. [DOI] [PubMed] [Google Scholar]
- 42.Schuetz C, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood. 2014 Jan 9;123:281. doi: 10.1182/blood-2013-01-476432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamani NR, et al. Malignancies after hematopoietic cell transplantation for primary immune deficiencies: a report from the Center for International Blood and Marrow Transplant Research. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2011 Dec;17:1783. doi: 10.1016/j.bbmt.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eapen M, et al. Long-term survival and late deaths after hematopoietic cell transplantation for primary immunodeficiency diseases and inborn errors of metabolism. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2012 Sep;18:1438. doi: 10.1016/j.bbmt.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah AJ, Kohn DB. Neurocognitive function of patients with severe combined immunodeficiency. Immunology and allergy clinics of North America. 2010 Feb;30:143. doi: 10.1016/j.iac.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Titman P, et al. Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies. Blood. 2008 Nov 1;112:3907. doi: 10.1182/blood-2008-04-151332. [DOI] [PubMed] [Google Scholar]
- 47.Walter AW, et al. Survival and neurodevelopmental outcome of young children with medulloblastoma at St Jude Children's Research Hospital. Journal of Clinical Oncology. 1999 Dec;17:3720. doi: 10.1200/JCO.1999.17.12.3720. [DOI] [PubMed] [Google Scholar]
- 48.Mathisen SE, Glavin K, Lien L, Lagerlov P. Prevalence and risk factors for postpartum depressive symptoms in Argentina: a cross-sectional study. International journal of women's health. 2013;5:787. doi: 10.2147/IJWH.S51436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin M, et al. Long-term neurocognitive function of pediatric patients with severe combined immune deficiency (SCID): pre- and post-hematopoietic stem cell transplant (HSCT) Journal of clinical immunology. 2009 Mar;29:231. doi: 10.1007/s10875-008-9250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science. 2007 Nov 23;318:1296. doi: 10.1126/science.1149726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straathof KC, et al. Haemopoietic stem-cell transplantation with antibody-based minimal-intensity conditioning: a phase 1/2 study. Lancet. 2009 Sep 12;374:912. doi: 10.1016/S0140-6736(09)60945-4. [DOI] [PubMed] [Google Scholar]
- 52.Hassan A, et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood. 2012 Oct 25;120:3615. doi: 10.1182/blood-2011-12-396879. [DOI] [PubMed] [Google Scholar]
- 53.Honig M, et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood 109. 2007 Apr 15;:3595. doi: 10.1182/blood-2006-07-034678. [DOI] [PubMed] [Google Scholar]
- 54.O'Marcaigh AS, et al. Bone marrow transplantation for T-B- severe combined immunodeficiency disease in Athabascan-speaking native Americans. Bone Marrow Transplantation. 2001 Apr;27:703. doi: 10.1038/sj.bmt.1702831. [DOI] [PubMed] [Google Scholar]
- 55.Bertrand Y, et al. Influence of severe combined immunodeficiency phenotype on the outcome of HLA non-identical, T-cell-depleted bone marrow transplantation: a retrospective European survey from the European group for bone marrow transplantation and the european society for immunodeficiency. The Journal of pediatrics. 1999 Jun;134:740. doi: 10.1016/s0022-3476(99)70291-x. [DOI] [PubMed] [Google Scholar]
- 56.Dalal I, et al. Matched unrelated bone marrow transplantation for combined immunodeficiency. Bone Marrow Transplantation. 2000 Mar;25:613. doi: 10.1038/sj.bmt.1702215. [DOI] [PubMed] [Google Scholar]
- 57.Knutsen AP, Wall DA. Umbilical cord blood transplantation in severe T-cell immunodeficiency disorders: two-year experience. Journal of clinical immunology. 2000 Nov;20:466. doi: 10.1023/a:1026463900925. [DOI] [PubMed] [Google Scholar]
- 58.Bhattacharya A, et al. Single centre experience of umbilical cord stem cell transplantation for primary immunodeficiency. Bone Marrow Transplantation. 2005 Aug;36:295. doi: 10.1038/sj.bmt.1705054. [DOI] [PubMed] [Google Scholar]