
Figure 4.
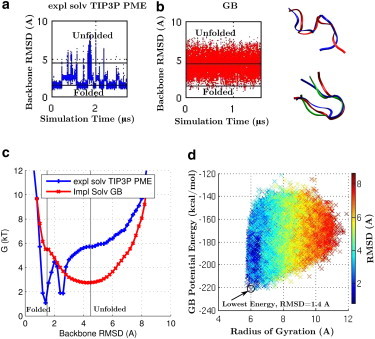
CLN025 miniprotein folding at its experimental melting temperature of 340 K. (a and b) RMSD of backbone heavy atoms relative to the starting structure for the explicit-solvent (TIP3P) PME simulation (a) and the GB simulation (b). The horizontal lines represent RMSD = 1.5 and 4.5 Å. Folded states are states with RMSD < 1.5 Å and unfolded states are states with RMSD > 4.5 Å. The trajectory is sampled every 100 ps for calculation of the RMSD values shown here. (c) Free-energy landscape for the explicit-solvent TIP3P PME and the GB simulations. (d) Potential energy, including solvation free energy, from the GB simulation, as a function of the distance (RMSD) from the experimental native structure. The lowest-energy structure approximates the correct folded state, as indicated by the low RMSD values. (Inset) Images of protein backbone conformations from representative snapshots for folded and unfolded states from the explicit-solvent (TIP3P) PME (blue) and GB (red) simulations. The starting structure (green) is shown for comparison. Images rendered using VMD (100). To see this figure in color, go online.