

Abstract
Megaesophagus is defined as the abnormal enlargement or dilatation of the esophagus, characterized by a lack of normal contraction of the esophageal walls. This is called achalasia when associated with reduced or no relaxation of the lower esophageal sphincter (LES). To date, there are few naturally occurring models for this disease. A colony of transgenic (Pvrl3-Cre) rats presented with megaesophagus at 3 to 4 months of age; further breeding studies revealed a prevalence of 90% of transgene-positive animals having megaesophagus. Affected rats could be maintained on a total liquid diet long term and were shown to display the classic features of dilated esophagus, closed lower esophageal sphincter, and abnormal contractions on contrast radiography and fluoroscopy. Histologically, the findings of muscle degeneration, inflammation, and a reduced number of myenteric ganglia in the esophagus combined with ultrastructural lesions of muscle fiber disarray and mitochondrial changes in the striated muscle of these animals closely mimic that seen in the human condition. Muscle contractile studies looking at the response of the lower esophageal sphincter and fundus to electrical field stimulation, sodium nitroprusside, and L-nitro-L-arginine methyl ester also demonstrate the similarity between megaesophagus in the transgenic rats and patients with achalasia. No primary cause for megaesophagus was found, but the close parallel to the human form of the disease, as well as ease of care and manipulation of these rats, makes this a suitable model to better understand the etiology of achalasia as well as study new management and treatment options for this incurable condition.
Keywords: achalasia, esophagus, imaging, immunohistochemistry, megaesophagus, muscle studies, Pvrl3, ultrastructure, rat
Megaesophagus is defined as the abnormal enlargement or dilatation of the esophagus, characterized by a lack of normal contraction of the esophageal walls. This is called achalasia when associated with reduced or no relaxation of the lower esophageal sphincter (LES). Primary achalasia is very rare, with an annual incidence of approximately 1/100 000 and a prevalence rate of 1/10 000.22 In the United States, the prevalence is 10 cases per 100 000, with an incidence of 6 cases per 100 000 each year.54
Achalasia is known to affect any age, although it is more commonly observed in an older population, usually secondary to other diseases. The familial or syndromic form of achalasia often presents in childhood.34 Due to the nonspecificity of symptoms and the rarity of the disease, many patients go years before a diagnosis of achalasia is made.25 The main clinical observation of achalasia is dysphagia to solids and liquids; in people, symptoms also include chest pain, heartburn, and regurgitation.12 Aspiration pneumonia is a common sequela, as is weight loss, although this typically manifests later in the disease process. Patients with chronic achalasia are also at increased risk of developing esophageal cancer,8,25 likely due to food stasis leading to bacterial overgrowth and nitrosamine production that causes chronic inflammation and dysplasia.24
There is no cure for achalasia, as normal motor function cannot be restored; clinical intervention is based on alleviating the symptoms of dysphagia and associated complications. The success of each therapeutic and management option currently available varies widely depending on the type of patient.22 Early diagnosis and treatment are important to reduce the risk of serious complications such as aspiration pneumonia and esophageal perforations.22 Many management options target reducing LES pressure through medications such as calcium channel blockers and nitrates, injections of botulinum toxin, or surgical intervention such as pneumatic dilations and Heller myotomies.12 Unfortunately, these are palliative treatments, and there is a tendency for quality of life to decline with time, leading to the constant search for better treatment and management options.53
In animals, achalasia is better known as megaesophagus. It is most often described in dogs, although a number of cases have also been cited in cats and ferrets.5,17,39 Some well-characterized causes in animals include myasthenia gravis,21,72,83 vascular ring anomalies,9,50,52 and hypothyroidism.30,44 Megaesophagus may also be inherited—one study demonstrated that a line of affected Wire Fox Terriers with megaesophagus were traced back to 1 sire.60
Achalasia as a cause of megaesophagus is often not well documented, and the etiology is still unknown.59 The current leading hypothesis is that achalasia is due to inflammation of the myenteric plexus in the esophagus triggered by an initial insult such as a familial, infectious, or autoimmune cause.14,34,61,64 The denervation and loss of ganglion cells in the myenteric plexus both in the esophageal body and the LES is a common feature directly linked to abnormal esophageal function36,61 but is not a consistent finding in all achalasia cases.61 Similarly, in dogs, it is debatable if the difference in the numbers of myenteric ganglion cells plays a significant role in this disease.15,16,42 The human esophagus consists of skeletal muscle proximally and gradually transitions into smooth muscle distally.49 In contrast, the entire esophagus is made up of skeletal muscle in many animals, including the rat.84 In addition, the LES of a rat consists of smooth muscle.7 Nonetheless, despite these anatomical differences, the presentation of achalasia (or megaesophagus) in both humans and animals is the same.
There are no suitable naturally occurring animal models for achalasia. Nonetheless, achalasia can be induced and is often done experimentally to study this condition.4,28,48,68,69,73,79,88 Using gauze soaked with benzalkonium chloride (BAC) wrapped around the abdominal portion of the esophagus in rats, investigators demonstrated that the esophageal diameter was increased and the number of myenteric plexus neurons was reduced while the histological changes and contractile characteristics similar to that in humans with achalasia were noted.28,68 One group successfully used this model to study preneoplastic lesions associated with achalasia.82
BAC has also been successfully applied in the opossum32,73 and dog.88 A drawback to BAC studies is that it takes 3 months or more for esophageal dilatation to occur. Other studies have used mechanical obstruction of the esophagus by banding46,51,69 or by placing a pressure cuff around the gastroesophageal junction.57,79 These models mimic distal esophageal obstruction and demonstrate that esophageal aperistalsis can be secondary to the obstruction, although these findings are insufficient to explain the entire presentation of achalasia.61 Surgical vagotomy has also been attempted in opossums,48 dogs,40 and primates,4 but achalasia is not consistently reproducible.
Here we present a model of achalasia with high penetrance in a colony of transgenic rats. At the time of megaesophagus presentation, no rats had undergone experimental manipulation. This article presents the clinical presentation, diagnosis, management, and pathology of these rats with megaesophagus and demonstrates the validity of this transgenic rat strain as a model for achalasia.
Materials and Methods
Animals and Clinical Monitoring
Transgenic rats were initially generated for a neuroscience laboratory at the Massachusetts Institute of Technology (MIT) by RIKEN, Japan, with the intention of studying the poliovirus receptor–related 3 (Pvrl3) gene in the CA1 region of the hippocampus according to previously published methods.76 Briefly, the Pvrl3-Cre gene was inserted into a bacterial artificial chromosome clone, which was subsequently linearized, purified, and injected into the pronucleus of Crlj:WI embryos (Charles River Japan, Yokohama, Japan). Injected embryos were surgically transferred to pseudopregnant female rats. Transgene-positive offspring were identified by standard polymerase chain reaction (PCR) genotyping methods (Transnetyx, Cordova, TN). Four founder lines were generated; however, all animals received at MIT were from the same founder male. In-house breeding at MIT crossed the transgenic animals with Crl:WI rats (Charles River Laboratories, Raleigh, NC), and 18 stud males were retained. This initial group of rats was between the ages of 3 and 4 months when they presented with megaesophagus and are categorized as group I. There were also 3 transgenic negative age-matched males in the colony that were used as controls. Animals affected with megaesophagus were transitioned onto the PMI Micro-stabilized rodent liquid diet LD101 (TestDiet, St Louis, MO) and maintained exclusively on this, while the unaffected rats were provided the regular commercial rodent diet Prolab RMH 3000, PMI (LabDiet, St Louis, MO). All diets were provided ad libitum.
Two of the stud males were backcrossed to their wild-type mothers and their offspring categorized as group II. Offspring from these matings were randomly divided in a blinded fashion into 2 subgroups—one maintained exclusively on the PMI liquid diet and the other on a half liquid and half regular diet. One of the transgene-negative rats was also mated with a wild-type mother to generate age-matched controls for group II.
The weights and clinical presentation of group I animals were intensively monitored for the first month after diagnosis until the weights were stabilized and maintained for a year. Group II animals were also weighed weekly from 4 to 11 weeks of age to tabulate weight changes.
Contrast Radiography and Fluoroscopy
For radiography, rats from group I were anesthetized with isoflurane (Forane; Baxter, Deerfield, IL) and then gavaged with Omnipaque 140 mg/ml (GE Healthcare, Core, Ireland). Radiographs were taken at 0, 30, and 60 minutes after administration. Group II animals were radiographed once at 0 minutes initially at 4 and 8 weeks of age. For fluoroscopy, a subset of group I rats were trained to voluntarily drink Omnipaque mixed with Nutri-Cal (Vétoquinol, Vineland, NJ) prior to imaging with the OEC9800 C-arm (GE Healthcare, Waukesha, WI).
Complete Blood Counts, Serum Biochemistry, and Acetylcholine Receptor Antibodies
Complete blood counts and serum biochemistry for a subset of animals were sent to Idexx Laboratories (North Grafton, MA). Serum was analyzed for the presence of acetylcholine receptor (AChR) antibodies to rule out myasthenia gravis as a cause. Antibodies were measured by immunoprecipitation of 125I-α bungarotoxin (125I-α Bgt)–labeled AChRs and expressed as nmol of toxin binding sites/L serum (nM). Serum from a Long Evans rat with confirmed megaesophagus (unpublished data) was included as a positive control. Titers less than 0.2 nM were considered negative.
Histopathology and Immunohistochemistry
Tissues collected at necropsy were fixed in 10% neutral buffered formalin (Sigma-Aldrich, St Louis, MO) for at least 24 hours and processed for embedding in paraffin, followed by sectioning of paraffin blocks at 5-μm sections and routinely stained with hematoxylin and eosin (HE). For immunohisto-chemistry, formalin-fixed sections of the esophagus and stomach were stained for neuronal nitric oxide synthase (nNOS) using rabbit polyclonal nNOS antibody (#61-7000; Invitrogen, Camarillo, CA) and secondary goat anti–rabbit antibody (#559286; BD Pharmingen, San Diego, CA) before development with SA-HRP and DAB. Methods for antigen retrieval, blocking steps, and counterstaining were as previously described.66 A board-certified veterinary pathologist (S.M.) evaluated all slides. The number of myenteric ganglia per ten 20× high-powered fields was counted and analyzed with the unpaired t-test using GraphPad Prism (GraphPad Software, La Jolla, CA). P < .05 was considered significant.
Transmission Electron Microscopy
Sections (1 mm) of skeletal muscle from the thoracic esophagus of 2 rats with megaesophagus and 1 control were fixed in 2.5% glutaraldehyde and 3% paraformaldehyde with 5% sucrose in 0.1 M sodium cacodylate buffer. The tissue was postfixed in 1% OsO4 in veronal acetate buffer. The tissue was stained en bloc overnight with 0.5% uranyl acetate in veronal acetate buffer, then dehydrated and embedded in Spurr’s resin. Sections were cut on a microtome (Reichert Ultracut E; Reichert Technologies, Buffalo, NY) with a diamond knife and stained with 2% uranyl acetate followed by 0.1% lead citrate. Samples were examined using the Technai Spirit Transmission Electron Microscope (FEI Company, Hillsboro, OR) at 80 kV and the ultrastructural images interpreted by S.M.
Muscle Contractile Studies
At necropsy, the esophagus and stomach of 2 transgenic and 2 normal rats were excised and placed in cold Kreb’s solution (120 mM NaCl, 5.9 mM KCl, 25 mM NaHCO3, 1.2 mM Na2H2PO4, 1.2 mM MgCl·6H2O, 2.5 mM CaCl2, and 11.5 mM dextrose). Three longitudinal smooth muscle strips from the fundus without mucosa and 2 strips with intact mucosa corresponding to the middle and distal portions of the LES were obtained from each animal. Smooth muscle strips were mounted in a tissue chamber maintained at 37°C and continuously gassed with carbogen (95% O2 + 5% CO2). Muscle strips from the LES and fundus were stretched to 5 g and 1.5 g, respectively, and equilibrated for 1 hour in Kreb’s solution to allow tissues to reach a stable tone before starting the stimulation. Neurally mediated nitrergic relaxation was elicited by electrical field stimulation (EFS; 20 V, 0.5–64 Hz, 0.5-ms pulse duration, 5 seconds). Tissue from the fundus was precontracted with carbachol (10−5 M) prior to EFS. The effect of EFS-induced relaxation in the fundus was also investigated in the presence of L-nitro-L-arginine methyl ester (L-NAME, 100 μM). In addition, nitrergic relaxation of smooth muscle was induced in both the LES and fundus by the addition of sodium nitroprusside (SNP, 1 μM).
Relaxation responses of smooth muscle strips were expressed as a percent change from the basal tone in the LES or from the active tension achieved by carbachol precontraction in the fundus. Comparison of contractile or relaxation responses between rats with megaesophagus and normal rats was analyzed by an unpaired t-test followed by Holm-Sidak post hoc analysis. The effect of L-NAME on fundus relaxation between the groups of rats was analyzed by a paired t-test. Data are reported as mean ± standard error of the mean (SEM). P < .05 was considered significant.
Results
Rats With Megaesophagus Can Be Maintained Long Term on a Total Liquid Diet
Of the 18 rats diagnosed with megaesophagus in group I, 7 showed clinical signs, including respiratory distress, porphyrin staining, excessive salivation, a hunched posture, and dehydration or did not gain weight after transitioning to the liquid diet. These animals were euthanized. The remaining 11 rats were maintained on the total liquid diet and did not present with clinical signs. These rats showed a mean ± SEM weight gain of 38.3 ± 9.7 g over the first month. Of the 11 rats, 6 were successfully maintained in the colony for more than a year and were clinically normal throughout. Radiography at 1 year showed the persistence of achalasia in all remaining group I animals.
Group II animals were placed on various modified diets to determine if the diets were appropriate in maintaining the rats and reducing clinical signs. Genotyping results at the end of the study revealed a total of 10 transgene-positive rats (4 males, 6 females) and 10 transgene-negative rats (6 males, 4 females) that were placed on the modified diets. Fifteen wild-type rats were produced and maintained on the regular diet. The weights between transgene-positive and transgene-negative animals on the same diet regime showed little difference; however, as a whole, rats maintained on the half liquid and half pellet diet showed higher weights than those on a liquid diet alone, with the former being more comparable to the wild-type animals (Table 1). None of the group II animals demonstrated any clinical signs of megaesophagus.
Table 1.
Average Weights of Rats at 4 and 11 Weeks Old.
| Sex | Transgenic Status | No. in Group | Diet | Average Weight at 4 wk, g | Average Weight at 11 wk, g | Weight Gained, Mean ± SEM, g |
|---|---|---|---|---|---|---|
| Male | Tg+ | 2 | Liquid | 95.1 | 361.4 | 266.4 ± 6.5 |
| 2 | Half | 108.1 | 406.4 | 298.4 ± 52.9 | ||
| Tg− | 3 | Liquid | 111.4 | 303.9 | 192.5 ± 36.1 | |
| 3 | Half | 125.6 | 453.3 | 327.7 ± 3.7 | ||
| Wild type | 10 | Regular | 124.5 | 462.9 | 338.4 ± 41.8 | |
| Female | Tg+ | 5 | Liquid | 97.9 | 225.9 | 127.9 ± 5.2 |
| 1 | Half | 110.1 | 278.5 | 168.4 ± 0.0 | ||
| Tg− | 1 | Liquid | 98.5 | 244.3 | 145.8 ± 0.0 | |
| 3 | Half | 107.4 | 261.5 | 154.1 ± 5.1 | ||
| Wild type | 5 | Regular | 114.4 | 293.9 | 179.5 ± 4.9 |
Rats were randomly assigned to different diet groups; age-matched wild-type rats were maintained on the regular diet. Animals on the half regular and half liquid diet maintained similar weights to the controls; animals on a complete liquid diet still gained weight but not to the same extent as the other 2 groups. SEM, standard error of the mean.
Radiography and Fluoroscopy Demonstrate the Classic Features of Megaesophagus
Using contrast radiography, all group I animals showed an enlarged esophagus with little clearance of contrast into the stomach at time 0 (Fig. 1). At 30 and 60 minutes, clearance was minimal, with most of the contrast agent still present within the esophagus after 60 minutes (Fig. 2). In comparison, the age-matched transgenic negative and wild-type animals all showed significant clearance into the stomach at time 0 (Fig. 3) and complete clearance from the esophagus by 30 minutes, with all contrast agent residing in the stomach at 60 minutes (Fig. 4).
Figure 1.
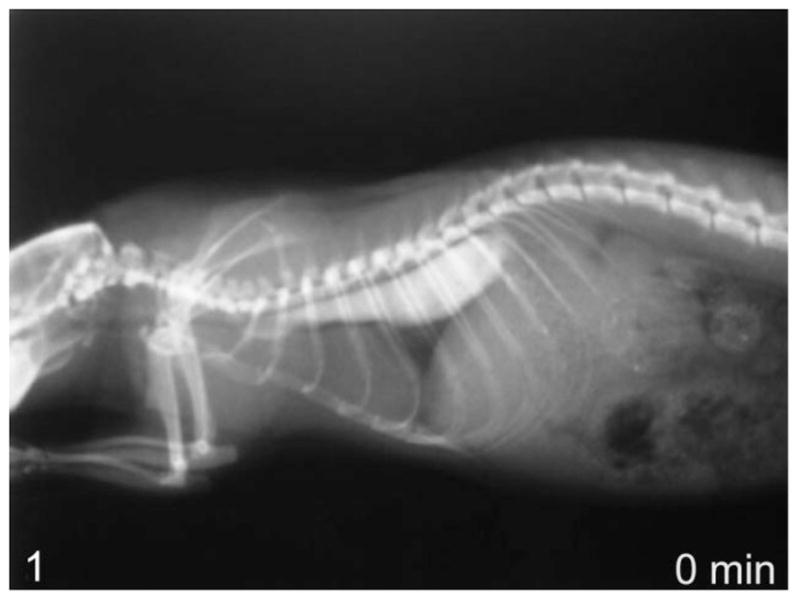
Lateral view of whole-body contrast radiography at 0 minutes in a rat with megaesophagus—pooling of contrast agent in esophagus.
Figure 2.
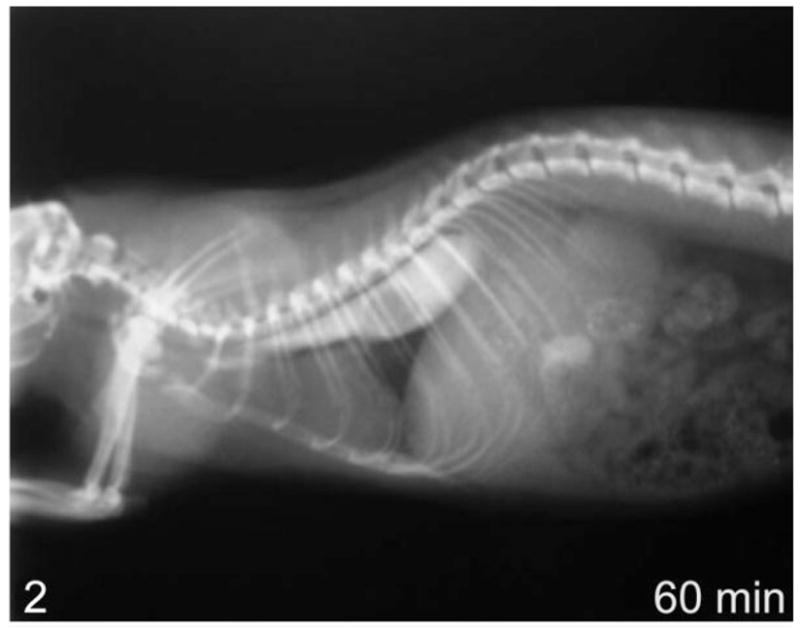
Lateral view of whole-body contrast radiography at 60 minutes in a rat with megaesophagus—no clearance of contrast agent into the stomach.
Figure 3.
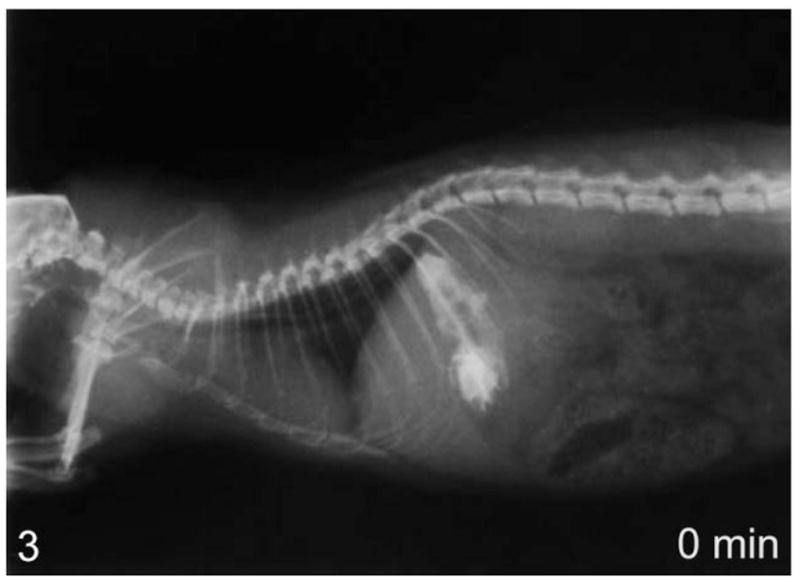
Lateral view of whole-body contrast radiography at 0 minutes in a normal rat—contrast agent starts to clear from the esophagus immediately after administration.
Figure 4.
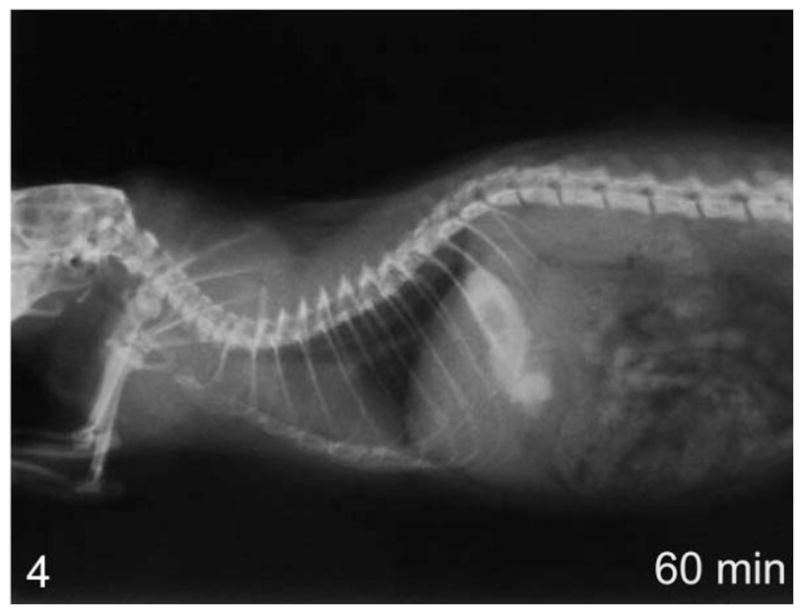
Lateral view of whole-body contrast radiography at 60 minutes in a normal rat—complete clearance of contrast agent into the stomach.
Of the rats belonging to group II, 7 had an enlarged esophagus when viewed using contrast radiography at 4 weeks of age. At 8 weeks, an additional 2 rats presented similarly. All 9 rats were transgene positive. The remaining animals showed normal esophageal sizes, although 1 was transgene positive. In total, 9 of 10 transgene-carrying animals (90%) presented radiographically with megaesophagus while no (0%) rats absent for the transgene showed any esophageal abnormalities.
Fluoroscopy revealed uncoordinated peristalsis and lack of motility in the esophageal walls of rats with megaesophagus. The LES demonstrated no relaxation, leading to the pooling of contrast within the esophagus (Fig. 5). In comparison, the rats with normal esophagi demonstrated smooth peristaltic movement from the pharynx to the stomach (Fig. 6). Clearance was within seconds after swallowing.
Figure 5.
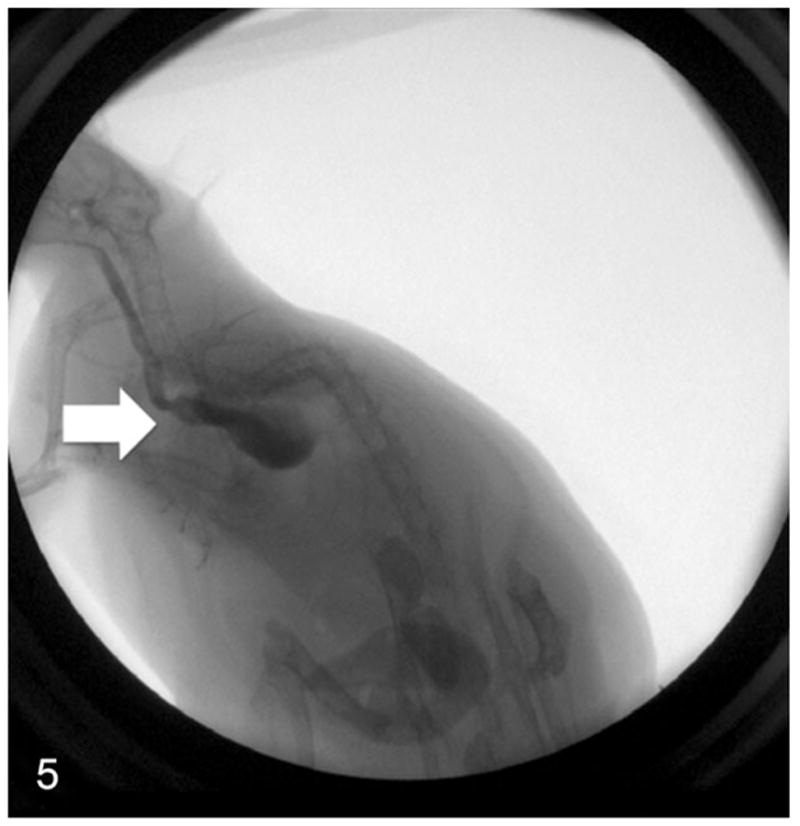
Real-time fluoroscopy image of a rat with megaesophagus swallowing contrast agent—abnormal contractions of the esophagus (arrow) and no relaxation of the lower esophageal sphincter lead to the retention of contrast agent in the esophagus.
Figure 6.
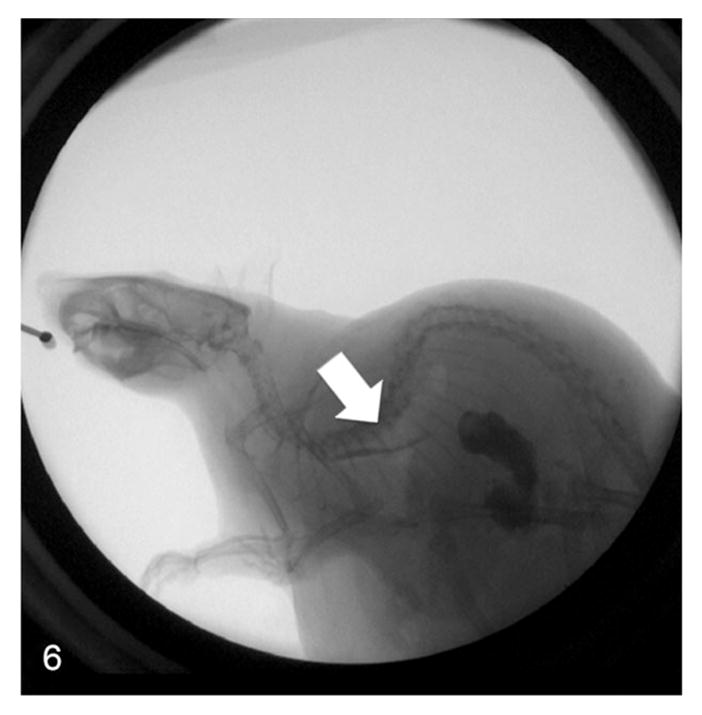
Real-time fluoroscopy image of a normal rat swallowing contrast agent—coordinated contractions of the esophagus (arrow) lead to clearance of contrast agent into the stomach.
No Primary or Concurrent Disease Identified in Rats With Megaesophagus
No significant deviations from the normal ranges in both the complete blood counts and serum biochemistry panels were noted in any affected rats. Rats were also negative for acetylcholine receptor antibodies (Fig. 7).
Figure 7.
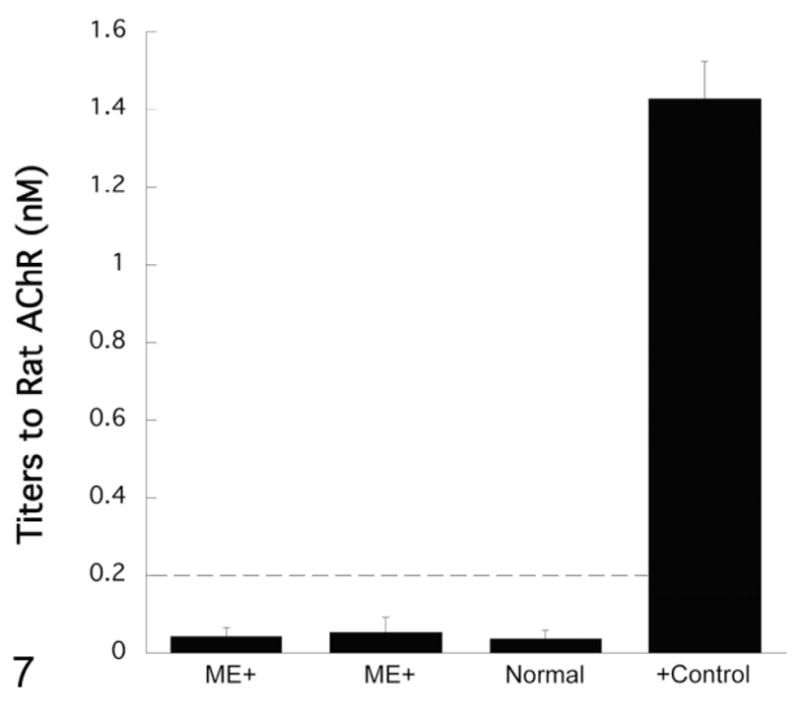
Antibody titers to muscle acetylcholine receptors (AChR). Representative samples from 2 transgene-positive rats with megaesophagus (ME+) and a normal transgene-negative rat from the same colony showing negative titers (<0.2 nM). A Long Evans rat with confirmed megaesophagus and a high AChR antibody titer (unpublished data) was used as a positive (+) control.
Megaesophagus is Associated with a Range of Histopathological Alterations in Affected Esophagi
All major organs were grossly and histologically evaluated in the first 10 cases diagnosed; no significant findings were noted except those in the esophagus. During the initial course of the study, it was determined that the esophagus was the primary organ that showed variable histological changes, and with the addition of more experimental animals, the esophagi of all animals were reevaluated in a blinded manner by a board-certified comparative pathologist (S.M.).
All transgene-negative animals had esophagi of normal diameters (0.2–0.3 cm; Fig. 8), while the esophagi of transgene-positive animals were grossly enlarged, particularly in the thoracic portion. Many had diameters ranging from 0.4 to 1.6 cm (Fig. 9).
Figure 8.
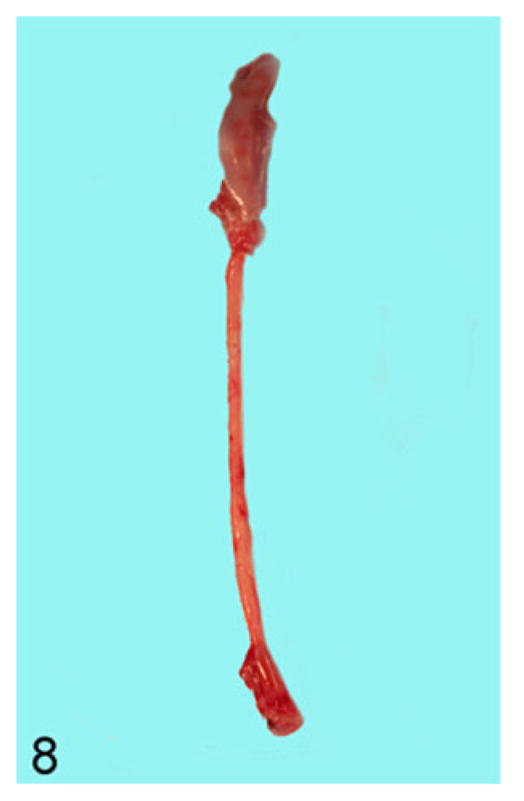
Esophagus; rat. Entire length of esophagus from a transgene-negative animal showing a normal esophageal diameter of 0.2 cm.
Figure 9.
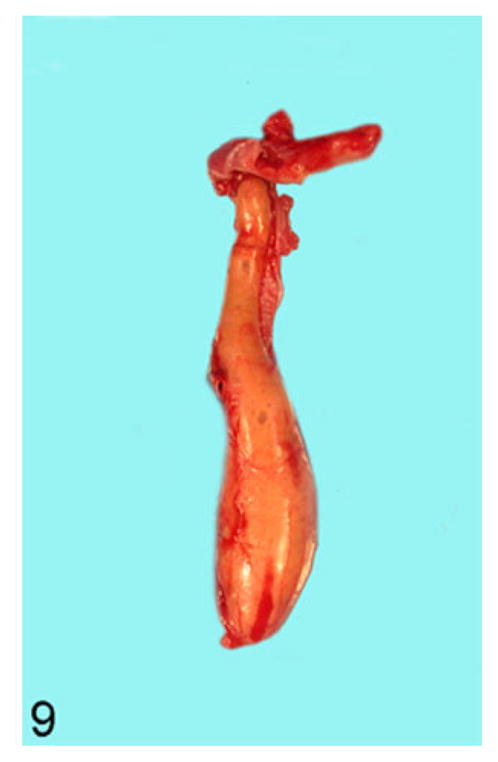
Esophagus; rat. A transgene-positive animal with megaesophagus showing an esophageal diameter of 1.6 cm. The entire esophagus is filled with compacted food and saliva.
The presence of any histologic changes was recorded separately for the epithelium, submucosa, muscularis proper, and adventitia, as well as nerve fibers whenever present in the plane of section. Histologically, all transgene-negative animals had essentially normal esophagi (Figs. 10, 11) except for rare instances of scattered mast cells (not shown). The esophagi of transgenic positive animals exhibited a broad range of morphological alterations that varied in type and severity. The most frequent finding was the presence of epithelial hyperkeratosis of varying severity (Fig. 12). There was a consistent increase in intraluminal debris (Fig. 12) frequently seen with intraluminal bacteria adherent to superficial keratin strands in the esophagus of affected rats (Fig. 13). Occasionally, epithelial ballooning degeneration was also noted, and in 1 instance, a subcorneal intraepithelial microabscess was also observed (Fig. 13). Epithelial and subepithelial inflammation was common, albeit minimal. Occasional mild epithelial hyperplasia was also noted (Fig. 14).
Figure 10.
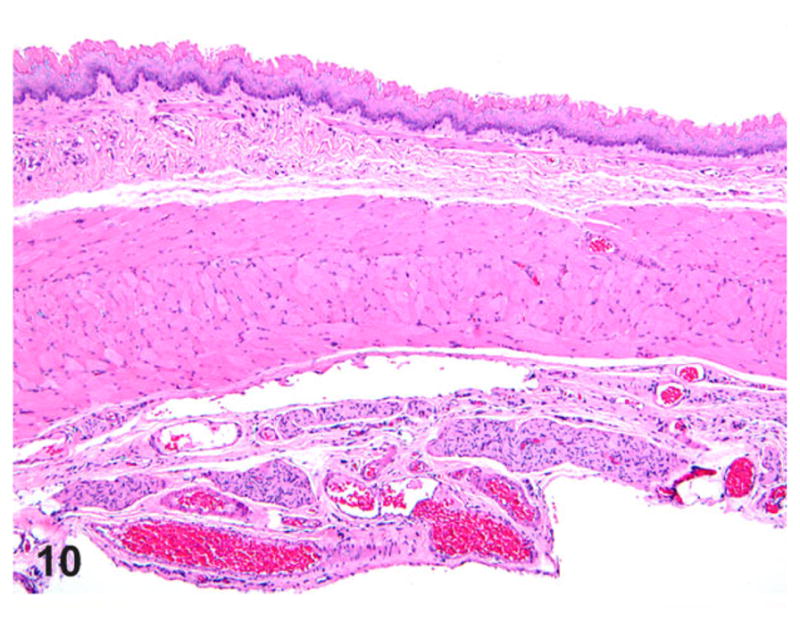
Esophagus; rat. Normal thoracic esophagus showing the stratified squamous epithelium, musculature, and adventitia. Hematoxylin and eosin (HE).
Figure 11.
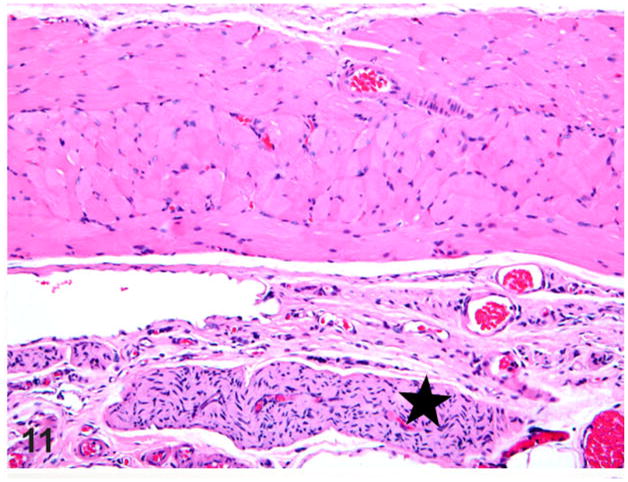
Esophagus; rat. Normal thoracic esophagus showing an associated vagus nerve branch (star) and vasculature. HE.
Figure 12.
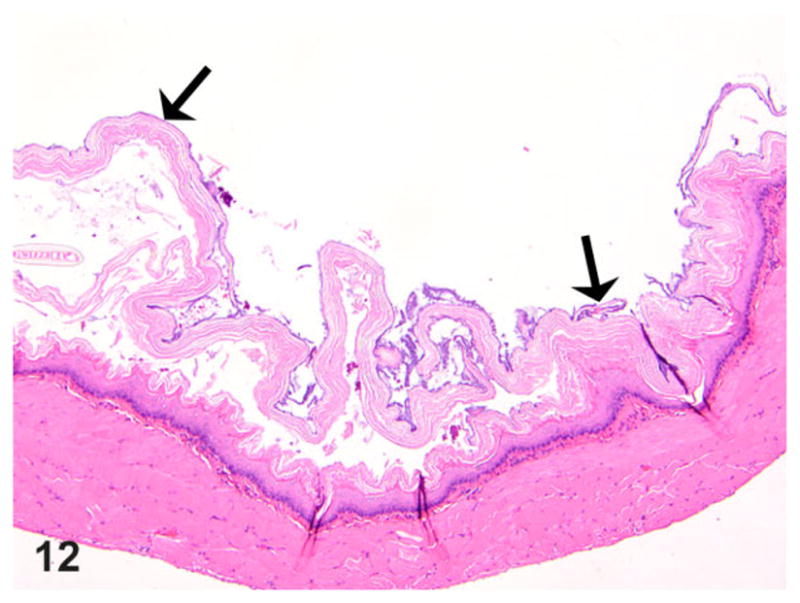
Esophagus; rat. Thoracic esophagus of a rat with megaesophagus showing increased intraluminal debris with epithelial hyperkeratosis (arrows). HE.
Figure 13.
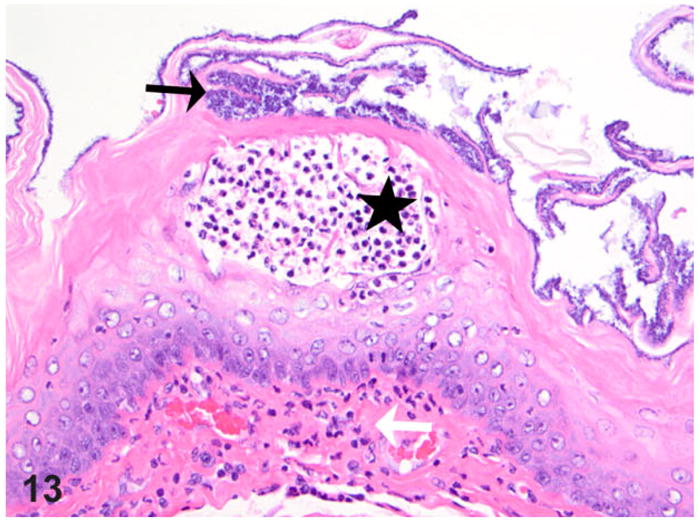
Esophagus; rat. Thoracic esophagus from a rat with megaesophagus showing adherent bacteria (black arrow) and focal subcorneal intraepithelial micro-abscess (star); note also focal subepithelial inflammatory cells present (granulocytes, white arrow). HE.
Figure 14.
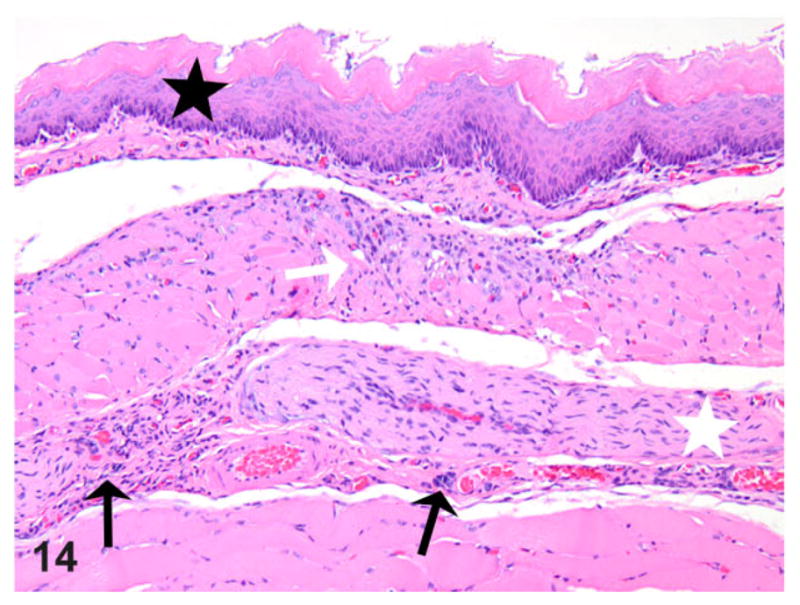
Esophagus; rat. Thoracic esophagus from a rat with megaesophagus showing mild epithelial hyperplasia (black star) above a focal area of muscle degeneration/necrosis (white arrow) and associated inflammation (black arrows, mast cells); also note the mild hypertrophy of a nerve branch (white star) within the affected segment. HE.
Nerve fibers (presumably branches of vagus), when present in the plane of section, were also assessed for any pathological alterations. Due to the variability in the plane of section between different animals, the thickness of the nerve fibers could not be accurately assessed, although at least in 1 case, there was minimal nerve hypertrophy and hypercellularity of the nerve fascicles in a region of muscle necrosis and/or inflammation (Figs. 14, 15). Inflammatory cells, chiefly mast cells and macrophages, were seen immediately adjacent to and occasionally within the nerve fibers (Figs. 14, 15). In a couple of cases, mast cells were also seen in association with the myenteric plexus (not shown).
Figure 15.
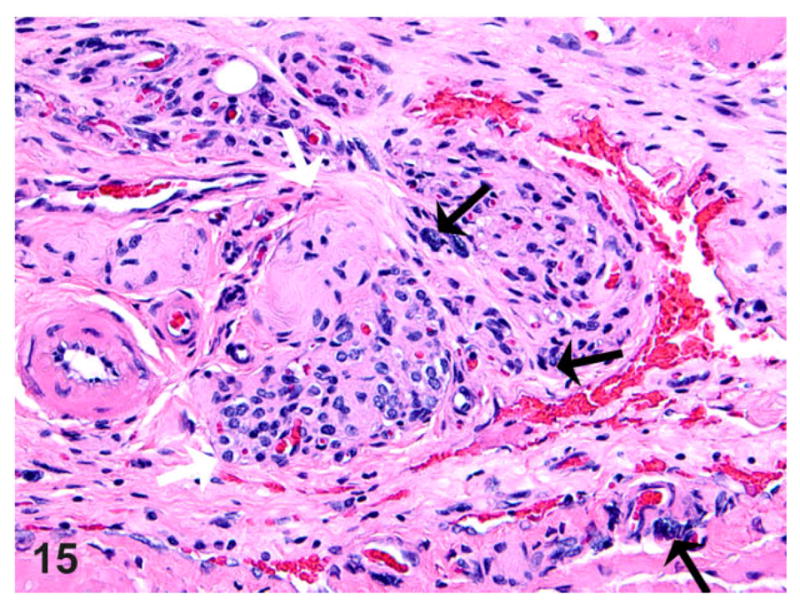
Esophagus; rat. An abdominal esophagus-associated nerve (white arrows) in the same animal as Fig. 14 showing hypercellularity of the nerve fascicles with the presence of mast cells (black arrows) within and surrounding the nerve fibers, together with prominent vascular channels with mild fibrosis. HE.
Skeletal muscle thickness was also carefully evaluated in at least 10 independent 10× objective fields from all segments of the esophagus, and in almost half of the megaesophagus cases, there was mild atrophy from either myofiber necrosis/loss or reduction in the number/thickness of fibers. In some, either focal muscle degeneration/necrosis (Fig. 14) or occasional segmental muscle necrosis, clearing, and fibrosis (Fig. 16) was present. Inflammation within the muscularis was variable, ranging from sparse to minimal, and was frequently noted in the areas of muscle degeneration/necrosis. Mast cells and macrophages were the cells commonly seen in the areas of muscle degeneration/necrosis.
Figure 16.
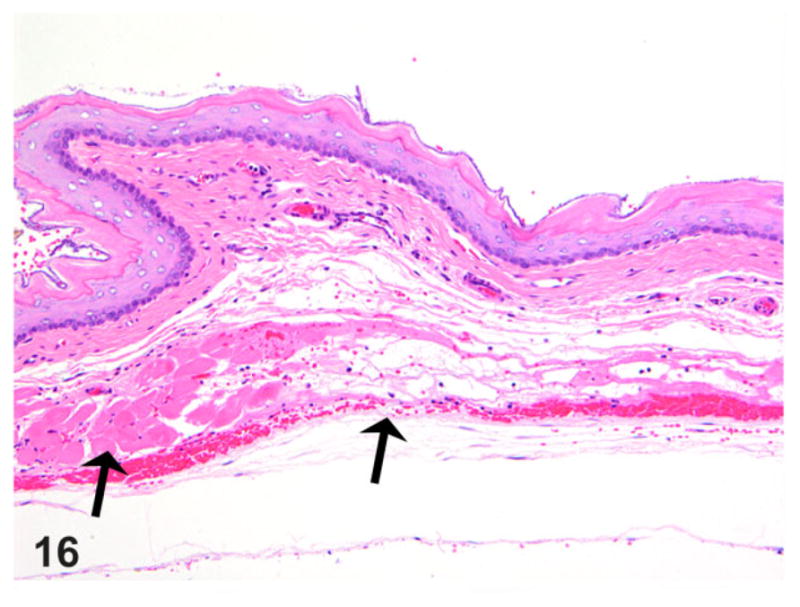
Esophagus; rat. Thoracic esophageal musculature from a rat with megaesophagus showing segmental muscle necrosis and atrophy (arrows) with associated minimal inflammation, fibrosis and hemorrhage. Hematoxylin and eosin (HE).
In addition, many affected rats and 1 control also showed minimal to mild multifocal myofiber pallor and/or clearing, as well as distinct sarcoplasmic vacuolation in the muscularis proper (Fig. 17). These sarcoplasmic changes were considered a true pathological alteration, especially when noticed in sections with no sectioning artifacts or when not present along the edges of the sections. The adventitia of affected cases in a few instances showed minimal to mild inflammation with a frequent occurrence of mast cells. Many of the muscle abnormalities noted were attributed to secondary change rather than considered a primary cause.
Figure 17.
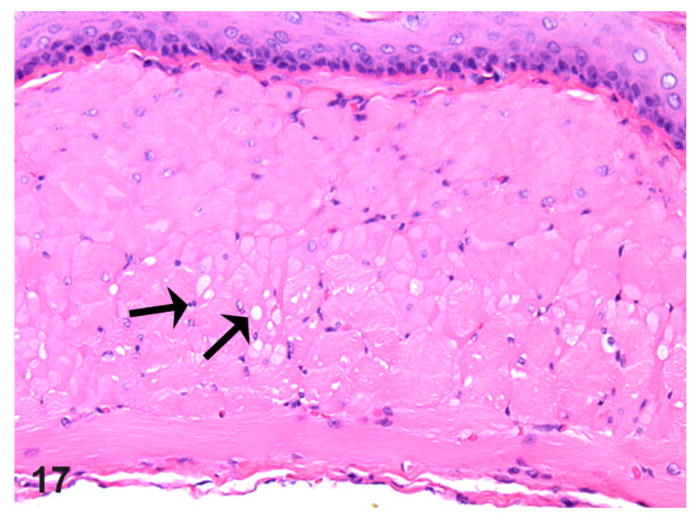
Esophagus; rat. Thoracic esophagus musculature from a rat with megaesophagus showing multifocal myofiber sarcoplasmic pallor and mild vacuolation (arrows). HE.
Dilated Esophagi have Reduced Myenteric Ganglion Cell Numbers with Ultrastructural Abnormalities
On nNOS immunohistochemistry, esophageal sections from normal rats had more nNOS-positive myenteric ganglia that were also larger in size (Fig. 18). The rats with megaesophagus had significantly lower numbers of nNOS-positive myenteric ganglia (P < .01; Figs. 19, 20). No significant difference in numbers of nNOS staining myenteric ganglia in the stomach was noted between the affected and normal rats (Fig. 20).
Figure 18.
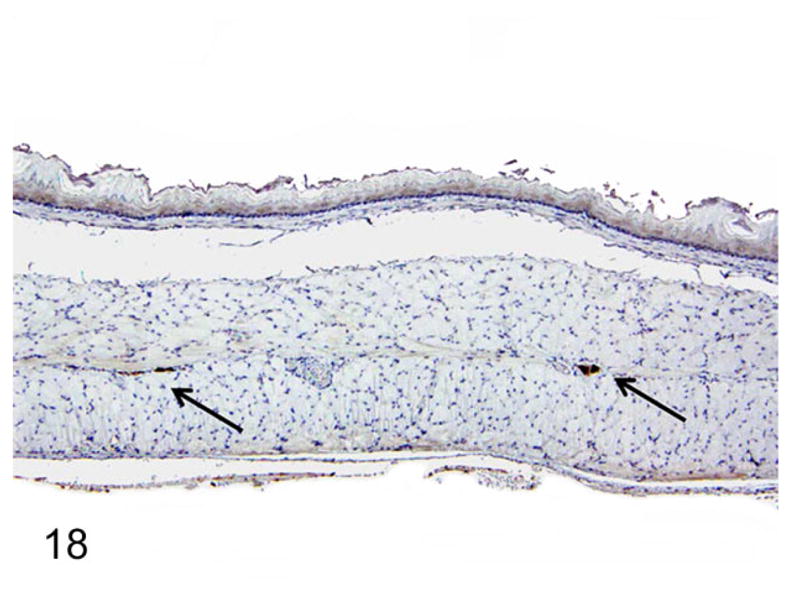
Normal rats had a greater number and larger neuronal nitric oxide synthase (nNOS)–positive ganglion cells (arrows). Immunohistochemistry-DAB, hematoxylin counterstain.
Figure 19.
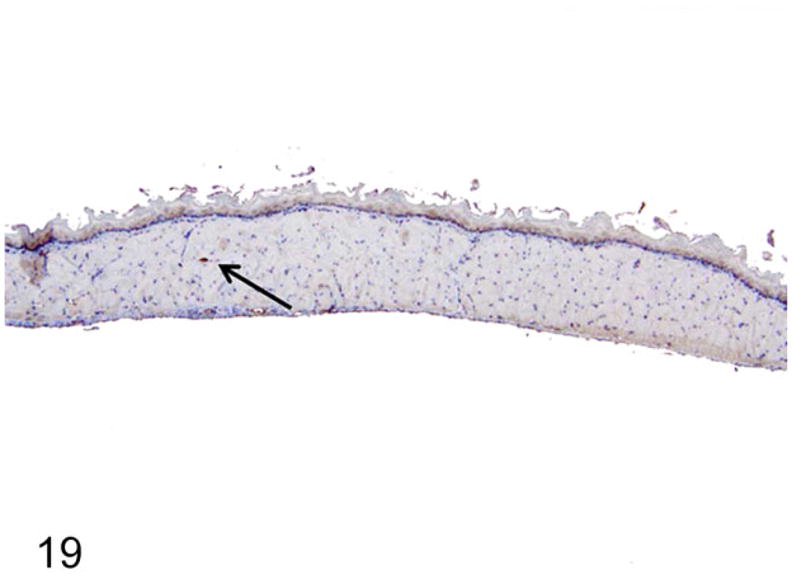
Rats with megaesophagus had fewer and smaller nNOS-positive ganglion cells (arrow). Immunohistochemistry-DAB, hematoxylin counterstain.
Figure 20.
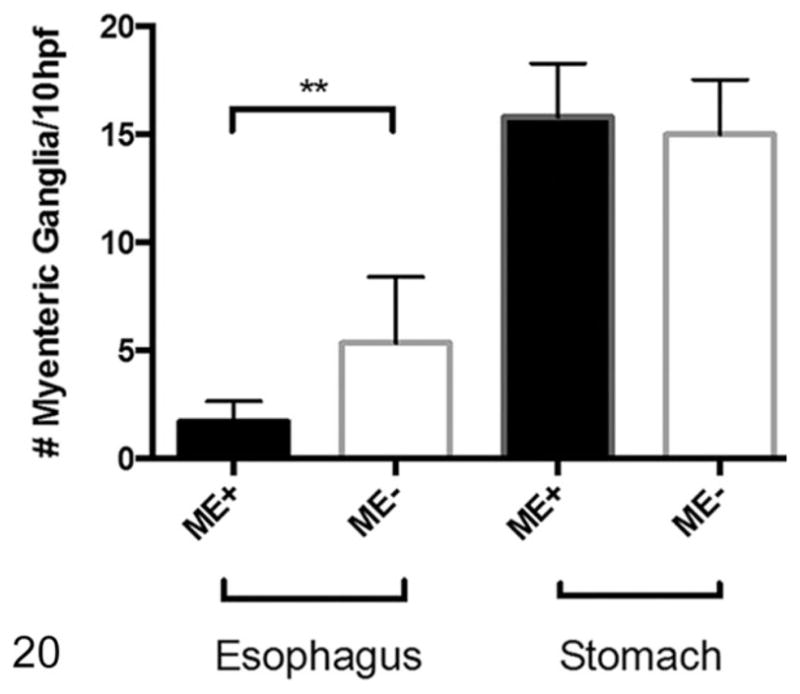
Number of myenteric ganglion units in the thoracic esophagus and stomach. Rats with megaesophagus (ME+) have significantly less neuronal nitric oxide synthase (nNOS)–staining myenteric ganglion units in the esophagus than normal rats (ME−) but equivalent numbers in the stomach; n = 5 in each group (**P < .01). hpf, high-power field.
Ultrastructurally, prominent mitochondrial changes were observed in the esophageal musculature of the rats with mega-esophagus. Compared with normal rats (Fig. 21), the esophageal skeletal muscle fibers of the affected rats frequently showed a prominent variation in the size and shape of the mitochondria that spanned most of the breadth of the I and A bands with associated fiber architectural disarray (Fig. 22). Rarely, vacuoles in the mitochondria were also present (Fig. 23).
Figure 21.
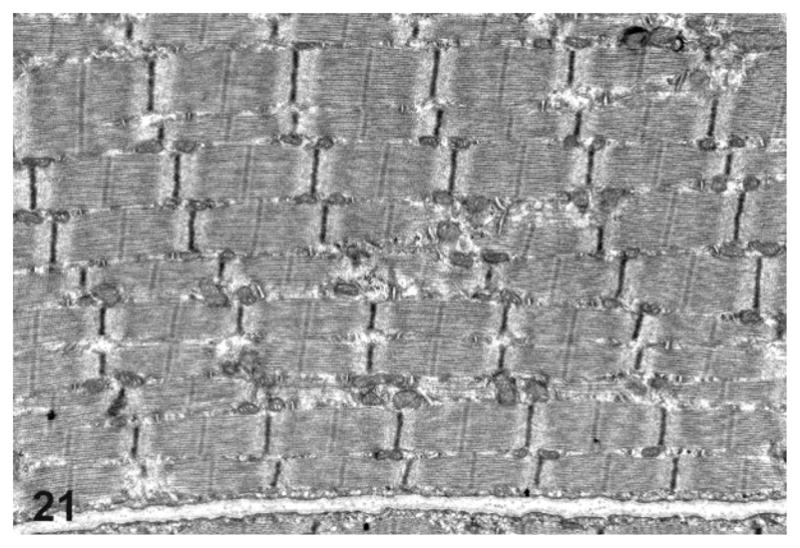
Transmission electron microscopy of the thoracic esophagus from a normal rat showing normal esophageal architecture.
Figure 22.

Transmission electron microscopy of the thoracic esophagus from a rat with megaesophagus showing muscle fiber disarray.
Figure 23.
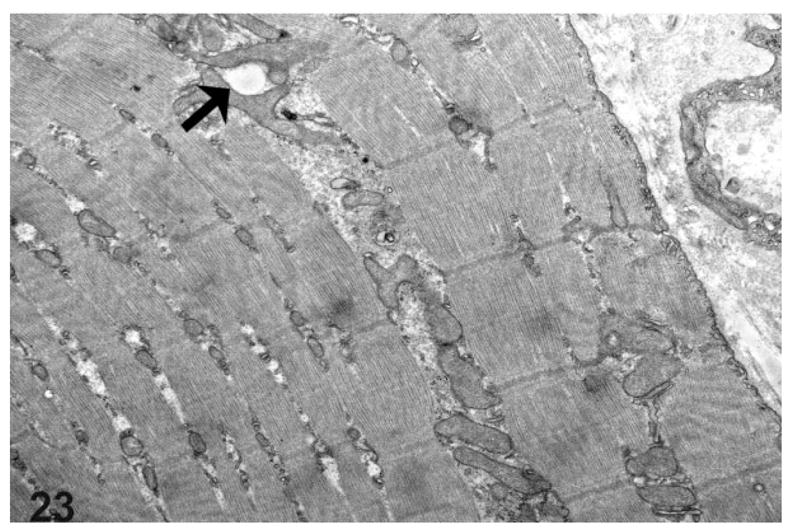
Transmission electron microscopy of the thoracic esophagus from a rat with megaesophagus showing prominent mitochondria of varying sizes and shapes with rare mitochondrial vacuoles visible (arrow).
Loss of Inhibition and Impaired Relaxation in the LES Muscle of Rats With Megaesophagus
By induction with EFS, the rats with megaesophagus had a lower percentage of in vitro relaxation of muscle strips from the LES compared with the normal rats at all frequencies, which was significant at frequencies of 0.5, 1, and 2 Hz (Fig. 24). The addition of SNP caused relaxation in the LES of rats with megaesophagus to a similar degree as the control animals (not shown). EFS-induced relaxation of the muscle strips from the fundus in affected rats was significantly lower than normal rats at frequencies greater than 2Hz (Fig. 25). In normal rats, L-NAME treatment significantly reduced relaxation in response to EFS in the fundus at frequencies higher than 2 Hz (Fig. 26). In contrast, rats with megaesophagus showed lower relaxation percentages to begin with and no significant changes following L-NAME treatment (Fig. 27).
Figure 24.
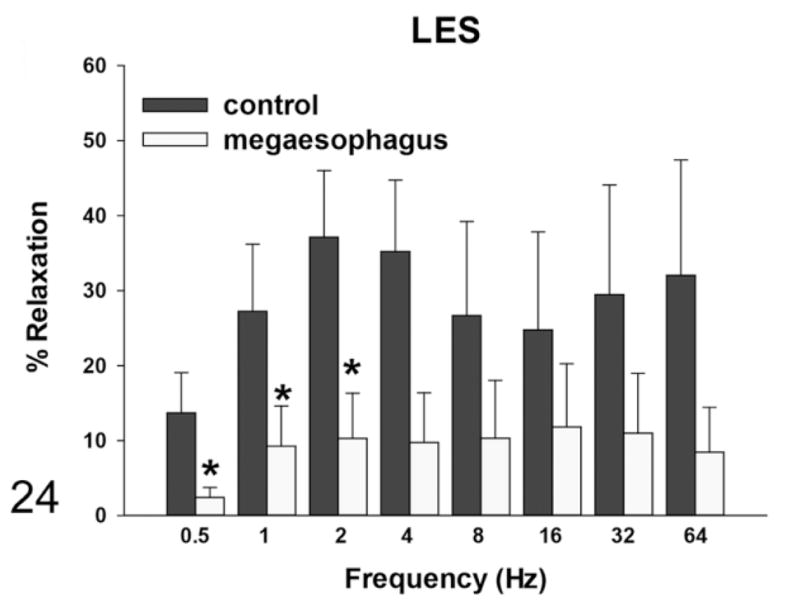
Muscle relaxation responses of the lower esophageal sphincter (LES)—rats with megaesophagus show less relaxation with electrical field stimulation (EFS) induction than normal rats (*P < .05).
Figure 25.
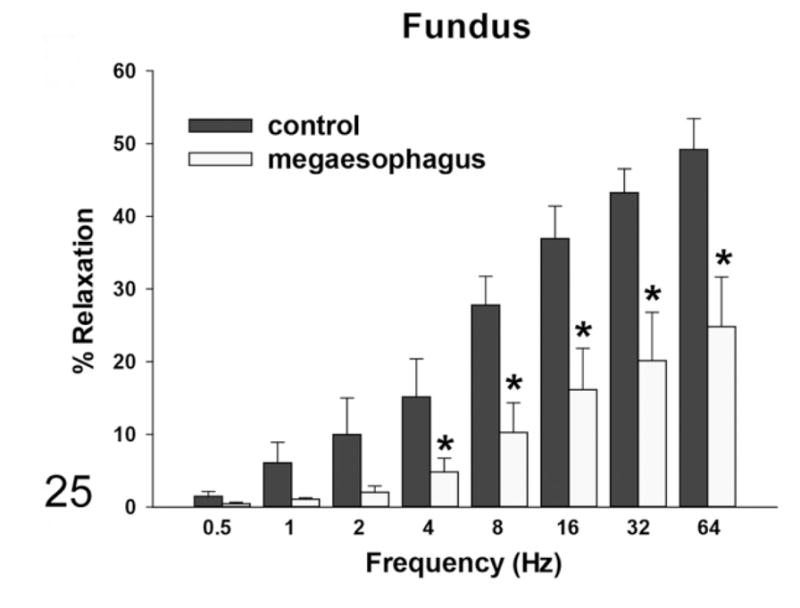
Muscle relaxation responses of the fundus—rats with megaesophagus show less relaxation with EFS induction than normal rats (*P < .05).
Figure 26.
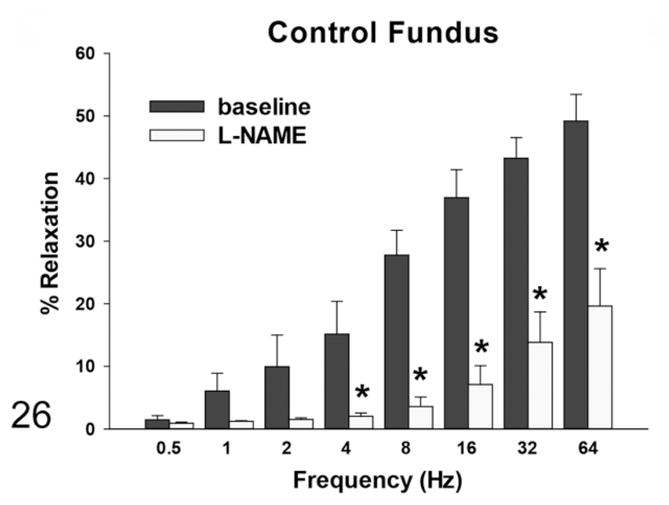
Muscle relaxation responses of the fundus—in normal rats, the relaxation response induced by EFS was significantly reduced in the presence of L-nitro-L-arginine methyl ester (L-NAME) (*P < .05).
Figure 27.
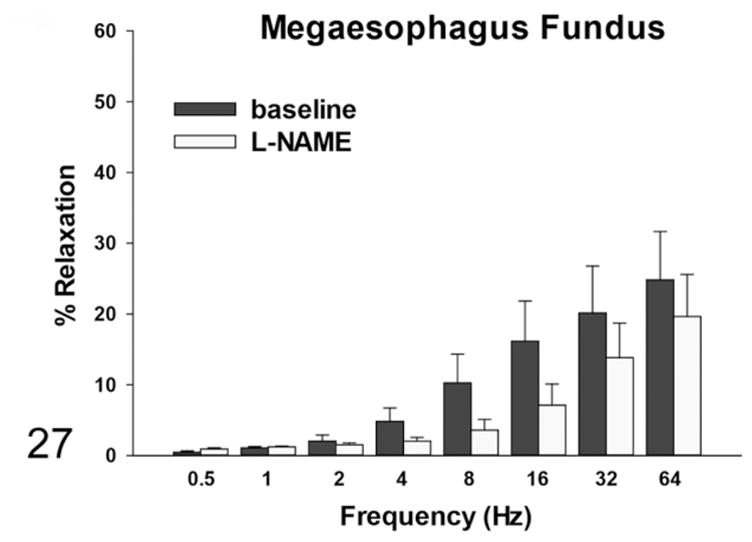
Muscle relaxation responses of the fundus—in rats with megaesophagus, there are lower baseline relaxation percentages with EFS induction and no significant changes after L-NAME treatment.
Discussion
Achalasia or megaesophagus in rats has been published infrequently. In 1958, Schulte70 commented that changes in the esophagus are rare in laboratory rodents. The first full publication by von Deerberg and Pittermann20 described megaesophagus in the inbred rat strain BDE/HAN. Approximately 20% of this colony was affected, and the authors concluded that megaesophagus in these rats was most likely genetic in origin. Megaesophagus was subsequently characterized in a 4-month-old male Wistar rat descendant Hla:(WI)BR2. The animal exhibited clinical signs similar to what we observed in our first few presenting cases (weight loss, dehydration, hunching, and saliva mixed with porphyrin staining the face, chin, and paws). The rat was found dead in its cage 1 month after being placed on a pelleted diet. Necropsy and histopathological findings mimicked those seen in other cases of megaesophagus.38 No obvious causes could be identified in this case, and the etiology was considered idiopathic.38
A stock colony of Srl:BHE rats aged 6 to 26 months also presented with esophageal impaction. About 21% of deaths (~1.3% of the total number of animals) in the colony were attributed to megaesophagus, and those affected included both sexes. Some rats also had recurrent episodes consistent with megaesophagus.67 Finally, Long-Evans rats on an aging study were observed with signs of achalasia.3 Thirty-nine percent of the colony obtained from a commercial source presented similarly between the ages of 3 and 32 months. Esophageal inflammation was not a key feature identified, but reduction in the myenteric ganglion cell numbers was demonstrated.3
Historically, the North American opossum (Didelphis virginiana) has been an excellent model for human esophageal physiology as it is similar to that of humans in terms of the proportion of smooth and skeletal muscle and intrinsic motor function.71 It is also large enough to evaluate manometrically and radiographically. Many other animals such as the dog have also been employed in a similar manner. While idiopathic cases have been noted in many species, no naturally occurring lines of achalasia have been identified.
However, in mice, several genetically modified lines have proven valuable. Megaesophagus in the ICRC/HiCri mouse is known to be an autosomal recessive trait, histologically characterized by decreased myenteric ganglia and muscular abnormality in the abdominal esophagus,65 suggesting involvement of the skeletal muscle. Similar to our findings, megaesophagus affected both sexes. More recently, the Lsc/p115−/− mouse showed that the lack of Lsc leads to an achalasia-like phenotype. These mice first present around 4 months of age and progress to more severe achalasia as they age.90 Other lines such as the Sprouty2−/− mouse demonstrate esophageal dilation but also feature other intestinal motility disorders.77 Rassf1a−/− mice reveal that inflammation of the myenteric plexus can lead to achalasia.81 The advantage of this strain is that the Rassf1a gene is a known tumor suppressor gene, and hence this may be the suggestive link between achalasia and cancer.
Given the lack of evidence that megaesophagus in this colony of rats was secondary to other conditions, our transgenic model is more consistent with the evidence that certain types of achalasia can be attributed to genetic influences,89 as shown by the association that achalasia has to other genetic diseases.34 For example, primary idiopathic achalasia is often seen in children with Allgrove, Alport, and Down syndromes.22,34
Isolated (nonsyndromic) achalasia may be the most common form of achalasia, but familial achalasia is also reported fairly frequently.34 However, no study has been comprehensive or large enough to study the genetic effect on achalasia. As the rats presented in this study both carry the same transgene and are related, this may provide an opportunity to analyze the genetic basis of this disease. To date, we have been unable to determine if the association between transgenic status and the presentation of megaesophagus is due to the insertion of the transgene or whether this was a result of a random mutation in a founder that was subsequently fixed in the population.
One of the biggest challenges for patients with achalasia is maintaining adequate nutrition;22 all the therapies and management options focus on this critical issue. Similarly with animals, providing nutritional support is an important part of any treatment plan. Animals with megaesophagus are unable to survive on the regular commercial rodent diet, but with the alternative diet regimen used in this study, group I rats were successfully maintained long term without clinical signs. Despite the slower weight gain on the total liquid diet, the common significant clinical signs due to esophageal impaction and aspiration pneumonia were completely eliminated in group II rats by placing them on modified diets immediately after weaning. Thus, even though the animals still demonstrated megaesophagus, the condition was subclinical.
Esophageal manometry is considered the gold standard for diagnosing achalasia. Symptomatic scoring has also been described,29,85 although this is not an option in animal studies. Overall, most agree that a barium esophagogram coupled with fluoroscopy is the best approach for an accurate diagnosis of achalasia.26 Characteristics noted in humans include the loss of primary peristalsis in the distal two-thirds of the esophagus and poor esophageal emptying with retained food. Absent or abnormal peristaltic waves have been observed in both humans and dogs,60 findings consistent with our analysis of esophageal motility in the affected rats under fluoroscopy. A smooth tapering of the lower esophagus leading to a closed LES, resembling a bird’s beak,59 is considered a classic presentation and is a feature consistently noted with rats in this current study. A timed barium esophagogram can also assist in assessing improvements in esophageal emptying after therapy.13,58,80
Our study did not use esophageal manometry, but the method has been used to measure LES pressure in rats.63 Nonetheless, by performing esophagograms and fluoroscopy, megaesophagus was diagnosed antemortem in the transgenic rats, with comparable findings and features to that in humans. To our knowledge, this is the first time fluoroscopy has been used on unanesthetized rats, which not only confirms the abnormal motility of the esophagus in affected rats without the complications of anesthesia but will also serve as a useful tool in analyzing treatments and progression of the disease in the future. The evidence of megaesophagus from a young age also provides a unique opportunity for studying the features of congenital achalasia and, combined with the ease of monitoring, opens the door to longitudinal studies.
It is currently accepted that the degree of neuronal loss in the myenteric plexus is dependent on the extent of the disease; human patients at an earlier stage of the disease tend to have a more functional esophagus and similar numbers of ganglion cells with minimal inflammation compared with healthy individuals.10,35,87 On the converse, a reduction or lack of ganglion cell numbers has been noted in congenital achalasia,2,23 and our study further supports this. The presence of mast cells is an additional feature that has been described, although their function remains unknown.6,87
Many of the changes in humans occur in the smooth muscle, where hypertrophy and muscle degeneration are both common findings.36 However, myodegeneration and cytoplasmic vacuolation in the esophagus have also been noted in rats with esophageal impaction.67 Hence, even though rats have different esophageal musculature than humans, this study shows that muscle degeneration, inflammation, and in particular mast cell infiltration are still common features of achalasia regardless of species.
With achalasia, the primary issue is the inability of the LES to completely relax. This involves the complex interplay of nerves, smooth muscle, interstitial cells of Cajal, and hormones. Peristalsis is also dependent on the complex interaction between inhibitory and excitatory neurotransmitters to propel food boluses toward the stomach. Inhibitory neurotransmitters cause relaxation of muscles distally, and excitatory neurotransmitters cause contraction of muscles proximal to the bolus.
Postganglionic cholinergic LES innervation in patients with achalasia is either normal or only minimally impaired, in contrast to the marked impairment of the inhibitory nerves governing LES relaxation.43 While there are a variety of mediators involved, the main inhibitory neurotransmitter is nitric oxide.1,41 nNOS is constitutively present and is responsible for producing nitric oxide (NO), and it is the deficiency of NO that leads to an increased resting tone of the LES.47
Studies investigating the knockout mouse strain nNOS−/− on a 129/Sv background confirmed a higher resting LES pressure and suppressed LES relaxation to varying degrees with swallowing and vagal stimulation.74 While this is a good model for LES hypertension, this knockout mouse lacks significant esophageal dilatation. In contrast, the transgenic rat model has significant enlargement of the esophagus and thus may be able to better elucidate questions about pathophysiology. The only human association study between achalasia and NOS gene polymorphisms was inconclusive,55 and further study on a larger sample size is warranted.
When subjected to EFS, there is contraction of the circular muscle strips of the LES in patients with achalasia instead of relaxation as noted in healthy humans.78,86 Comparably, transgenic rats with achalasia had a decrease in relaxation with EFS. Sodium nitroprusside, a NO donor given to elicit LES relaxation, was able to exert similar effects on both normal and affected rats. This is consistent with other experimental models of achalasia that demonstrate that the LES muscle is still functional, but the impairment of relaxation is due to neuronal loss rather than a smooth muscle function deficit.32,56
Gastric disturbances have also been noted in patients with achalasia,18,19 and as such, the fundus of the stomach was included in this part of the study to examine if the rats with megaesophagus showed any signs of extraesophageal involvement. Even though the numbers of nNOS ganglion units were comparable in the stomach of affected and control rats, there was a functional reduction in the rats with megaesophagus in response to EFS and L-NAME, a NO inhibitor known to reduce relaxation of the LES in healthy humans86 and rats.45 No clinical significance for gastric involvement has been recognized, but our study suggests that nNOS may not be as important for relaxation in the stomach as it is in the esophagus.
The loss of myenteric ganglion cells,10 degeneration of the vagus nerve,11 and infiltration of mast cells87 are easily visible with electron microscopy. However, changes in muscle are often nonspecific. In humans, prominent alterations include the detachment of myofilaments from the surface membranes of the cells and cellular atrophy with or without associated increases in cytoplasmic ribosomes.10 There may also be cellular hypertrophy10 and nuclear and cytoplasmic inclusions.31 Fibrosis among and inside muscle bundles has also been noted.27 A study examining denervation effects on rat skeletal muscle indicated that a loss of striation in the Z lines occurred first, prior to the filamental disorder and detachment,62 which is consistent with what was noted in this study.
Mitochondria also tend to disappear as the muscle atrophies.62 All the findings indicate that the muscle abnormalities, smooth or skeletal, result from denervation. Similar results were documented in dogs, including complete loss of the normal shape of mitochondria.75 These organelles were swollen and condensed with thick electron-dense material, as was seen in the rats with megaesophagus. Due to the limitations on the esophageal sections taken in this study, the vagus nerve could not be closely studied. However, the changes in mitochondria and the muscle as a whole align closely with the literature, once again highlighting the advantage for comparative use of this model.
In conclusion, this transgenic rat line has the advantage of a high penetrance rate (~90%) with significant esophageal dilation and a closed LES, both confirmed functionally and thus a convincing model for achalasia.37 The transgenic rat with achalasia presents with similar clinical signs to that observed in humans, and it is amenable to manipulation, evaluation, and management. It is comparable to congenital achalasia in humans, which makes it valuable for studying the genetic basis of the disease. It is also suitable for studying the progression of achalasia as the animal ages, with no need for additional manipulation or lag time to clinical presentation. Sustaining this colony long term is also convenient, allowing validation of current and identification of new long-term management strategies. Furthermore, it may be beneficial for studying carcinogenesis in a manner similar to the ICRC/HiCri mouse.33 It is more cost-effective to maintain a colony of rats long term compared with the larger animal models currently employed to study achalasia.
While determining why the transgenic rats developed achalasia without any other clinical findings is beyond the scope of our current study, we postulate that either the insertion of the trans-gene caused a disruption in neuronal pathways specific to the esophagus and parts of the stomach, or a random mutation in one of the parental animals and subsequent fixation of this gene in the population led to the widespread presentation of megaesophagus in this colony. Further studies are required to establish the particular location of insertion or mutation and the genes involved and should prove pivotal in understanding the etiology of this disease and ultimately provide more treatment and management options for patients with achalasia.
Acknowledgments
We thank Susumu Tonegawa for generously providing the transgenic rats to DCM and Raj Goyal for his collaboration and expert review on this study. Thanks also to Nicki Watson at the W. M. Keck Microscopy Facility for her assistance with electron microscopy and to Wener Lv and Richard Cohen for the use of the fluoroscope. Special thanks to Elaine Robbins, Lucy Wilhelm, and Alyssa Terestre for the preparation of this manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Institutes of Health (grant numbers T32OD011141 and P30ES002109).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Aggestrup S, Uddman R, Sundler F, et al. Lack of vasoactive intestinal polypeptide nerves in esophageal achalasia. Gastroenterology. 1983;84(5 pt 1):924–927. [PubMed] [Google Scholar]
- 2.Badrawy R, Abou-Bieh A. Congenital achalasia of the oesophagus in children. J Laryngol Otol. 1975;89(7):697–706. doi: 10.1017/s0022215100080919. [DOI] [PubMed] [Google Scholar]
- 3.Baiocco A, Boujon CE, Häfeli W, et al. Megaoesophagus in rats: a clinical, pathological and morphometrical study. J Comp Pathol. 1993;108:269–281. doi: 10.1016/s0021-9975(08)80290-3. [DOI] [PubMed] [Google Scholar]
- 4.Binder HJ, Bloom DL, Stern H, et al. The effect of cervical vagectomy on esophageal function in the monkey. Surgery. 1968;64(6):1075–1083. [PubMed] [Google Scholar]
- 5.Blanco MC, Fox JG, Rosenthal K, et al. Megaesophagus in nine ferrets. J Am Vet Med Assoc. 1994;205(3):444–447. [PubMed] [Google Scholar]
- 6.Bohl JR, Junginger T, Eckardt VF, et al. Electron microscopic studies of esophageal wall structures in patients with achalasia: casting more light on unresolved aspects of pathogenesis. Hepatogastroenterology. 2010;57(99–100):507–512. [PubMed] [Google Scholar]
- 7.Brown HR, Hardisty JF. Oral cavity, esophagus and stomach. In: Boorman GA, Eustis SL, Elwell MR, et al., editors. Pathology of the Fischer Rat: Reference and Atlas. San Diego, CA: Academic Press; 1990. pp. 9–30. [Google Scholar]
- 8.Brücher BL, Stein HJ, Bartels H, et al. Achalasia and esophageal cancer: incidence, prevalence, and prognosis. World J Surg. 2001;25(6):745–749. doi: 10.1007/s00268-001-0026-3. [DOI] [PubMed] [Google Scholar]
- 9.Buchanan JW. Tracheal signs and associated vascular anomalies in dogs with persistent right aortic arch. J Vet Intern Med. 2004;18(4):510–514. doi: 10.1892/0891-6640(2004)18<510:tsaava>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 10.Cassella RR, Brown AL, Jr, Sayre GP, et al. Achalasia of the esophagus: pathologic and etiologic considerations. Ann Surg. 1964;160(3):474–485. doi: 10.1097/00000658-196409000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassella RR, Ellis FH, Jr, Brown AL., Jr Fine-structure changes in achalasia of the esophagus, I: vagus nerves. Am J Pathol. 1965;46:279–288. [PMC free article] [PubMed] [Google Scholar]
- 12.Cheatham JG, Wong RKH. Current approach to the treatment of achalasia. Curr Gastroenterol Rep. 2011;13:219–225. doi: 10.1007/s11894-011-0190-z. [DOI] [PubMed] [Google Scholar]
- 13.Chuah SK, Hu TH, Wu KL, et al. The role of barium esophagogram measurements in assessing achalasia patients after endoscope-guided pneumatic dilation. Dis Esophagus. 2008;22(2):163–168. doi: 10.1111/j.1442-2050.2008.00888.x. [DOI] [PubMed] [Google Scholar]
- 14.Clark SB, Rice TW, Tubbs RR, et al. The nature of the myenteric infiltrate in achalasia. Am J Surg Pathol. 2000;24(8):1153–1158. doi: 10.1097/00000478-200008000-00014. [DOI] [PubMed] [Google Scholar]
- 15.Clifford DH, Gyorkey F. Myenteric ganglial cells in dogs with and without achalasia of the esophagus. J Am Vet Med Assoc. 1967;150(2):205–211. [PubMed] [Google Scholar]
- 16.Clifford DH, Pirsch JG. Myenteric ganglial cells in dogs with and without hereditary achalasia of the esophagus. Am J Vet Res. 1971;32(4):615–619. [PubMed] [Google Scholar]
- 17.Clifford DH, Soifer FK, Freeman RG. Stricture and dilatation of the esophagus in the cat. J Am Vet Med Assoc. 1970;156(8):1007–1014. [PubMed] [Google Scholar]
- 18.Csendes A, Smok G, Braghetto I, et al. Histological studies of Auerbach’s plexuses of the oesophagus, stomach, jejunum, and colon in patients with achalasia of the oesophagus: correlation with gastric acid secretion, presence of parietal cells and gastric emptying of solids. Gut. 1992;33(2):150–154. doi: 10.1136/gut.33.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Giorgio R, Simone MP, Stanghellini V, et al. Esophageal and gastric nitric oxide synthesizing innervation in primary achalasia. Am J Gastroenterol. 1999;94(9):2357–2362. doi: 10.1111/j.1572-0241.1999.01357.x. [DOI] [PubMed] [Google Scholar]
- 20.Deerberg von F, Pittermann W. Megaesophagus als todesurasche bei BDE/Han Rattan. Z Versuchstierk Bd. 1972;14:177–182. [PubMed] [Google Scholar]
- 21.Dewey CW, Bailey CS, Shelton GD, et al. Clinical forms of acquired myasthenia gravis in dogs: 25 cases (1988–1995) J Vet Intern Med. 1997;11(2):50–57. doi: 10.1111/j.1939-1676.1997.tb00073.x. [DOI] [PubMed] [Google Scholar]
- 22.Dughera L, Chiaverina M, Cacciotella L, et al. Management of achalasia. Clin Exp Gastroenterol. 2011;4:33–41. doi: 10.2147/CEG.S11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Earlam RJ, Zollman PE, Ellis FH., Jr Congenital oesophageal achalasia in the dog. Thorax. 1967;22:466–472. doi: 10.1136/thx.22.5.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckardt AJ, Eckardt VF. Current clinical approach to achalasia. World J Gastroenterol. 2009;15(32):3969–3975. doi: 10.3748/wjg.15.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckardt VF, Kohne U, Junginger T, et al. Risk factors for diagnostic delay in achalasia. Dig Dis Sci. 1997;42(3):580–585. doi: 10.1023/a:1018855327960. [DOI] [PubMed] [Google Scholar]
- 26.Farrokhi F, Vaezi MF. Idiopathic (primary) achalasia. Orphanet J Rare Dis. 2007;2:38. doi: 10.1186/1750-1172-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faussone-Pellegrini MS, Cortesini C. The muscle coat of the lower esophageal sphincter in patients with achalasia and hypertensive sphincter: an electron microscopic study. J Submicrosc Cytol. 1985;17(4):673–685. [PubMed] [Google Scholar]
- 28.Febrônio LH, Britto-Garcia S, de Oliveira JS, et al. Megaesophagus in rats. Res Exp Med. 1997;197:109–115. doi: 10.1007/s004330050060. [DOI] [PubMed] [Google Scholar]
- 29.Ferri LE, Cools-Lartigue J, Cao J, et al. Clinical predictors of achalasia. Dis Esophagus. 2010;23(1):76–81. doi: 10.1111/j.1442-2050.2009.01006.x. [DOI] [PubMed] [Google Scholar]
- 30.Fracassi F, Tamborini A. Reversible megaoesophagus associated with primary hypothyroidism in a dog. Vet Rec. 2011;168(12):329b. doi: 10.1136/vr.c6348. [DOI] [PubMed] [Google Scholar]
- 31.Friesen DL, Henderson RD, Hanna W. Ultrastructure of the esophageal muscle in achalasia and diffuse esophageal spasm. Am J Clin Pathol. 1983;79(3):319–325. doi: 10.1093/ajcp/79.3.319. [DOI] [PubMed] [Google Scholar]
- 32.Gaumnitz EA, Bass P, Osinki MA, et al. Electrophysiological and pharmacological responses of chronically denervated lower esophageal sphincter of the opossum. Gastroenterology. 1995;109:789–799. doi: 10.1016/0016-5085(95)90386-0. [DOI] [PubMed] [Google Scholar]
- 33.Ghaisas S, Saranath D, Deo MG. ICRC mouse with congenital mega-esophagus as a model to study esophageal tumorigenesis. Carcinogenesis. 1989;10(10):1847–1854. doi: 10.1093/carcin/10.10.1847. [DOI] [PubMed] [Google Scholar]
- 34.Gockel HR, Schumacher J, Gockel I, et al. Achalasia: will genetic studies provide insights? Hum Genet. 2010;128(4):353–364. doi: 10.1007/s00439-010-0874-8. [DOI] [PubMed] [Google Scholar]
- 35.Goldblum JR, Rice TW, Richter JE. Histopathologic features in esophagomyotomy specimens from patients with achalasia. Gastroenterology. 1996;111(3):648–654. doi: 10.1053/gast.1996.v111.pm8780569. [DOI] [PubMed] [Google Scholar]
- 36.Goldblum JR, Whyte RI, Orringer MB. Achalasia: amorphologic study of 42 resected specimens. Am J Surg Pathol. 1994;18(4):327–337. [PubMed] [Google Scholar]
- 37.Goyal RK, Chaudhury A. Pathogenesis of achalasia: lessons from mutant mice. Gastroenterology. 2010;139(4):1086–1090. doi: 10.1053/j.gastro.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 38.Harkness JE, Ferguson FG. Idiopathic megaesophagus in a rat. Lab Anim Sci. 1979;29(4):495–498. [PubMed] [Google Scholar]
- 39.Harms CA, Andrews GA. Megaesophagus in a domestic ferret. Lab Anim Sci. 1993;43(5):506–508. [PubMed] [Google Scholar]
- 40.Higgs B, Ellis FH., Jr The effect of bilateral supranodosal vagotomy on canine esophageal function. Surgery. 1965;58(5):828–834. [PubMed] [Google Scholar]
- 41.Hirano I. Pathophysiology of achalasia and diffuse esophageal spasm. In: Goyal RK, Shaker R, editors. GI Motility Online. New York, NY: Nature Publishing Group; 2006. [Google Scholar]
- 42.Hoffer RE, Valdes-Dapena A, Baue AE. A comparative study of naturally occurring canine achalasia. Arch Surg. 1967;95(1):83–88. doi: 10.1001/archsurg.1967.01330130085017. [DOI] [PubMed] [Google Scholar]
- 43.Holloway RH, Dodds WJ, Helm JF, et al. Integrity of cholinergic innervation to the lower esophageal sphincter in achalasia. Gastroenterology. 1986;90(4):924–929. doi: 10.1016/0016-5085(86)90869-3. [DOI] [PubMed] [Google Scholar]
- 44.Huber E, Armbrust W, Forster JL, et al. Resolution of megaesophagus after treatment of concurrent hypothyroidism in a dog. Schweiz Arch Tierheilkd. 2001;143(10):512–514. [PubMed] [Google Scholar]
- 45.Kawahara H, Blackshaw LA, Lehmann A, et al. Responses of the rat lower oesophageal sphincter (LOS) to vagal efferent activation. Neurogastroenterol Motil. 1997;9(2):85–97. doi: 10.1046/j.1365-2982.1997.d01-24.x. [DOI] [PubMed] [Google Scholar]
- 46.Khajanchee YS, vanAndel R, Jobe BA. Electrical stimulation of the vagus nerve restores motility in an animal model of achalasia. J Gastrointest Surg. 2003;7(7):843–849. doi: 10.1007/s11605-003-0028-6. [DOI] [PubMed] [Google Scholar]
- 47.Konturek JW, Thor P, Lukaszyk A, et al. Endogenous nitric oxide in the control of esophageal motility in humans. J Physiol Pharmacol. 1997;48(2):201–209. [PubMed] [Google Scholar]
- 48.Kravitz JJ, Snape WJ, Jr, Cohen S. Effect of thoracic vagotomy and vagal stimulation on esophageal function. Am J Physiol Gastrointest Liver Physiol. 1978;234(4):G359–G364. doi: 10.1152/ajpendo.1978.234.4.E359. [DOI] [PubMed] [Google Scholar]
- 49.Kuo B, Urma D. Esophagus—anatomy and development. In: Goyal RK, Shaker R, editors. GI Motility Online. New York, NY: Nature Publishing Group; 2006. [Google Scholar]
- 50.Larcher T, Abadie J, Roux FA, et al. Persistent left cranial vena cava causing oesophageal obstruction and consequent megaoesophagus in a dog. J Comp Pathol. 2006;135(2–3):150–152. doi: 10.1016/j.jcpa.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 51.Little AG, Correnti FS, Calleja IJ, et al. Effect of incomplete obstruction on feline esophageal function with a clinical correlation. Surgery. 1986;100(2):430–436. [PubMed] [Google Scholar]
- 52.Loughin CA, Marino DJ. Delayed primary surgical treatment in a dog with a persistent right aortic arch. J Am Anim Hosp Assoc. 2008;44(5):258–261. doi: 10.5326/0440258. [DOI] [PubMed] [Google Scholar]
- 53.Massey BT. Management of idiopathic achalasia: short-term and long-term outcomes. Curr Gastroenterol Rep. 2000;2:196–200. doi: 10.1007/s11894-000-0061-5. [DOI] [PubMed] [Google Scholar]
- 54.Mayberry JF. Epidemiology and demographics of achalasia. Gastrointest Endosc Clin North Am. 2001;11(2):235–248. [PubMed] [Google Scholar]
- 55.Mearin F, García-González MA, Strunk M, et al. Association between achalasia and nitric oxide synthase gene polymorphisms. Am J Gastroenterol. 2006;101(9):1979–1984. doi: 10.1111/j.1572-0241.2006.00762.x. [DOI] [PubMed] [Google Scholar]
- 56.Mearin F, Mourelle M, Guarner F, et al. Patients with achalasia lack nitric oxide synthase in the gastrooesophageal junction. Eur J Clin Invest. 1993;23(11):724–728. doi: 10.1111/j.1365-2362.1993.tb01292.x. [DOI] [PubMed] [Google Scholar]
- 57.Mittal RK, Ren J, McCallum RW, et al. Modulation of feline esophageal contractions by bolus volume and outflow obstruction. Am J Physiol Gastrointest Liver Physiol. 1990;258(2 pt 1):G208–G215. doi: 10.1152/ajpgi.1990.258.2.G208. [DOI] [PubMed] [Google Scholar]
- 58.Montazeri G, Nouri N, Estakhri A, et al. Lower oesophageal sphincter pressure and timed barium oesophagogram: two objective parameters in the non-invasive assessment of primary achalasia. Aliment Pharmacol Ther. 2005;22(3):261–265. doi: 10.1111/j.1365-2036.2005.02557.x. [DOI] [PubMed] [Google Scholar]
- 59.O’Neill OM, Johnston BT, Coleman HG. Achalasia: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. 2013;19(35):5806–5812. doi: 10.3748/wjg.v19.i35.5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Osborne CA, Clifford DH, Jessen C. Hereditary esophageal achalasia in dogs. J Am Vet Med Assoc. 1967;151(5):572–581. [PubMed] [Google Scholar]
- 61.Park W, Vaezi MF. Etiology and pathogenesis of achalasia: the current understanding. Am J Gastroenterol. 2005;100(6):1404–1414. doi: 10.1111/j.1572-0241.2005.41775.x. [DOI] [PubMed] [Google Scholar]
- 62.Pellegrino C, Franzini C. An electron microscope study of denervation atrophy in red and white skeletal muscle fibers. J Cell Biol. 1963;17(2):327–349. doi: 10.1083/jcb.17.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qi B, Diez-Pardo JA, Soto C, et al. Transdiaphragmatic pressure gradients and the lower esophageal sphincter after tight abdominal wall plication in the rat. J Pediatr Surg. 1996;31(12):1666–1669. doi: 10.1016/s0022-3468(96)90044-5. [DOI] [PubMed] [Google Scholar]
- 64.Quidute AR, Freitas EV, Lima TG, et al. Achalasia and thyroid disease: possible autoimmune connection? Arq Bras Endocrinol Metabol. 2012;56(9):677–682. doi: 10.1590/s0004-27302012000900013. [DOI] [PubMed] [Google Scholar]
- 65.Randelia HP, Deo MG, Lalitha VS. Megaesophagus in mouse—histochemical studies. Gastroenterology. 1988;94(5 pt 1):1243–1244. doi: 10.1016/0016-5085(88)90033-9. [DOI] [PubMed] [Google Scholar]
- 66.Rogers AB, Cormier KS, Fox JG. Thiol-reactive compounds prevent nonspecific antibody binding in immunohistochemistry. Lab Invest. 2006;86(5):526–533. doi: 10.1038/labinvest.3700407. [DOI] [PubMed] [Google Scholar]
- 67.Ruben Z, Rohrbacher E, Miller JE. Esophageal impaction in BHE rats. Lab Anim Sci. 1983;33(1):63–65. [PubMed] [Google Scholar]
- 68.Sabirov AG, Raginov IS, Burmistrov MV, et al. Morphofunctional analysis of experimental model of esophageal achalasia in rats. Bull Exp Biol Med. 2010;149(4):466–470. doi: 10.1007/s10517-010-0972-6. [DOI] [PubMed] [Google Scholar]
- 69.Schneider JH, Peters JH, Kirkman E, et al. Are the motility abnormalities of achalasia reversible? An experimental outflow obstruction in the feline model. Surgery. 1999;125(5):498–503. [PubMed] [Google Scholar]
- 70.Schulte F. Speiseröhre Magen, Darm und Bauchfell. In: Cohrs P, Jaffé R, Meessen H, editors. Pathologie der Laboratoriumstiere. Berlin, Germany: Springer Verlag; 1958. [Google Scholar]
- 71.Schulze K, Dodds WJ, Christensen J, et al. Esophageal manometry in the opossum. Am J Physiol. 1977;233(3):E152–E159. doi: 10.1152/ajpendo.1977.233.3.E152. [DOI] [PubMed] [Google Scholar]
- 72.Shelton GD, Schule A, Kass PH. Risk factors for acquired myasthenia gravis in dogs: 1,154 cases (1991–1995) J Am Vet Med Assoc. 1997;211(11):1428–1431. [PubMed] [Google Scholar]
- 73.Singaram C, Sweet MA, Gaumnitz EA, et al. Evaluation of early events in the creation of amyenteric opossum model of achalasia. Neurogastroenterol Motil. 1996;8(4):351–361. doi: 10.1111/j.1365-2982.1996.tb00273.x. [DOI] [PubMed] [Google Scholar]
- 74.Sivarao DV, Mashimo HL, Thatte HS, et al. Lower esophageal sphincter is achalasic in nNOS(−/−) and hypotensive in W/W(v) mutant mice. Gastroenterology. 2001;121(1):34–42. doi: 10.1053/gast.2001.25541. [DOI] [PubMed] [Google Scholar]
- 75.Suryawanshi RV, Raghavender KBP, Gireeshkumar V, et al. Scanning and electron microscopic study of canine megaesophagus and its management. Int J Plant Anim Env Sci. 2011;1(3):116–122. [Google Scholar]
- 76.Takahashi R, Hirabayashi M, Ueda M. Production of transgenic rats using cryopreserved pronuclear staged zygotes. Transgenic Res. 1999;8:397–400. doi: 10.1023/a:1008910629235. [DOI] [PubMed] [Google Scholar]
- 77.Taketomi T, Yoshiga D, Taniguchi K, et al. Loss of mammalian Sprouty2 leads to enteric neuronal hyperplasia and esophageal achalasia. Nat Neurosci. 2005;8(7):855–857. doi: 10.1038/nn1485. [DOI] [PubMed] [Google Scholar]
- 78.Tłottrup A, Forman A, Funch-Jensen P, et al. Effects of postganglionic nerve stimulation in oesophageal achalasia: an in vitro study. Gut. 1990;31(1):17–20. doi: 10.1136/gut.31.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tung HN, Schulze-Delrieu K, Shirazi S, et al. Hypertrophic smooth muscle in the partially obstructed opossum esophagus. The model: histological and ultrastructural observations. Gastroenterology. 1991;100(4):853–864. doi: 10.1016/0016-5085(91)90256-k. [DOI] [PubMed] [Google Scholar]
- 80.Vaezi MF, Baker ME, Achkar E, et al. Timed barium oesophagram: better predictor of long term success after pneumatic dilation in achalasia than symptom assessment. Gut. 2002;50(6):765–770. doi: 10.1136/gut.50.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van der Weyden L, Happerfield L, Arends MJ, et al. Megaoesophagus in Rassf1a-null mice. Int J Exp Pathol. 2009;90(2):101–108. doi: 10.1111/j.1365-2613.2008.00635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vespúcio MV, Paschoal RM, Zucoloto S, et al. A new experimental model to study preneoplastic lesions in achalasia of the esophagus. Acta Cir Bras. 2005;20(6):418–421. doi: 10.1590/s0102-86502005000600004. [DOI] [PubMed] [Google Scholar]
- 83.Webb AA, Taylor SM, McPhee L. Focal myasthenia gravis in a dog. Can Vet J. 1997;38(8):493–495. [PMC free article] [PubMed] [Google Scholar]
- 84.Wörl J, Neuhuber WL. Enteric co-innervation of motor endplates in the esophagus: state of the art ten years after. Histochem Cell Biol. 2005;123(2):117–130. doi: 10.1007/s00418-005-0764-7. [DOI] [PubMed] [Google Scholar]
- 85.Yaghoobi M, Mikaeli J, Montazeri G, et al. Correlation between clinical severity score and the lower esophageal sphincter relaxation pressure in idiopathic achalasia. Am J Gastroenterol. 2003;98(2):279–283. doi: 10.1111/j.1572-0241.2003.07266.x. [DOI] [PubMed] [Google Scholar]
- 86.Yamato S, Saha JK, Goyal RK. Role of nitric oxide in lower esophageal sphincter relaxation to swallowing. Life Sci. 1992;50(17):1263–1272. doi: 10.1016/0024-3205(92)90326-k. [DOI] [PubMed] [Google Scholar]
- 87.Zarate N, Wang XY, Tougas G, et al. Intramuscular interstitial cells of Cajal associated with mast cells survive nitrergic nerves in achalasia. Neurogastroenterol Motil. 2006;18(7):556–568. doi: 10.1111/j.1365-2982.2006.00788.x. [DOI] [PubMed] [Google Scholar]
- 88.Zhu YQ, Cheng YS, Li MH, et al. Temporary self-expanding cardia stents for the treatment of achalasia: an experimental study in dogs. Neurogastroenterol Motil. 2010;22:e1240–e1322. doi: 10.1111/j.1365-2982.2010.01573.x. [DOI] [PubMed] [Google Scholar]
- 89.Zilberstein B, de Cleva R, Gabriel AG, et al. Congenital achalasia: facts and fantasies. Dis Esophagus. 2005;18(5):335–337. doi: 10.1111/j.1442-2050.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- 90.Zizer E, Beilke S, Bäuerle T, et al. Loss of Lsc/p115 protein leads to neuronal hypoplasia in the esophagus and an achalasia-like phenotype in mice. Gastroenterology. 2010;139(4):1344–1354. doi: 10.1053/j.gastro.2010.06.041. [DOI] [PubMed] [Google Scholar]