

ABSTRACT
In strains of Neisseria gonorrhoeae harboring the mtr and penB determinants that decrease permeation of antibiotics into the periplasm, mutation or deletion of the PilQ secretin of type IV pili increases resistance to penicillin by ∼3-fold, indicating a role for PilQ in antibiotic permeation. In this study, we examined spontaneously arising mutants with decreased susceptibility to penicillin. One class of mutants had a phenotype indistinguishable from that of a previously characterized pilQ2 mutation that interfered with the formation of SDS-resistant PilQ multimers. A second class of mutants contained frameshift mutations in genes upstream of pilQ in the pilMNOPQ operon that increased resistance to levels similar to those of the pilQ2 mutation. In-frame deletions of these genes were constructed, but only the frameshift mutations increased antibiotic resistance, suggesting that the mutations had polar effects on PilQ. Consistent with this result, titration of wild-type PilQ levels revealed a direct correlation between resistance and expression levels of PilQ. To determine which form of PilQ, the monomer or the multimer, was responsible for antibiotic permeation, we manipulated and quantified these forms in different mutants. Deletion of PilW, which is responsible for the maturation of PilQ into SDS-resistant multimers, had no effect on resistance. Moreover, Western blot analysis revealed that while SDS-resistant multimer levels were decreased by 26% in frameshift mutants, the levels of PilQ monomers were decreased by 48%. These data suggest that immature, SDS-labile complexes, not mature, SDS-resistant PilQ complexes, serve as the route of entry of antibiotics into the periplasm.
IMPORTANCE The capacity of antibiotics to reach their target is crucial for their activity. In Neisseria gonorrhoeae, the PilQ secretin of type IV pili plays an important role in antibiotic influx when diffusion of antibiotics through porins is limited (e.g., in most resistant strains). On Western blots, PilQ exists both as a mature higher-order multimer and an immature, SDS-labile monomer. In this study, we examined spontaneously arising mutations in PilQ and in the genes upstream of PilQ in the pilMNOPQ operon that increase resistance to penicillin. We provide evidence that PilQ monomers associate by mass action to form immature multimers and that these complexes likely mediate the diffusion of antibiotics across the outer membrane.
INTRODUCTION
Gonorrhea, the sexually transmitted infection (STI) caused by Neisseria gonorrhoeae, is the most prevalent STI worldwide, with an estimated 106 million infections per year (1). There is no vaccine against N. gonorrhoeae, and because patients do not develop immunological memory, there is a high rate of reinfection (2). Because of the absence of an adequate immune response, antibiotic therapy is the standard of care for curing gonorrheal infections, but antibiotic resistance has rendered many of the available therapeutics ineffective. Historically, gonorrhea has been treated with penicillin, tetracycline, spectinomycin, or fluoroquinolones, but these antibiotics were discontinued because of widespread resistance (3, 4). The current CDC recommendation for the treatment of gonorrhea in the United States is dual therapy with ceftriaxone and azithromycin or doxycycline; however, strains resistant to each of these antibiotics have been reported (5–8), eliciting concern that we are ushering in an era of untreatable gonorrhea (9).
The gonococcus develops resistance to penicillin either through the acquisition of a plasmid encoding a TEM-1-like β-lactamase or by acquiring multiple chromosomal mutations that incrementally increase resistance in a stepwise manner (10). Because N. gonorrhoeae is naturally competent, chromosomal resistance alleles can be transferred from a penicillin-resistant strain to a susceptible strain by transformation and homologous recombination (11). Four known resistance determinants (and a fifth, unknown determinant) contribute to penicillin resistance; three of these determinants (penA, mtr, and penB) have been characterized extensively and have well-documented effects on resistance, while the contributions of the remaining determinants (ponA and the unknown determinant) are more complicated or have not been identified molecularly (12). Each determinant by itself increases resistance by only 2- to 8-fold; however, the combination of all five determinants results in a 400-fold increase in resistance to penicillin and subsequent treatment failure (13).
The first resistance determinant acquired by a susceptible strain is the penA allele, which encodes altered forms of penicillin-binding protein 2 (PBP 2), the lethal target of penicillin, with a decreased rate of acylation by penicillin (14, 15). While the penA alleles that contribute to high-level penicillin resistance have only 4 to 8 mutations, the recent emergence and spread of strains with intermediate- and high-level resistance to ceftriaxone and cefixime are due in part to the acquisition of highly mosaic alleles of penA with up to 70 amino acid changes that result in markedly lower rates of acylation to expanded-spectrum cephalosporins (9, 16). The second resistance determinant, mtr, encompasses mutations located either in the overlapping promoter regions of mtrR and mtrCDE or within the coding sequence of mtrR that increase the expression of the MtrC-MtrD-MtrE efflux pump (17, 18). mtr mutations confer only a 2-fold increase in the MIC of penicillin but are necessary for high-level penicillin resistance in clinical isolates (19). The third resistance determinant, penB, encodes mutations in the outer membrane porin, PorB1B. These mutations decrease the influx of penicillin (and other antibiotics) through porin channels, thereby reducing the concentration of the antibiotic in the periplasm (20–22). Surprisingly, penB mutations are phenotypically silent (i.e., they confer no resistance) unless there is a coresident mtr mutation (22).
Given the importance of antibiotic resistance in gonococci, it is important to understand all aspects of antibiotic action in the organism. To this end, we have described a spontaneously occurring mutation in PilQ (pilQ2) that increases resistance to penicillin and other antibiotics by 2- to 3-fold in a strain (FA19 penA mtrR penB) harboring the first three resistance determinants (12, 23). PilQ is a major component of the outer membrane and is essential for type IV pilus (tfp) function, including twitching motility, uptake of extracellular DNA, and cell attachment and invasion (24–26). The pilus is composed of a polymer of pilin subunits that extends and retracts through a pore in the outer membrane formed by a multimeric PilQ complex (27). When gonococcal membranes are analyzed by SDS-PAGE and Western blotting, PilQ is found in two forms: a mature, SDS-resistant multimer that runs near the top of the running gel and an immature, SDS-labile form that runs at the size of a monomer (28, 29). The mutation in the pilQ2 allele (E666K) prevents maturation of the complex so that all of the PilQ subunits migrate as a monomer on SDS-PAGE; this strain also exhibits a nonpiliated colony morphology and a 50-fold decrease in transformation efficiency (23). These data were consistent with a model in which the mature, SDS-resistant multimer functions as a pore through which antibiotics and small molecules diffuse into the periplasm, with the pilQ2 mutation preventing the formation of the mature oligomer and leading to an increase in antibiotic resistance.
In this study, we further investigated the influence of PilQ on antibiotic permeability. We isolated a number of spontaneously arising mutants displaying a nonpiliated colony morphology and reduced susceptibility to penicillin. Some of these mutants contained amino acid substitutions in the C-terminal portion of PilQ and had a phenotype identical to that of the pilQ2 mutant, i.e., loss of the SDS-resistant multimer on SDS-PAGE. We also isolated resistant mutants with near-normal levels of the PilQ multimer but with frameshift mutations in the genes upstream of pilQ in the pilMNOPQ operon. The effects of these frameshift mutations on resistance were due to polar effects on PilQ, however, as in-frame deletions of the pilMNOP genes had no effect on penicillin resistance. Further examination of these mutants and strains by manipulating either the levels of PilQ expression or the formation of stable SDS-resistant multimers led us to develop a model in which PilQ monomers associate in the outer membrane to form immature, SDS-labile complexes that act as channels for antibiotics. In this model, the frameshift mutants result in marked reduction of PilQ monomers, thereby decreasing the association of these subunits into immature oligomers that allow antibiotics to diffuse into the periplasm and increasing antibiotic resistance.
MATERIALS AND METHODS
Bacterial strains.
N. gonorrhoeae strains FA19 (a penicillin-susceptible laboratory strain; penicillin MIC = 0.01 μg/ml) was kindly provided by Fred Sparling, University of North Carolina at Chapel Hill (UNC) (30). PR100 (FA19 penA mtrR penB) and SZ3 (RM11.2 penA mtrR penB), both of which harbor three penicillin resistance determinants from FA6140, have been described previously (12, 23). RM11.2 (FA1090 recA6) has the recA gene under regulation by the lac operator/promoter (31). These strains and others used in this study are listed in Table 1.
TABLE 1.
N. gonorrhoeae strains used in this study
| Straina | Description | Reference |
|---|---|---|
| FA19 | Penicillin-sensitive clinical isolate | 30 |
| PR100 | FA19 penA mtrR penB | 12 |
| RM11.2 | FA1090 containing recA under IPTG regulation | 31 |
| SZ3 | RM11.2 penA mtrR penB | 23 |
| PR100 pilQ2 | PR100 containing a PilQ G666L mutation | 23 |
| PR100 pilW::kan | PR100 containing pilW disrupted with a Kanr cassette | This study |
| PR100 proAB-pilW pKH-pilW | PR100 containing two IPTG-inducible copies of pilW | This study |
| PR100 pKH35-pilQHA | PR100 containing an IPTG-inducible copy of pilQ | This study |
| PR100 pKH35-pilQHA pilQ::Ω | PR100 pKH35-pilQHA with a disrupted endogenous copy of pilQ | This study |
PilQ point mutants and the frameshift and in-frame pilM, pilN, pilO, and pilP mutants not listed here were constructed in either PR100 or SZ3, as described in the text.
We used SZ3 and PR100 interchangeably in this study. When the three resistance determinants were transferred into SZ3 and PR100, the resulting strains had nearly identical properties. Because the pilE allele can affect transformation efficiency, SZ3 was initially used in transformation efficiency experiments to prevent recA-dependent changes in pilE (23), but PR100 was the parental strain in later experiments examining the complementation of pilM, pilN, pilO, and pilP in-frame deletions. The only difference we observed between the two strains was that the penicillin MIC for PR100 was slightly higher (0.75 μg/ml) than that for SZ3 (0.5 μg/ml); introduction of the various mutations described here resulted in similar increases in the MIC of penicillin regardless of whether the parental strain was PR100 or SZ3.
Bacterial media, growth conditions, and MIC determinations.
N. gonorrhoeae strains were grown at 37°C in a humidified 4% CO2 incubator on GC medium base (GCB) agar plates fortified with supplements I and II (32). Escherichia coli strains were grown on Luria-Bertani plates or in 2× YT medium, as described previously (12). For cloning in E. coli, genes were cloned into pUC18us (pUC18 containing the 10-bp gonococcal uptake sequence, GCCGTCTGAA [33]) or pGCC4, a complementation vector that puts genes under the control of the isopropyl-β-d-thiogalactoside (IPTG)-inducible lac promoter/operator and allows recombination into the silent intergenic region between lctP and aspC (34) in N. gonorrhoeae. E. coli transformants were selected on either 100 μg/ml carbenicillin (pUC18us) or 350 μg/ml erythromycin (pGCC4). Sequence-verified plasmids were used to transform gonococci using the protocol outlined below.
MICs were determined by the spot method, as described previously (12). Briefly, cells were resuspended at a density of 1 × 107 cells/ml, and 5 μl (50,000 colonies) was spotted onto GCB agar plates containing increasing concentrations of the indicated antibiotics. The MIC was defined as the lowest concentration at which no more than 5 colonies grew following incubation for 24 h. MICs were determined for multiple transformants and represent the averages and standard errors of the mean of at least three independent experiments.
Transformations and transformation efficiency assays.
Transformations were carried out essentially as described previously (12). Because pilQ mutations and pilM, pilN, pilO, and pilP frameshift mutations conferred resistance to penicillin, these transformants were selected on penicillin plates. For pilM, pilN, pilO, and pilP in-frame constructs that did not confer increased penicillin resistance, the transformation mixture was plated on GCB plates with no selection, and colonies with a nonpiliated morphology were screened by colony PCR using gene-specific primers. All strains were verified by PCR amplification of the appropriate regions and sequencing the resulting DNA fragments. Transformants of pGCC4 plasmids into PR100 were selected on 6 μg/ml erythromycin. Because many of the constructs rendered the recipient strain nontransformable upon recombination, it was necessary to transform cells with the complementation vector first, followed by transformation of the mutant construct.
Quantification of transformation efficiency was carried out exactly as described by Zhao et al. (23). IPTG was added to GCB+ (GCB with supplements I and II, 10 mM MgCl2, and 20 mM sodium bicarbonate) at 1 mM for transformations in SZ3 to induce expression of recA (23).
Isolation of spontaneously arising penicillin-resistant clones.
To identify spontaneously arising mutations conferring resistance to penicillin, piliated PR100 or SZ3 cells were prepared as described for the transformation protocol, except that cells were plated directly (without addition of DNA) on GCB agar plates containing concentrations of penicillin just above their respective MICs (0.75 and 0.5 μg/ml, respectively). Spontaneously arising penicillin-resistant clones, all of which were nonpiliated by colony morphology, were passaged onto fresh selective media for further analysis. A total of 615 clones were isolated and characterized.
All of the clones were analyzed by Western blotting with anti-PilQ antibody as described below. Those colonies that showed a phenotype similar to that of a pilQ2 mutant (i.e., loss of the SDS-resistant oligomer) were prepared for colony PCR with pilQ-specific primers. Briefly, cells were transferred to 25 μl water, the mixture was boiled for 5 min, and 5 μl of the cleared lysate was amplified with either Pfu or Taq polymerase. The PCR products were sequenced directly at the UNC sequencing facility. For resistant clones that showed a PilQ phenotype similar to that of the wild type, the genes upstream of pilQ (pilMNOP) were amplified and sequenced.
Scanning electron microscopy (SEM).
SZ3 and three spontaneously arising class 1 pilQ mutants of SZ3 (G668S, a mutant with a 7-amino-acid [7-aa] insertion at position 631, and E712K) were grown to mid-log phase in GCB broth, and 500 μl of the suspension was pelleted via centrifugation. The cells were resuspended in 500 μl of 3% glutaraldehyde, 0.15 M sodium phosphate buffer (pH 7.4); fixed at room temperature for 30 min; and stored at 4°C. The fixed cell suspension was then applied to 12-mm round poly-l-lysine-coated coverslips and incubated for 2 h at room temperature (RT) in a humid environment. After washing the coverslips three times with 0.15 M sodium phosphate buffer, pH 7.4, they were dehydrated with increasing concentrations of ethanol (30%, 50%, 75%, and 100%). The dehydrated coverslips were dried in a Samidri-795 critical-point drier with carbon dioxide as the transitional solvent (Tousimis Research Corporation, Rockville, MD). The coverslips were mounted on 13-mm aluminum stubs and sputter coated with 10 nm of a gold-palladium alloy (60% Au-40% Pd; Hummer X sputter coater; Anatech, Ltd., Alexandria, VA). The bacterial cells were observed using a Zeiss Supra 25 field emission scanning electron microscope operating at 5 kV, with a 5-mm working distance and a 10-μm aperture (Carl Zeiss SMT Inc., Peabody, MA).
Construction of pilM, pilN, pilO, and pilP frameshift and in-frame deletion mutant clones.
The pilM, pilN, pilO, and pilP genes were amplified from FA19 and cloned into pUC18us. Endogenous ClaI, BclI, and BsmI sites in pilM, pilN, and pilP, respectively, were eliminated by digesting with the specified endonuclease, filling in with Klenow fragment, and religating. For pilO, an XbaI site was introduced via Quik-Change mutagenesis (Agilent Technologies, Santa Clara, CA), digested with XbaI, filled in, and religated. All plasmids harboring frameshift mutations in pilM, pilN, pilO, and pilP were sequenced to confirm that the correct mutations were incorporated, and these plasmids were used to transform PR100 and SZ3 to decreased penicillin susceptibility. PCR amplification and digestion with the endonucleases listed above were used to confirm recombination of the mutant gene.
In-frame deletions in pilM, pilN, pilO, and pilP were created by amplifying 5′ and 3′ fragments of the gene of interest and cloning them into pUC18us. While the external primers contained different restriction sites at their 5′ ends, the internal primers contained a common restriction site that facilitated construction of the in-frame deletion. Each gene retained some 5′ and 3′ sequence, but approximately two-thirds of the interior sequence was deleted. Specifically, the pilM in-frame deletion deleted codons 88 to 317 (bp 262 to 951), the pilN construct deleted codons 31 to 155 (bp 91 to 465), pilO was missing codons 26 to 170 (bp 76 to 510), and pilP was missing codons 31 to 144 (bp 91 to 432). Each deletion construct also contained ∼300 bp of sequence both upstream and downstream of the deletion to facilitate successful recombination with chromosomal DNA. The in-frame deletion constructs were used to transform PR100, and because no selectable marker was transferred to the chromosome, colonies with a nonpiliated morphology were chosen for PCR screening to identify correct transformants. In-frame deletion clones were verified by sequencing and Western blotting.
Gel filtration.
Gel filtration was performed as previously described (23). Briefly, FA19 cells were grown in liquid culture, the cells were pelleted and lysed with an Emulsiflex-C5 homogenizer (Avestin, Ottawa, Canada), and the lysate was centrifuged at 100,000 × g to isolate the membranes. PilQ proteins were extracted from the membranes via Dounce homogenization in 30 mM Tris, 1.6 mM NaCl, 2 mM EDTA, 2% SB-10 [3-(decyldimethylammonio)propanesulfonate], pH 8.0, and submitted to gel filtration on a Sephacryl S-500 column in 20 mM Tris, 500 mM NaCl, 1 mM EDTA, 1.2% SB-10, pH 8. The eluted fractions were analyzed by Western blotting with PilQ antibody and quantified using QuantityOne software (Bio-Rad, Hercules, CA).
Overexpression of PilQ.
Expression of full-length pilQ in E. coli is lethal; therefore, the 5′ and 3′ ends of pilQ were introduced into the recipient strain in a stepwise manner. The 5′ end of pilQ (including its ribosomal binding site) through bp 1180 was amplified as a PacI-SacI fragment and ligated into pKH35, a complementation vector similar to pGCC4, except that it contains a more extensive multiple cloning site and a chloramphenicol acetyltransferase gene for selection. This construct was transformed into N. gonorrhoeae strain FA19, and recombinants were selected on 0.6 μg/ml chloramphenicol. The 3′ end of pilQ (bp 671 to 2196) was amplified using a 5′ primer containing a HindIII site and a 3′ primer containing an XbaI site and a hemagglutinin (HA) tag. A 400-bp fragment from pKH35 downstream of the multiple cloning site was amplified using primers that added a 5′ XbaI site and a 3′ BamHI site, and this fragment was cloned into the vector pUC18us in a three-way ligation with the 3′ fragment of pilQ. To allow selection of gonococcal transformants, a kanamycin resistance cassette was introduced into the XbaI site at the 3′ end of the pilQ sequence. This plasmid was then transformed into the FA19 strain that had been previously transformed with the plasmid containing the 5′ pilQ sequence.
Because strain PR100 has reduced transformation efficiency with pKH35 plasmids relative to FA19, the entire IPTG-inducible pilQ complementation locus was constructed in FA19, and genomic DNA was prepared from this strain and used to transform PR100 to give PR100 pKH35-pilQHA, which contains two copies of pilQ. This strain was then transformed with a construct that disrupted the endogenous copy of pilQ via the insertion of a spectinomycin resistance cassette (35). This strain, PR100 pKH35-pilQHA pilQ::Ω, contained only an IPTG-inducible copy of pilQ.
pilW knockout and overexpression.
An insertionally inactivated mutant of pilW was created by cloning pilW into pUC18us, digesting with BstXI and HpaI, filling in with T4 DNA polymerase, and ligating in a kanamycin resistance cassette. The kanamycin phosphotransferase gene was cloned in the same direction as the pilW gene. PR100 was transformed with the pUC18us-pilW::kan plasmid, and transformants were selected on GCB agar containing 50 μg/ml kanamycin. For overexpression of PilW, we utilized a plasmid (pUC18K-proAB-Q19e) containing the lacIq gene from pMJR200, the tac promoter/operator and multiple cloning site from pTTQ18, the erythromycin resistance gene, and flanking sequence from the gonococcal proAB operon to facilitate homologous transformation. pilW was cloned under the transcriptional control of the tac promoter, and the resulting plasmid was used to transform PR100 to erythromycin resistance (selected on 4 μg/ml erythromycin). To further increase PilW expression, pilW was cloned into pKH35, and this construct was used to transform the preceding strain. The different selectable markers allowed both expression constructs to be introduced into the same cell. Expression of PilW was induced by 0.1 to 10 mM IPTG.
SDS-PAGE and Western blotting.
Cells were swabbed from GCB plates, resuspended in GCB+, and diluted to an optical density at 560 nm (OD560) of 0.18 (∼1 × 108 cells/ml). An aliquot (1 ml) of the diluted cells was centrifuged at 8,000 rpm for 3 min to pellet the cells, the supernatants were aspirated, and the pellets were resuspended in 100 to 200 μl of SDS-PAGE loading buffer. The samples were heated in a boiling water bath for 5 min, and 5 to 10 μl was loaded onto SDS-polyacrylamide gels (8% for PilQ and 12% for all other proteins) and separated by electrophoresis. The proteins were then transferred onto nitrocellulose membranes overnight in 25 mM Tris, 192 mM glycine, 10% methanol, and 0.02% SDS at 4°C. The lower methanol concentration and presence of SDS appeared to increase the efficiency of transfer of the PilQ oligomer (23). Following transfer, the membrane was rinsed in phosphate-buffered saline containing 0.1% Tween 20 (PBS-T), followed by blocking with 5% nonfat dry milk in PBS-T for 1 h at RT, and then probed with a 1:10,000 dilution of a rabbit PilM, PilN, PilO, PilP, or PilQ polyclonal antibody for 2 h at RT. Anti-PilP and PilQ antibodies were made by Charles E. Wilde III, Indiana University School of Medicine, and obtained from Hank Seifert, Northwestern University. PilM, PilN, and PilO antibodies were produced by Covance (Princeton, NJ) from the purified recombinant proteins. The membrane was washed three times with PBS-T and incubated for another hour with horseradish peroxidase (HRP)-conjugated mouse anti-rabbit secondary antibody (Amersham Biosciences, Piscataway, NJ). After washing the membrane three times with PBS-T, the membrane was treated with SuperSignal West Pico (Pierce Chemical, Rockford, IL) for 5 min at RT, and expression of Pil protein bands was visualized by exposing the membrane to X-ray film (Kodak).
For quantitative Western blots, 4 to 6 independent cell lysates from PR100 and PR100 pilMFS (where pilMFS represents the pilM frameshift mutation) were prepared as described above, and 15 μl of each sample was loaded onto an SDS-8% PAGE gel and run at 80 V for approximately 150 min. Proteins were transferred to low-fluorescence polyvinylidene difluoride (LF-PVDF) membranes with a semidry transfer apparatus at 155 mA for 45 min in the presence of the transfer buffer described above. The membranes were blocked in PBS-T containing 5% nonfat dry milk and incubated with primary antibody overnight at 4°C. The membranes were washed 3 times in PBS-T and incubated with an anti-rabbit Cy3-conjugated secondary antibody (GE Healthcare, Piscataway, NJ) for 2 h. The blots were washed twice with PBS-T and once with PBS before imaging on a Typhoon 9400 imager (GE Healthcare). PilQ and PBP 1 bands were quantified using ImageQuant software.
For statistical analyses, the mean densitometry values for the PilQ multimer and monomer were set at 100% for PR100 for each blot, with the levels of each lane of PR100 calculated individually based on the mean (to provide an indication of variance between the same samples). The levels of multimer and monomer from PR100 pilMFS were then calculated as a percentage of the mean values for the respective bands from PR100. Three separate Western blots containing 4 to 6 independent samples from each strain were analyzed, with the values calculated separately for each blot (to account for differences in arbitrary densitometry numbers). This resulted in 10 to 13 independent determinations for the two strains. Statistical significance was determined by a one-way analysis of variance (ANOVA) with multiple comparisons in GraphPad Prism (San Diego, CA).
RESULTS
Phenotypic identification of spontaneously arising penicillin-resistant clones.
We have shown previously that a spontaneously arising mutation in pilQ, pilQ-E666K (the E-to-K change at position 666 encoded by pilQ; also referred to as pilQ2), increases resistance to penicillin and other antibiotics by 2- to 3-fold, provided that the strain also harbors the mtrR and penB resistance determinants (23) that collaborate to decrease permeation through the outer membrane porin (22). The increase in resistance conferred by this mutation was nearly identical to that conferred by an insertionally inactivated allele. The pilQ2 mutation causes the PilQ secretin, which normally runs on SDS-PAGE as both a mature, SDS-resistant multimer and an immature, SDS-labile monomer, to migrate entirely as a 72-kDa SDS-labile monomer.
To examine in more detail how different mutations in pilQ regulate the formation and expression of SDS-resistant PilQ oligomers, we plated two intermediate-resistant strains, PR100 (FA19 penA mtrR penB [12]) and SZ3 (RM11.2 penA mtrR penB [23]) (Table 1), on GCB plates containing concentrations of penicillin slightly above the MICs of penicillin for the two strains (0.6 and 0.75 μg/ml for PR100 and SZ3, respectively). Spontaneously arising penicillin-resistant colonies were obtained with SZ3 and PR100 at frequencies ranging from 2 × 10−6 to 5 × 10−6. These colonies, all of which had a nonpiliated morphology, were analyzed by SDS-PAGE and Western blotting with an anti-PilQ antibody, which revealed three distinct classes of mutants (a representative blot is shown in Fig. 1A): (i) class 1 mutants had a phenotype identical to that of the pilQ2 mutants, i.e., PilQ ran entirely as an SDS-labile monomer (Fig. 1A, lanes 4, 5, and 13 from the left), (ii) class 2 mutants appeared to be similar to the parental strain (Fig. 1A, lanes 3 and 6 to 10), and (iii) class 3 mutants had either smaller fragments (i.e., unstable mutants) or a complete loss of pilQ (Fig. 1A, lanes 11, 12, and 14). Of the 615 mutants analyzed, 14.9% were class 1, 69.4% were class 2, and 15.7% were class 3.
FIG 1.

Western blots of PilQ from spontaneously arising penicillin-resistant mutants of PR100. (A) Spontaneous mutations in PilQ were isolated by plating PR100 cells on GCB plates containing penicillin at concentrations just above the MIC. A representative Western blot with PilQ antibody is shown for 12 out of 615 nonpiliated colonies with increased penicillin resistance. The arrows indicate bands corresponding to the SDS-resistant multimer and the SDS-labile monomer. Three classes of mutations (class 1, pilQ2-like; class 2, normal PilQ; and class 3, PilQ deletion or truncation) were identified. The mutant classes of the 12 mutant strains are shown below the blot. (B) Thirteen different class 1 pilQ mutants were backcrossed into SZ3 and analyzed by Western blotting using a PilQ antibody. All but four mutants showed complete loss of the SDS-resistant PilQ multimer; these four—G668S, G441C, D472N, and P719T—had a small amount of multimer.
Characterization of class 1 mutants.
Class 1 mutants had a pilQ phenotype identical to that of the pilQ2 mutant; therefore, we amplified and sequenced the pilQ gene from each of the mutants to identify the mutation(s) responsible for the loss of the SDS-resistant oligomer. Sequence analysis identified 13 distinct mutations (several were obtained multiple times) that clustered solely within the C-terminal domain (aa 400 to 731) of PilQ: G441C, D472N, R483P, A550V, A585V, A585T, G617W, ΔT619, a 7-aa insertion after I631 (I631-7aa), G668S, D709G, E712K, and P719T. We did not identify the previously described E666K mutation in pilQ2, although we did isolate a G668S mutation 2 amino acids distant. To ensure that other potential mutations in non-pilQ genes would not confound subsequent analyses, the mutant pilQ genes were backcrossed into SZ3, and clean mutants were selected for increased penicillin resistance and were verified by sequencing.
Western blots of lysates from the 13 backcrossed mutants (Fig. 1B) revealed that PilQ from these strains migrated as an SDS-labile monomer, although in four mutants, G688S, G441C, P719T, and D472N, the level of the SDS-resistant oligomer was markedly reduced but not eliminated. To determine if the mutations interfered with the transport of PilQ to the outer membrane, we isolated outer membranes (36) from each of the 13 mutants and examined PilQ expression by Western blotting. In each case, PilQ was present and showed the same banding pattern as in Fig. 1B (data not shown). Therefore, these mutant PilQ variants, much like pilQ2, are transported to the outer membrane but are unable to mature into SDS-resistant multimers.
The effects of the different pilQ mutations on penicillin resistance and transformation efficiency were also determined. All of the mutant pilQ alleles increased the MIC of penicillin by 2.5- to 3-fold, the same level as the previously identified pilQ2 mutation (Fig. 2A). Increased MIC values were also observed for several other antibiotics, including ceftriaxone, vancomycin, and rifampin (data not shown). Although each mutation conferred nearly identical increases in the MIC of penicillin, their effects on transformation competence were much more varied. Four of the mutations (G617W, I631-7aa, R483P, and ΔT619) completely ablated transformation, five others (G441C, A550V, A585V, G668S, and P719T) had transformation efficiencies identical to the parental strain, and the remaining four mutations caused a disruption in transformation (∼50- to 100-fold) similar in magnitude to that of the pilQ2 mutation (Fig. 2B). These data reflect a lack of correlation between antibiotic influx and transformation efficiency.
FIG 2.

Phenotypes of spontaneously arising pilQ mutants. (A) The MIC of penicillin G (MICpenG) was determined for strains harboring one of the 13 different class 1 mutations in pilQ, as well as the parental strain (SZ3) and SZ3 pilQ2. The error bars indicate standard errors of the mean. (B) Transformation efficiencies of the 13 class 1 pilQ-backcrossed mutants, SZ3, and SZ3 pilQ2. Each strain was transformed with a plasmid (pSY6) carrying a gyrB allele that confers resistance to nalidixic acid (Nalr). SZ3 is a variant of strain RM11.2 that contains an inducible recA allele that allows IPTG control of pilin antigenic variation. Each of the strains used in this experiment contained the same pilE sequence to minimize the influence of different PilE sequences on transformation efficiency. (C) Scanning electron microscopy was used to examine SZ3 and three pilQ mutants with normal, ablated, and reduced transformation efficiencies (left to right).
Although the spontaneously isolated mutants exhibited a nonpiliated colony morphology, the observation that many of the mutants remained competent for transformation prompted us to examine several of the mutants by SEM (Fig. 2C). Three mutants with very different transformation efficiencies were chosen: (i) G668S, which exhibited a transformation efficiency equivalent to that of the parental strain, SZ3; (ii) the mutant with a 7-amino-acid insertion at position 631, which had a transformation efficiency below the limit of detection; and (iii) E712K, which has a transformation efficiency similar to that of the pilQ2 mutant. Despite their nonpiliated colony morphology, each of these three mutants expressed the tfp, albeit at markedly reduced levels compared to the parental strain. Many fewer cells expressed pili, and mutant cells that possessed pili had fewer pili than individual wild-type cells. However, no correlation was observed between the numbers of pili produced by the mutants and their transformation efficiencies.
Analysis of class 2 mutants.
The class 2 mutants were interesting in that the strains appeared to have a PilQ phenotype identical to that of the parental strains. Because these strains were nonpiliated by colony morphology, we suspected that the mutations affected PilQ in a way that did not alter its migration on SDS-PAGE or that they mapped to a different gene in the pilus-biosynthetic pathway. Sequencing of the pilQ genes from two of the strains revealed a complete absence of mutations. Because pilQ is the last gene of a large operon (pilMNOPQ), we sequenced the genes upstream of pilQ, which revealed that the pilM genes from both mutants contained a single-base insertion after codon 19 that altered the reading frame of pilM. Analysis of additional class 2 mutants with pilN and pilO antibodies also revealed several mutants with frameshift mutations in pilN.
To aid in subsequent analyses, we generated our own frameshift mutations in pilM, pilN, pilO, and pilP by digesting and filling in a unique endogenous restriction site in plasmids harboring each of the genes and then religating. These mutants were then used to transform SZ3 to increased penicillin resistance. All of the transformants were morphologically nonpiliated, as expected, and appeared identical to the original frameshift mutation that arose spontaneously. When these transformants were analyzed by Western blotting with an antibody against PilQ, the levels of the mature, SDS-resistant oligomer were similar to those in the parental strain, whereas there was a marked decrease in the levels of the immature, SDS-labile form and its breakdown products (Fig. 3A). To examine whether the frameshift mutations could be complemented, SZ3 was transformed with constructs that inserted an IPTG-inducible copy of the wild-type gene into a silent region of the gonococcal genome, followed by transformation with the appropriate frameshift pil mutant (because the pil mutants prevent subsequent transformation, this order of transformation was necessary). However, the expression of the intact gene with IPTG had no effect on penicillin resistance, suggesting that the effects of the frameshift mutations were polar (data not shown).
FIG 3.
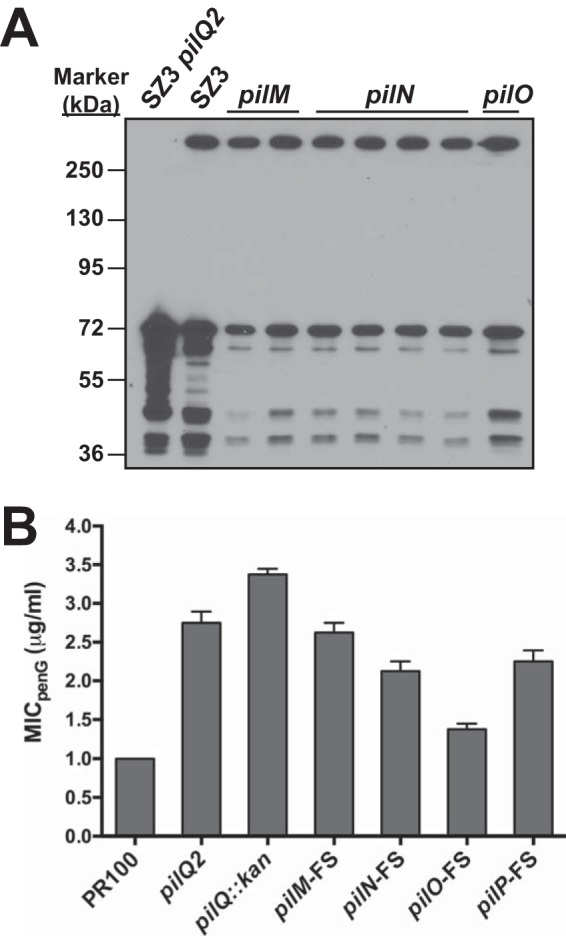
MICs of penicillin and levels of PilQ for strains containing pilM, pilN, and pilO frameshift mutations. (A) The levels of PilQ in strains harboring frameshift mutations in pilM, pilN, and pilO (parental strain, SZ3) were determined by Western blotting. (B) The MICs of penicillin for the different frameshift mutants transformed into PR100 were determined as described in Materials and Methods and compared to those of PR100 and PR100 harboring either pilQ2 or pilQ::kan. The error bars indicate standard errors of the mean.
Effects of in-frame pilM, pilN, pilO, and pilP mutations on penicillin resistance, transformation efficiency, and expression of the other Pil proteins.
In-frame deletions in each of the four pil genes were constructed and used to transform SZ3 or PR100 to increased penicillin resistance, but no transformants could be selected. This necessitated transforming PR100 with each of the constructs and, after plating on GCB, analyzing colonies with a nonpiliated morphology by PCR for the presence of the deletion. When we examined the sequence-verified transformants, MICs for strains harboring three of the four in-frame deletions (pilM, pilN, and pilO) were identical to that for the parental strain (Fig. 4A), while the strain containing the pilP deletion displayed a small increase in the MIC. These data again suggested that the original frameshift mutations we isolated in the screen had a polar effect. As previously established in Neisseria meningitidis, the in-frame deletions of pilM, pilN, pilO, and pilP completely ablated transformation (37), which could be restored by expressing the corresponding wild-type gene (Fig. 4B and C).
FIG 4.
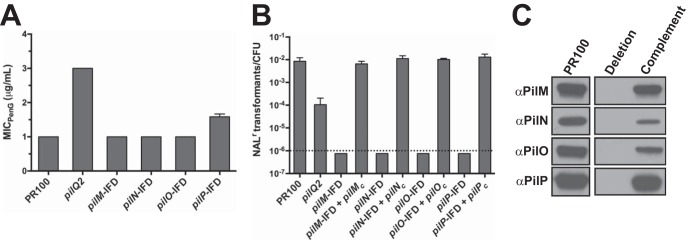
MICs of penicillin and transformation efficiencies for strains harboring in-frame deletions of pilM, pilN, pilO, and pilP. (A) In-frame deletions (IFD) of pilM, pilN, pilO, and pilP were constructed and transformed into PR100, and the MIC of penicillin for each strain, together with PR100 and PR100 pilQ2, was determined. The error bars indicate standard errors of the mean. (B) Effects on transformation efficiencies of PR100 with an in-frame deletion of pilM, pilN, pilO, or pilP and of the corresponding complemented strains (indicated by a subscript c). (C) Western blots of PilM, PilN, PilO, and PilP showing the wild-type strain, individual deletions, and the mutants complemented by expressing an IPTG-inducible allele of the deleted gene.
Polyclonal antibodies against purified PilM, PilN, and PilO were generated and used to determine the effects of the in-frame mutations on other Pil proteins. The pilM mutation had no noticeable effect on expression of any of the other proteins (Fig. 5, lanes 3 and 4 from the left). In contrast, deletion of pilN, pilO, or pilP had profound effects on the stability of some or all of the other proteins. For example, deletion of PilN caused the loss of PilO, and deletion of PilO caused the loss of PilN (Fig. 5, lanes 5 to 8), with levels of the other proteins similar to those in the parental strain. These data are consistent with more recent reports showing that the two proteins form a complex, and thus, we concluded that loss of one of the two proteins results in the misfolding, instability, or degradation of the other. Interestingly, whereas deletion of PilN or PilO caused only a minor decrease in the PilP levels, deletion of PilP resulted in the loss of PilN and PilO (Fig. 5, lanes 9 and 10). These data suggest that PilP acts to stabilize PilN and PilO but that PilP is stable in the absence of PilN and PilO.
FIG 5.
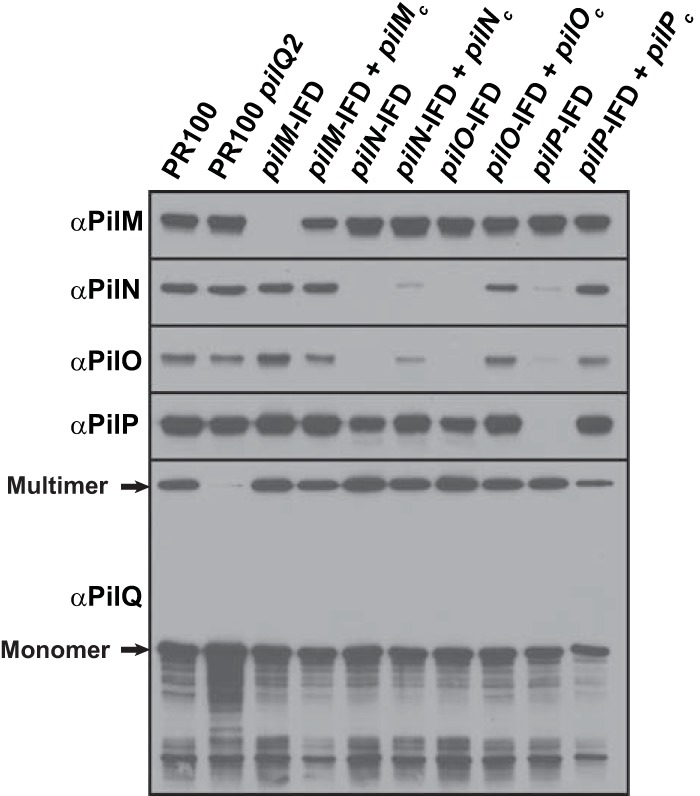
Effects of in-frame deletions of pilM, pilN, pilO, and pilP on the stability of the proteins encoded by other genes in the operon. The Western blots show the effects of deletion of pilM, pilN, pilO, or pilP on the levels of other proteins encoded by the pilMNOPQ operon relative to wild-type PR100 and the pilQ2 mutant.
Analysis of the frameshift mutations with the different Pil antibodies (data not shown) showed the same banding pattern for PilM, PilN, PilO, and PilP that was observed with the in-frame deletions (Fig. 5), suggesting that increases in the MIC of penicillin were not due to changes in expression of these proteins. The similarity of the banding patterns in the frameshift and in-frame deletion mutants, together with the observation that in-frame deletions of these four Pil proteins had no effect on resistance levels, indicated that the absence of any of the proteins was not responsible for changes in antibiotic permeation. This conclusion focused our attention back on PilQ and on how the frameshift mutations (e.g., pilMFS) result in nearly the same level of resistance to penicillin as a complete deletion of the pilQ gene.
Effects of titration of PilQ levels on penicillin resistance and transformation efficiency.
To obtain direct evidence that levels of PilQ determine gonococcal sensitivity to penicillin, we created a strain with an IPTG-inducible, HA-tagged pilQ gene inserted into the chromosome of PR100, using the integration vector pKH35, to create PR100 pKH35-pilQHA. This strain was then transformed with an insertionally inactivated pilQ construct to disrupt the endogenous pilQ allele, yielding PR100 pKH35-pilQHA pilQ::Ω. The former strain expresses more PilQ than parental cells in the presence of IPTG, whereas the latter strain expresses PilQHA only following IPTG induction. We then used these two strains and the parental strain PR100 to evaluate the effects of different levels of PilQ expression on susceptibility to penicillin and transformation efficiency. As expected, Western blots using both anti-HA and anti-PilQ antibodies revealed that the levels of PilQHA in the pKH35-pilQ strains varied with the amount of IPTG added (Fig. 6A) and that PR100 pKH35-pilQ produced higher levels of PilQ than PR100 pKH35-pilQHA pilQ::Ω.
FIG 6.

Effects of different levels of PilQ on transformation efficiency and the MIC of penicillin. PR100 was transformed in a stepwise manner to insert an IPTG-inducible copy of pilQHA at a transcriptionally silent locus on the gonococcal genome (PR100 pKH35-pilQHA). The endogenous copy of pilQ was subsequently disrupted to create PR100 pKH35-pilQHA pilQ::Ω. (A) Western blot showing the effects of 0.01, 0.1, and 1.0 mM IPTG on the expression of PilQ in the strains containing an IPTG-inducible copy of pilQ. (B) The MICs of penicillin for the IPTG-inducible PilQ strains were determined in the presence of three different concentrations of IPTG (0.01, 0.1, and 1.0 mM). The error bars indicate standard errors of the mean. (C) The transformation efficiencies of the IPTG-inducible PilQ strains were determined in the presence of three different concentrations of IPTG (0.01, 0.1, and 1.0 mM). NT, not tested. The dotted line represents the limit of detection.
The MICs of penicillin were determined for the three strains on GCB-penicillin plates containing increasing amounts of IPTG (Fig. 6B). As expected, the MIC of penicillin for PR100 did not change with the different concentrations of IPTG. For PR100 pKH35-pilQHA, the MIC for penicillin in the presence of 0.01 mM IPTG was identical to that of PR100, indicating a very low level of pilQHA induction, but as the concentration of IPTG increased, the MIC decreased below the level of the parental strain, strongly suggesting that the higher levels of PilQ enhance permeation of penicillin into the periplasm and increase susceptibility to the antibiotic. In contrast, the MIC of penicillin for PR100 pKH35-pilQHA pilQ::Ω in the presence of 0.01 mM IPTG was similar to that for a strain that produces no PilQ, but as the concentration of IPTG increased, the MIC of penicillin decreased, so that at 1.0 mM IPTG, it was identical to the MIC for PR100. These data strongly support our hypothesis that penicillin gains entry to the periplasm by diffusing through pores formed by PilQ.
We next examined the effects of manipulating levels of PilQ on transformation efficiency. The transformation efficiency in PR100 pKH35-pilQHA pilQ::Ω was below the level of detection at 0.01 mM IPTG, consistent with the absence of PilQ determined by Western blotting (Fig. 6A and C), but was essentially identical to that in PR100 at 0.1 and 1.0 mM IPTG, concentrations at which pilQ expression was similar to that in PR100. The transformation efficiency of PR100 pKH35-pilQHA, which contained the endogenous pilQ gene, was similar to that of PR100 at 0.01 mM IPTG and increased by ∼10-fold as the concentration of IPTG increased.
Analysis of PilQ levels in wild-type versus frameshift mutants.
There are three forms of PilQ that can be observed on a Western blot: (i) the mature, SDS-resistant multimer at the top of the running gel; (ii) the immature, SDS-labile monomer (72 kDa); and (iii) PilQ breakdown products. Theoretically, any (or all) of these forms could be responsible for allowing diffusion of antibiotics into the periplasm. One observation that was consistent over multiple blots was that the pilM frameshift mutation resulted in decreased levels of all three forms of PilQ but most notably affected the levels of the monomer and breakdown products (Fig. 3A). To examine the levels of the different PilQ forms in the parental strain versus the pilM frameshift mutant, we quantified the amounts of PilQ multimer and monomer present in lysates of wild-type and pilM mutant cells. Averaging over several blots with multiple independently prepared samples, we found that the level of the PilQ monomer in the pilM frameshift mutant was decreased by 48% compared to that in the wild-type strain, whereas the level of multimer in the frameshift mutant was decreased by only 26% (Fig. 7), with the decrease in the monomer significantly different from the decrease in the multimer (P < 0.05). These data suggested that the immature, SDS-labile form of PilQ (or its degradation products), and not the mature multimer, might be the entity responsible for forming the pores through which antibiotics diffuse into the periplasm.
FIG 7.
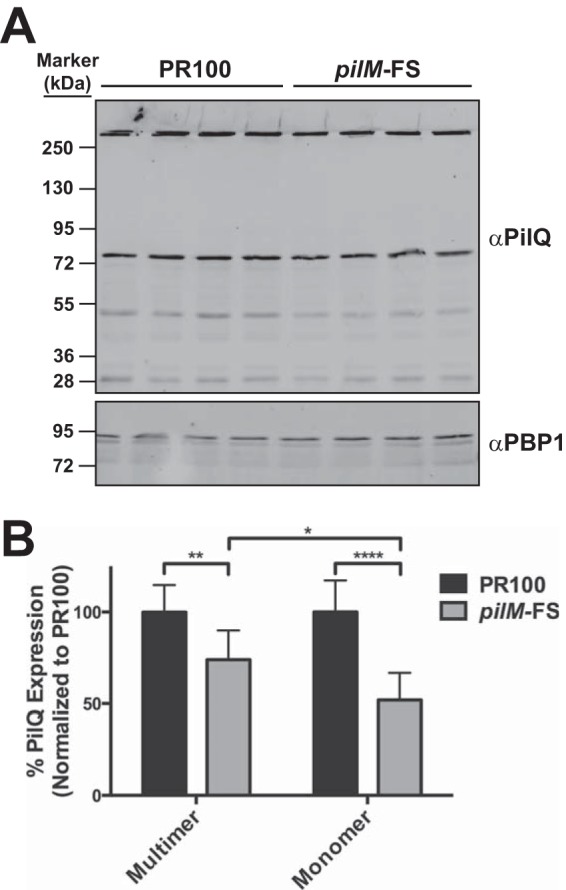
Quantitative Western blot for PilQ in PR100 and PR100 pilMFS. (A) Representative quantitative Western blot showing the effect of the pilM frameshift mutation on the expression of PilQ multimers and monomers. Anti-PilQ antibody was detected with a Cy3-conjugated anti-rabbit secondary antibody using a Typhoon fluorescence imager. (B) The levels of PilQ multimers and monomers from 10 to 13 independent samples from 3 separate blots were quantified using ImageQuant software, as described in Materials and Methods. The mean decreases for the multimer and monomer in the pilMFS mutant relative to the parental strain were 26% and 48%, respectively. *, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001. The error bars indicate standard errors of the mean.
For the SDS-labile monomers of PilQ to be responsible for antibiotic influx, they would likely need to assemble into immature oligomers (i.e., similar in size and structure to mature PilQ oligomers but not yet SDS resistant) that form a pore in the outer membrane. To investigate this possibility, PilQ proteins were detergent extracted from membranes of FA19 and chromatographed on Sephacryl S-500, and fractions were assessed for the levels of the different forms of PilQ. As reported previously, the SDS-resistant multimer eluted as a broad single peak centered at 1 MDa (23). In contrast, SDS-labile PilQ monomers eluted in two peaks: a shoulder peak that coeluted at the same molecular mass as the SDS-resistant PilQ oligomers and a large, broader peak that eluted with a molecular mass of ∼350 kDa (Fig. 8) (23). These large oligomers, like the SDS-resistant oligomer, could potentially mediate both the diffusion of small molecules through their central pore and the extrusion of the pilus through the outer membrane.
FIG 8.

Gel filtration of PilQ extracted from FA19 membranes. FA19 membranes were extracted with 2% SB-10 and subjected to gel filtration on a Sephacryl S-500 column as described in Materials and Methods. The eluted fractions were assessed by SDS-PAGE and Western blotting for PilQ. The amounts of PilQ multimer and monomer were quantified using QuantityOne software. The arrows and molecular weights above the graph show the positions of protein standards. (A portion of this graph was reprinted from reference 23 with permission of the publisher.)
Manipulation of PilW expression to regulate levels of the SDS-resistant PilQ multimer.
To further investigate the roles of the different forms of PilQ in antibiotic permeation, we manipulated levels of PilW, a protein required for formation of the SDS-resistant PilQ multimer (37–39). We reasoned that if the SDS-resistant multimer of PilQ was responsible for antibiotic influx, then the absence of the oligomer should result in an increase in the MIC of penicillin to a level similar to that of the pilQ2 mutant. Therefore, we insertionally inactivated pilW with a kanamycin resistance cassette, which resulted in complete conversion of the SDS-resistant PilQ oligomer to the immature monomeric form on SDS-PAGE (Fig. 9A). However, conversion of the multimeric PilQ to the monomeric form on SDS-PAGE had no effect on penicillin resistance (Fig. 9B), suggesting that the SDS-resistant multimer is not the species that forms pores in the outer membrane that allow diffusion of antibiotics into the periplasm. FA19 pilW::kan cells demonstrated only a slight decrease in transformation efficiency (Fig. 9C), consistent with our gel filtration data showing that the SDS-labile species of PilQ that runs as a monomer on SDS-PAGE can still oligomerize and form a pore through which the pilus can extrude.
FIG 9.

Effects of deletion of PilW on the levels of PilQ multimer, the MIC of penicillin, and transformation efficiency. (A) Western blot using a fluorescent secondary antibody to PilW showing the impact of insertional inactivation of pilW on the PilQ multimer/monomer ratio relative to wild-type cells, the pilQ2 mutant, and the pilM frameshift mutant. (B and C) Effects of disruption of pilW on the MIC of penicillin (B) and transformation efficiency (C). The error bars indicate standard errors of the mean.
We next attempted to decrease the levels of SDS-labile PilQ monomers by overexpressing PilW, which we hypothesized would convert more of the monomers to the mature form. Two additional inducible copies of pilW were inserted onto the chromosome while leaving the endogenous pilW gene intact, and Western blotting confirmed that PilW was indeed overexpressed (Fig. 10A). However, there was no corresponding increase in the PilQ multimer, and we did not observe a decrease in the PilQ monomer compared to the PR100 parental strain (Fig. 10A). Moreover, overexpression of PilW had essentially no effect on the MIC of penicillin (Fig. 10B).
FIG 10.
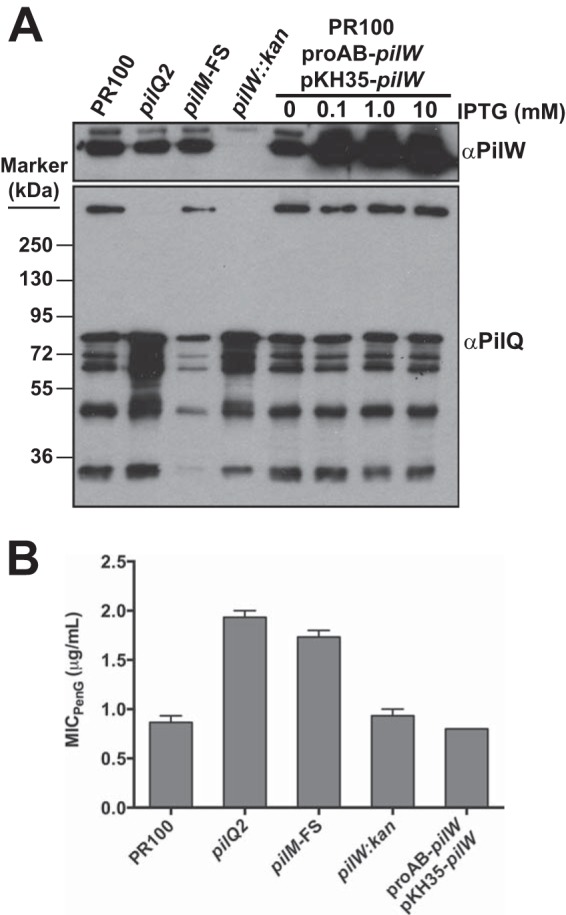
Effects of overexpression of PilW on the levels of PilQ multimer and the MIC of penicillin. Wild-type cells were transformed with a proAB-targeted expression construct (pUC18K-proAB-Q19e) containing pilW under the control of the tac promoter. The resulting strain was also transformed with the plasmid pKH35-pilW, which introduced an additional copy of the IPTG-inducible pilW gene into the gonococcal genome. The resulting strain contained the native pilW gene, as well as two IPTG-inducible copies in separate regions of the chromosome. (A) Western blot showing the effect of expressing native PilW and IPTG-inducible PilW at various concentrations of IPTG compared to wild-type cells, the pilQ2 mutant, the pilM frameshift mutant, and the pilW::kan mutant. (B) Effects of two IPTG-inducible copies of pilW on the MIC of penicillin. The error bars indicate standard errors of the mean.
When taken together, the data presented above are consistent with a model in which an overabundance of PilQ subunits, only some of which mature to the SDS-resistant multimer, associate to form immature PilQ multimers that mediate antibiotic influx into the cell. Thus, frameshift mutations in pilMNOP exert their polar effects on penicillin resistance by decreasing the amounts of PilQ monomers.
DISCUSSION
The contribution of PilQ to antibiotic susceptibility was first identified in a search for genes involved in high-level penicillin resistance. A spontaneously arising penicillin-resistant mutation, termed pilQ2, resulting from an amino acid change of Glu-666 to Lys, interfered with the formation of SDS-resistant PilQ oligomers; decreased transformation competency; and, in the presence of other resistance determinants, conferred a 2- to 3-fold increase in the MICs of penicillin and tetracycline (23). In this study, we followed up on these initial observations by identifying additional spontaneously arising mutations that increase the MIC of penicillin. Surprisingly, in addition to expected mutations in pilQ, we identified mutations in the genes upstream of pilQ in the pilMNOPQ operon that resulted in a resistance phenotype similar to that of the pilQ2 mutant, but the increased resistance of these mutants was due to polar effects on pilQ rather than the absence of the upstream genes. To verify that PilQ expression levels can alter sensitivity to penicillin, strains that allowed us to vary the level of PilQ were generated, and we observed that the MIC of penicillin was inversely proportional to the expression level of PilQ. These data implicate PilQ in antibiotic susceptibility and suggest that as the levels of PilQ increase, so does the formation of outer membrane pores through which penicillin can access the periplasm.
We considered two models that were most likely to explain our results. The first model, which is perhaps the most straightforward and is the one we favored at the start of these experiments, was that antibiotics gain entry to the periplasm through mature PilQ oligomeric pores. This model is supported by the observation that the total amount of PilQ is decreased in the frameshift mutants, meaning that fewer pores that can allow the antibiotics to permeate the outer membrane exist. However, the SDS-resistant oligomer was the form of PilQ least affected by the frameshift mutations, and recent evidence suggests that similar secretins have mechanisms that allow them to close in vivo (28, 40, 41). In addition, deletion of pilW, the protein required for production of the stable SDS-resistant PilQ multimer (37, 39), did not increase penicillin resistance. Finally, if SDS-resistant oligomers were responsible for antibiotic influx, there should have been a linear relationship between the levels of the multimer and the MIC; however, the levels of the PilQ oligomer in strains containing a PilM frameshift mutation were decreased by only 26%, yet these strains had MICs that were nearly identical to those of strains with a pilQ2 mutation or PilQ deletion (Fig. 7 and 9 and data not shown). Taken together, these data were inconsistent with the model that the mature, SDS-resistant oligomer forms a pore for antibiotics and other small molecules.
In contrast, our data are entirely consistent with the second model, in which immature PilQ entities permit diffusion of antibiotics into the periplasm. This model is supported by experiments showing that there is a much greater decrease in the PilQ monomer in the frameshift mutants (46%) than in the PilQ multimer. This model also accounts for the absence of a resistant phenotype in a strain lacking PilW; when pilW is disrupted, the SDS-resistant PilQ multimer is completely absent and looks similar to the pilQ2 mutant by Western blotting, but without a corresponding increase in antibiotic resistance. These observations, together with gel filtration data on detergent extracts of gonococcal membranes (Fig. 8) (23) demonstrating that a portion of monomeric PilQ (by SDS-PAGE) exists in multimeric form, suggest that PilQ subunits oligomerize to form immature, SDS-labile multimers that allow antibiotic diffusion into the periplasm.
It is important to distinguish the effects of mutations in PilQ, which both increase resistance and disrupt multimer formation, from the effects of frameshift mutations in the upstream pilMNOP genes, which also increase resistance but have only minor effects on multimer formation. Previous data demonstrated that resistance-conferring PilQ mutations do not prevent oligomerization of PilQ subunits; instead, these mutations must affect the conformation of the SDS-labile oligomer so that the pore is essentially blocked, thereby preventing the diffusion of antibiotics. The observation that some class 1 PilQ mutants still undergo transformation suggests that some pilus strands are able to extrude through the immature secretin, but based on our SEM results, this occurs in only a small portion of the PilQ oligomers in the outer membrane. In contrast, there are no mutations in PilQ that prevent antibiotic diffusion in the frameshift mutations in pilMNOP; instead, these mutations are polar and decrease the level of PilQ monomers in the outer membrane, with the lower levels of PilQ decreasing the mass action association of PilQ monomers to form immature oligomeric complexes that we predict serve as a route of entry of antibiotics.
For reasons that are not entirely clear, N. gonorrhoeae expresses an overabundance of PilQ subunits, only some of which become mature multimers. Our model predicts that there is a threshold level at which immature subunits begin to associate and form pores, which is consistent with the nonlinearity observed between resistance and PilQ levels. Thus, if a threshold level of PilQ monomers is required to promote formation of immature complexes by mass action, then decreasing PilQ expression by >30% overall would be sufficient to reduce oligomerization and increase resistance. Our studies manipulating the levels of PilQ were entirely consistent with this hypothesis. Unfortunately, efforts to decrease the levels of SDS-labile monomers by overexpressing the PilW protein involved in maturing PilQ complexes (37, 39), which would be a direct test of this model, were unsuccessful.
We cannot rule out the possibility that proteolytic fragments of immature PilQ subunits, which are observed in Western blots of both N. gonorrhoeae and Pseudomonas aeruginosa membranes (42), are the entities that form pores. Proteolysis of the N-terminal region of PilQ that leaves the C-terminal beta-barrel region intact could in theory result in the formation of pores lacking a “plug” that full-length, loosely associated complexes possess. However, the difficulties of preventing proteolysis of PilQ, particularly when oligomer formation is impeded or eliminated, precluded us from fully examining this possibility, and thus, we cannot eliminate the possibility that proteolytic fragments of PilQ play a part in antibiotic entry.
Thus far, PilQ mutants have not been found in clinical isolates; indeed, nearly all clinical isolates are piliated at the time of isolation (32, 43). Whiley et al. examined the pilQ sequences of 63 N. gonorrhoeae clinical isolates and found that mutations in PilQ did not contribute to decreased susceptibility to cefixime or ceftriaxone (44). Because all of the mutations that we identified in PilQ or upstream in PilM, PilN, PilO, or PilP that increase resistance also decreased or disrupted piliation, it appears likely that antibiotic-resistant mutants that arise during the course of infection and antibiotic treatment would be unfit and outcompeted by other piliated strains. However, experimental infections of male volunteers using a nonpiliated variant of F62 or an FA1090 pilE mutant showed that the pilus was not required for infection, although the symptoms were less severe in the infections with the nonpiliated variant (45). Both of these strains possessed a normal copy of the pilQ gene, so it remains possible that PilQ is required for infection even in the absence of pili. Furthermore, the role of the pilus in disseminated gonococcal infections has not been examined, and it is possible that nonpiliated antibiotic-resistant mutants could cause serious complications.
Although the pilMNOPQ mutations described in this paper may not be clinically relevant, our comparisons of the frameshift and in-frame deletion mutants provide insight into the biogenesis of type IV pili, a complex process that has not yet been entirely elucidated. A screen of N. meningitidis pilus-deficient mutants had previously identified all five genes in the pilMNOPQ operon as essential for pilus assembly (37), and experiments performed in P. aeruginosa have shown that the proteins encoded by these genes form an inner membrane complex (37, 42, 46–48). Ayers et al. (42) showed that in P. aeruginosa, mutation of PilN resulted in the loss of PilO expression, and vice versa, consistent with our results and those of others (46) showing that PilN and PilO from P. aeruginosa form heterodimers. Ayers et al. also showed that deletion of PilM resulted in the complete loss of PilN, PilO, and PilP expression, whereas deletion of PilN, PilO, and PilP had no effect on PilM levels. In contrast, we showed that deletion of gonococcal PilM had no effect on PilN, PilO, and PilP expression. Another difference between our results and those from P. aeruginosa is that deletion of either PilN or PilO in N. gonorrhoeae had no effect on PilP expression, whereas in P. aeruginosa, deletion of PilP was reported to result in the loss of PilN and PilO. Moreover, deletion of any of the first four genes in the pilMNOPQ operon in P. aeruginosa decreased levels of PilQ (42), whereas we saw no difference in PilQ expression in strains with in-frame deletions of PilM, PilN, PilO, or PilP. The differences between the two organisms are surprising, given that the gonococcal proteins are ∼50% identical to their corresponding pseudomonal counterparts.
Despite these differences, our results are generally consistent with models for PilQ complexes proposed in both P. aeruginosa and N. meningitidis. These models have the cytoplasmic protein PilM interacting with the N terminus of the inner membrane protein PilN, PilN interacting with PilO, and PilP interacting with PilN to form PilNOP heterotrimers (42, 46–49). While relevant for pilus assembly, none of the studies on the complex formed by the first 4 proteins encoded by the pilMNOPQ operon explain how frameshift mutations upstream of pilQ affect antibiotic susceptibility. Also to be considered is the phenotype of a previously described F595L mutation in pilQ (pilQ1), which confers increased antibiotic susceptibility. The pilQ1 mutation was isolated as a spontaneous suppressor mutation that allowed a strain of FA1090 with a deletion in the HpuA hemoglobin receptor protein to again utilize hemoglobin as the sole source of iron, likely by allowing free heme to diffuse into the cell (35). The different effects of these pilQ mutations on antibiotic susceptibility highlight the role of PilQ in membrane permeability.
Although all of the class 1 PilQ mutations increased the MIC of penicillin to the same level, the effects of the different mutations on transformation efficiency varied dramatically. For example, of the 13 class 1 mutations, 9 had transformation efficiencies similar to those of the wild-type strain or the pilQ2 mutant and 4 had transformation efficiencies that were below the limit of detection (Fig. 2B). Thus, even though the strains harboring the 13 class 1 PilQ mutants appear identical by Western blotting and MIC, the function of their type IV pili is affected in different ways. Surprisingly, we observed type IV pili by SEM (albeit quite sparsely) for strains harboring three class 1 PilQ mutants that conferred normal, decreased, and ablated tranformation efficiencies. Since the capacity to undergo transformation depends on the ability of the tfp to extend and retract through the PilQ secretin, the PilQ in the 9 transformation-competent mutants must be able to form outer membrane pores, even though SDS-resistant oligomers are not observed by SDS-PAGE. This further suggests that pores can be formed through interactions of PilQ complexes that are not fully mature.
In conclusion, this study identifies immature PilQ complexes, and not SDS-resistant PilQ multimer complexes, as the entities involved in forming pores in the outer membrane of N. gonorrhoeae and allowing permeation of antibiotics into the periplasm. Although not yet observed clinically, it remains possible that mutations that block permeation but allow nearly normal piliation will arise in the future and provide additional resistance to antibiotics in strains that have decreased membrane permeability.
ACKNOWLEDGMENT
This work was supported by grant AI36901 from the National Institutes of Health.
REFERENCES
- 1.World Health Organization. 2012. Global action plan to control the spread and impact of antimicrobial resistance in Neisseria gonorrhoeae. http://www.who.int/reproductivehealth/publications/rtis/9789241503501/en/.
- 2.Hedges SR, Mayo MS, Mestecky J, Hook EW III, Russell MW. 1999. Limited local and systemic antibody responses to Neisseria gonorrhoeae during uncomplicated genital infections. Infect Immun 67:3937–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control. 1987. Antibiotic-resistant strains of Neisseria gonorrhoeae. Policy guidelines for detection, management, and control. MMWR Morb Mortal Wkly Rep 36(Suppl 5):1S–18S. [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2007. Update to CDC's sexually transmitted diseases treatment guidelines, 2006: fluoroquinolones no longer recommended for treatment of gonococcal infections. MMWR Morb Mortal Wkly Rep 56:332–336. [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2012. Update to CDC's sexually transmitted diseases treatment guidelines, 2010: oral cephalosporins no longer a recommended treatment for gonococcal infections. MMWR Morb Mortal Wkly Rep 61:590–594. [PubMed] [Google Scholar]
- 6.Ohnishi M, Saika T, Hoshina S, Iwasaku K, Nakayama S, Watanabe H, Kitawaki J. 2011. Ceftriaxone-resistant Neisseria gonorrhoeae, Japan. Emerg Infect Dis 17:148–149. doi: 10.3201/eid1701.100397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unemo M, Golparian D, Nicholas R, Ohnishi M, Gallay A, Sednaoui P. 2012. High-level cefixime- and ceftriaxone-resistant Neisseria gonorrhoeae in France: novel penA mosaic allele in a successful international clone causes treatment failure. Antimicrob Agents Chemother 56:1273–1280. doi: 10.1128/AAC.05760-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Unemo M, Golparian D, Syversen G, Vestrheim DF, Moi H. 2010. Two cases of verified clinical failures using internationally recommended first-line cefixime for gonorrhoea treatment, Norway, 2010. Euro Surveill 15:19721. [DOI] [PubMed] [Google Scholar]
- 9.Unemo M, Nicholas RA. 2012. Emergence of multidrug-resistant, extensively drug-resistant and untreatable gonorrhea. Future Microbiol 7:1401–1422. doi: 10.2217/fmb.12.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon JG, Sparling PF. 1984. The genetics of the gonococcus. Annu Rev Microbiol 38:111–133. doi: 10.1146/annurev.mi.38.100184.000551. [DOI] [PubMed] [Google Scholar]
- 11.Faruki H, Sparling PF. 1986. Genetics of resistance in a non-beta-lactamase-producing gonococcus with relatively high-level penicillin resistance. Antimicrob Agents Chemother 30:856–860. doi: 10.1128/AAC.30.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ropp PA, Hu M, Olesky M, Nicholas RA. 2002. Mutations in ponA, the gene encoding penicillin-binding protein 1, and a novel locus, penC, are required for high-level chromosomally mediated penicillin resistance in Neisseria gonorrhoeae. Antimicrob Agents Chemother 46:769–777. doi: 10.1128/AAC.46.3.769-777.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao S, Duncan M, Tomberg J, Davies C, Unemo M, Nicholas RA. 2009. Genetics of chromosomally mediated intermediate resistance to ceftriaxone and cefixime in Neisseria gonorrhoeae. Antimicrob Agents Chemother 53:3744–3751. doi: 10.1128/AAC.00304-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brannigan JA, Tirodimos IA, Zhang QY, Dowson CG, Spratt BG. 1990. Insertion of an extra amino acid is the main cause of the low affinity of penicillin-binding protein 2 in penicillin-resistant strains of Neisseria gonorrhoeae. Mol Microbiol 4:913–919. doi: 10.1111/j.1365-2958.1990.tb00664.x. [DOI] [PubMed] [Google Scholar]
- 15.Dowson CG, Jephcott AE, Gough KR, Spratt BG. 1989. Penicillin-binding protein 2 genes of non-beta-lactamase-producing, penicillin-resistant strains of Neisseria gonorrhoeae. Mol Microbiol 3:35–41. doi: 10.1111/j.1365-2958.1989.tb00101.x. [DOI] [PubMed] [Google Scholar]
- 16.Nicholas RA, Davies C. 2012. Structural mechanisms of β-lactam antibiotic resistance in penicillin-binding proteins, p 397–425. In Dougherty TJ, Pucci MJ (ed), Antibiotic discovery and development. Springer, New York, NY. [Google Scholar]
- 17.Pan W, Spratt BG. 1994. Regulation of the permeability of the gonococcal cell envelope by the mtr system. Mol Microbiol 11:769–775. doi: 10.1111/j.1365-2958.1994.tb00354.x. [DOI] [PubMed] [Google Scholar]
- 18.Hagman KE, Pan W, Spratt BG, Balthazar JT, Judd RC, Shafer WM. 1995. Resistance of Neisseria gonorrhoeae to antimicrobial hydrophobic agents is modulated by the mtrRCDE efflux system. Microbiology 141:611–622. doi: 10.1099/13500872-141-3-611. [DOI] [PubMed] [Google Scholar]
- 19.Veal WL, Nicholas RA, Shafer WM. 2002. Overexpression of the MtrC-MtrD-MtrE efflux pump due to an mtrR mutation is required for chromosomally mediated penicillin resistance in Neisseria gonorrhoeae. J Bacteriol 184:5619–5624. doi: 10.1128/JB.184.20.5619-5624.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill MJ, Simjee S, Al-Hattawi K, Robertson BD, Easmon CS, Ison CA. 1998. Gonococcal resistance to beta-lactams and tetracycline involves mutation in loop 3 of the porin encoded at the penB locus. Antimicrob Agents Chemother 42:2799–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olesky M, Hobbs M, Nicholas RA. 2002. Identification and analysis of amino acid mutations in porin IB that mediate intermediate-level resistance to penicillin and tetracycline in Neisseria gonorrhoeae. Antimicrob Agents Chemother 46:2811–2820. doi: 10.1128/AAC.46.9.2811-2820.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olesky M, Zhao S, Rosenberg RL, Nicholas RA. 2006. Porin-mediated antibiotic resistance in Neisseria gonorrhoeae: ion, solute, and antibiotic permeation through PIB proteins with penB mutations. J Bacteriol 188:2300–2308. doi: 10.1128/JB.188.7.2300-2308.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao S, Tobiason DM, Hu M, Seifert HS, Nicholas RA. 2005. The penC mutation conferring antibiotic resistance in Neisseria gonorrhoeae arises from a mutation in the PilQ secretin that interferes with multimer stability. Mol Microbiol 57:1238–1251. doi: 10.1111/j.1365-2958.2005.04752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helm RA, Barnhart MM, Seifert HS. 2007. pilQ missense mutations have diverse effects on PilQ multimer formation, piliation, and pilus function in Neisseria gonorrhoeae. J Bacteriol 189:3198–3207. doi: 10.1128/JB.01833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newhall WJ, Wilde CE III, Sawyer WD, Haak RA. 1980. High-molecular-weight antigenic protein complex in the outer membrane of Neisseria gonorrhoeae. Infect Immun 27:475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tonjum T, Koomey M. 1997. The pilus colonization factor of pathogenic neisserial species: organelle biogenesis and structure/function relationships—a review. Gene 192:155–163. doi: 10.1016/S0378-1119(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 27.Wolfgang M, van Putten JP, Hayes SF, Dorward D, Koomey M. 2000. Components and dynamics of fiber formation define a ubiquitous biogenesis pathway for bacterial pili. EMBO J 19:6408–6418. doi: 10.1093/emboj/19.23.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins RF. 2004. Structure of the Neisseria meningitidis outer membrane PilQ secretin complex at 12 A resolution. J Biol Chem 279:39750–39756. doi: 10.1074/jbc.M405971200. [DOI] [PubMed] [Google Scholar]
- 29.Collins RF, Davidsen L, Derrick JP, Ford RC, Tonjum T. 2001. Analysis of the PilQ secretin from Neisseria meningitidis by transmission electron microscopy reveals a dodecameric quaternary structure. J Bacteriol 183:3825–3832. doi: 10.1128/JB.183.13.3825-3832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maness MJ, Sparling PF. 1973. Multiple antibiotic resistance due to a single mutation in Neisseria gonorrhoeae. J Infect Dis 128:321–330. doi: 10.1093/infdis/128.3.321. [DOI] [PubMed] [Google Scholar]
- 31.Seifert HS. 1997. Insertionally inactivated and inducible recA alleles for use in Neisseria. Gene 188:215–220. doi: 10.1016/S0378-1119(96)00810-4. [DOI] [PubMed] [Google Scholar]
- 32.Kellogg DS, Peacock WL, Deacon WE, Brown L, Pirkle DI. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation J Bacteriol 85:1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elkins C, Thomas CE, Seifert HS, Sparling PF. 1991. Species-specific uptake of DNA by gonococci is mediated by a 10-base-pair sequence. J Bacteriol 173:3911–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stohl EA, Brockman JP, Burkle KL, Morimatsu K, Kowalczykowski SC, Seifert HS. 2003. Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J Biol Chem 278:2278–2285. doi: 10.1074/jbc.M210496200. [DOI] [PubMed] [Google Scholar]
- 35.Chen C-J, Tobiason DM, Thomas CE, Shafer WM, Seifert HS, Sparling PF. 2004. A mutant form of the Neisseria gonorrhoeae pilus secretin protein PilQ allows increased entry of heme and antimicrobial compounds. J Bacteriol 186:730–739. doi: 10.1128/JB.186.3.730-739.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heckels JE. 1977. The surface of Neisseria gonorrhoeae: isolation of the major components of the outer membrane. J Gen Microbiol 99:333–341. doi: 10.1099/00221287-99-2-333. [DOI] [PubMed] [Google Scholar]
- 37.Carbonnelle E, Helaine S, Prouvensier L, Nassif X, Pelicic V. 2005. Type IV pilus biogenesis in Neisseria meningitidis: PilW is involved in a step occurring after pilus assembly, essential for fibre stability and function. Mol Microbiol 55:54–64. doi: 10.1111/j.1365-2958.2004.04364.x. [DOI] [PubMed] [Google Scholar]
- 38.Koo J, Tang T, Harvey H, Tammam S, Sampaleanu L, Burrows LL, Howell PL. 2013. Functional mapping of PilF and PilQ in the Pseudomonas aeruginosa type IV pilus system. Biochemistry 52:2914–2923. doi: 10.1021/bi3015345. [DOI] [PubMed] [Google Scholar]
- 39.Trindade MB, Job V, Contreras-Martel C, Pelicic V, Dessen A. 2008. Structure of a widely conserved type IV pilus biogenesis factor that affects the stability of secretin multimers. J Mol Biol 378:1031–1039. doi: 10.1016/j.jmb.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 40.Burghout P, van Boxtel R, Van Gelder P, Ringler P, Müller SA, Tommassen J, Koster M. 2004. Structure and electrophysiological properties of the YscC secretin from the type III secretion system of Yersinia enterocolitica. J Bacteriol 186:4645–4654. doi: 10.1128/JB.186.14.4645-4654.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spagnuolo J, Opalka N, Wen WX, Gagic D, Chabaud E, Bellini P, Bennett MD, Norris GE, Darst SA, Russel M, Rakonjac J. 2010. Identification of the gate regions in the primary structure of the secretin pIV. Mol Microbiol 76:133–150. doi: 10.1111/j.1365-2958.2010.07085.x. [DOI] [PubMed] [Google Scholar]
- 42.Ayers M, Sampaleanu LM, Tammam S, Koo J, Harvey H, Howell PL, Burrows LL. 2009. PilM/N/O/P proteins form an inner membrane complex that affects the stability of the Pseudomonas aeruginosa type IV pilus secretin. J Mol Biol 394:128–142. doi: 10.1016/j.jmb.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 43.Kellogg DS, Cohen IR, Norins LC, Schroeter AL, Reising G. 1968. Neisseria gonorrhoeae. II. Colonial variation and pathogenicity during 35 months in vitro. J Bacteriol 96:596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whiley DM, Jacobsson S, Tapsall JW, Nissen MD, Sloots TP, Unemo M. 2010. Alterations of the pilQ gene in Neisseria gonorrhoeae are unlikely contributors to decreased susceptibility to ceftriaxone and cefixime in clinical gonococcal strains. J Antimicrob Chemother 65:2543–2547. doi: 10.1093/jac/dkq377. [DOI] [PubMed] [Google Scholar]
- 45.Hobbs MM, Sparling PF, Cohen MS, Shafer WM, Deal CD, Jerse AE. 2011. Experimental gonococcal infection in male volunteers: cumulative experience with Neisseria gonorrhoeae strains FA1090 and MS11mkC. Front Microbiol 2:123. doi: 10.3389/fmicb.2011.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sampaleanu LM, Bonanno JB, Ayers M, Koo J, Tammam S, Burley SK, Almo SC, Burrows LL, Howell PL. 2009. Periplasmic domains of Pseudomonas aeruginosa PilN and PilO form a stable heterodimeric complex. J Mol Biol 394:143–159. doi: 10.1016/j.jmb.2009.09.037. [DOI] [PubMed] [Google Scholar]
- 47.Tammam S, Sampaleanu LM, Koo J, Manoharan K, Daubaras M, Burrows LL, Howell PL. 2013. PilMNOPQ from the Pseudomonas aeruginosa type IV pilus system form a transenvelope protein interaction network that interacts with PilA. J Bacteriol 195:2126–2135. doi: 10.1128/JB.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tammam S, Sampaleanu LM, Koo J, Sundaram P, Ayers M, Andrew Chong P, Forman-Kay JD, Burrows LL, Howell PL. 2011. Characterization of the PilN, PilO and PilP type IVa pilus subcomplex. Mol Microbiol 82:1496–1514. doi: 10.1111/j.1365-2958.2011.07903.x. [DOI] [PubMed] [Google Scholar]
- 49.Georgiadou M, Castagnini M, Karimova G, Ladant D, Pelicic V. 2012. Large-scale study of the interactions between proteins involved in type IV pilus biology in Neisseria meningitidis: characterization of a subcomplex involved in pilus assembly. Mol Microbiol 84:857–873. doi: 10.1111/j.1365-2958.2012.08062.x. [DOI] [PubMed] [Google Scholar]