

Basic helix-loop-helix (bHLH) transcription factors recognize the canonical E-box (CANNTG) to regulate gene transcription, but it is unclear how individual bHLH proteins selectively recognize E-box sequences on their targets. Chang et al. found that the strict spatial configuration in the double E-box motif recognized by Twist aligns two Twist-E47 dimers on the same face of DNA, thus providing a high affinity site for a highly stable intramolecular tetramer. The WR domain of Twist dimerizes to mediate tetramer formation, which is functionally required for Twist-induced EMT.
Keywords: TWIST, bHLH transcription factor, WR domain, epithelial–mesenchymal transition
Abstract
Basic helix–loop–helix (bHLH) transcription factors recognize the canonical E-box (CANNTG) to regulate gene transcription; however, given the prevalence of E-boxes in a genome, it has been puzzling how individual bHLH proteins selectively recognize E-box sequences on their targets. TWIST is a bHLH transcription factor that promotes epithelial–mesenchymal transition (EMT) during development and tumor metastasis. High-resolution mapping of TWIST occupancy in human and Drosophila genomes reveals that TWIST, but not other bHLH proteins, recognizes a unique double E-box motif with two E-boxes spaced preferentially by 5 nucleotides. Using molecular modeling and binding kinetic analyses, we found that the strict spatial configuration in the double E-box motif aligns two TWIST–E47 dimers on the same face of DNA, thus providing a high-affinity site for a highly stable intramolecular tetramer. Biochemical analyses showed that the WR domain of TWIST dimerizes to mediate tetramer formation, which is functionally required for TWIST-induced EMT. These results uncover a novel mechanism for a bHLH transcription factor to recognize a unique spatial configuration of E-boxes to achieve target specificity. The WR–WR domain interaction uncovered here sets an example of target gene specificity of a bHLH protein being controlled allosterically by a domain outside of the bHLH region.
The basic helix–loop–helix (bHLH) transcription factor superfamily comprises a large number of transcriptional regulators that function in critical developmental processes and pathogenesis in organisms from yeast to humans. All members of the bHLH superfamily have two highly conserved and functionally distinct domains: the basic domain for DNA binding and the HLH domain to interact with another bHLH factor to form homodimeric or heterodimeric complexes. The consensus hexanucleotide sequence known as the E-box (CANNTG) is the canonical recognition sequence for all bHLH transcription factors. Two classes of bHLH proteins are known to preferentially recognize canonical E-box sites. Class I bHLH proteins—also known as E proteins such as E12, E47, and others—are expressed in most tissues and can form homodimers or heterodimers. Class II bHLH proteins, which include TWIST, NeuroD, and others, each show a tissue-restricted expression pattern and preferentially heterodimerize with the E proteins to bind to E-box sites (Massari and Murre 2000; Jones 2004). Given the prevalence of the canonical E-box sequences in a genome (one canonical E-box per 128 nucleotides [nt] by nucleotide frequency), a major unanswered question is how specificity is determined. It is hypothesized that class II bHLH proteins, as heterodimers with E proteins, might recognize additional specific nucleotides surrounding an E-box to increase binding site selectivity.
TWIST is a class II bHLH transcription factor that was originally found to be essential for initiating mesoderm formation in Drosophila (Thisse et al. 1987; Leptin and Grunewald 1990; Leptin 1991). This developmental transcription factor also plays a critical role in tumor progression, and its expression is associated with poor prognosis and distant metastasis in many human solid tumors (Peinado et al. 2007; Eckert et al. 2011; Tsai et al. 2012). TWIST is a key regulator of the epithelial–mesenchymal transition (EMT) program (Yang et al. 2004), which is reactivated during tumor progression to instruct stationary epithelial cells to lose cell–cell junctions and gain migratory and invasive capacities (Thiery and Morgan 2004; Tsai and Yang 2013). While the biological impact of TWIST on EMT has been well defined, little is known on how TWIST specifically binds to and regulates its specific target genes to induce EMT.
Our understanding of TWIST-mediated transcription is largely from studies on Drosophila Twist, the sole member of the Drosophila Twist family. Using early chromatin immunoprecipitation (ChIP)-on-chip technology, ∼500 DNA fragments containing Twist-binding sites were identified to contain E-box sequences (Sandmann et al. 2007; Zeitlinger et al. 2007). Similar findings using ChIP combined with high-throughput sequencing (ChIP-seq) technology again only identified the canonical E-box sequence in Twist-bound DNA (Ozdemir et al. 2011). However, as little additional sequence specificity outside the E-box was evident, it has been puzzling how such binding specificity is achieved because of the existence of enormous numbers of E-box sequences in both Drosophila and human genomes.
The TWIST protein is highly conserved from Drosophila to humans in two regions: the bHLH domain and the most C-terminal 20 residues, termed the WR domain (also known as the TWIST box) (Castanon and Baylies 2002), which is unique to the TWIST family of bHLH factors. However, there is also a key structural difference between Drosophila and human TWIST proteins: Drosophila Twist contains three glutamine and histidine-rich CAX domains at the N terminus that function as the canonical transactivation domain. In contrast, all vertebrate TWIST homologs lack this domain (Castanon and Baylies 2002) and instead appear to heterodimerize with E proteins to acquire the transactivation capability.
Given the differences in domain structure and cellular function between Drosophila and mammalian TWIST, this study set out to characterize the set of DNA elements bound by TWIST1 during EMT in human cells. By comparing the human and the Drosophila Twist-binding DNA patterns, we report the discovery of an evolutionarily conserved DNA architecture uniquely recognized by TWIST and present a novel molecular mechanism by which TWIST family bHLH transcription factors achieve target gene specificity.
Results
Human TWIST1 recognizes a double E-box motif with a unique spatial configuration
To determine the genome-wide binding pattern of TWIST1 in human cells, we performed ChIP coupled with high-throughput sequencing (ChIP-seq) for TWIST1-binding DNA elements in human mammary epithelial (HMLE) cells that have been induced to undergo TWIST1-mediated EMT (Casas et al. 2011). The specificity of the antibody used for immunoprecipitation was validated in Supplemental Figure S1C. More than 14,000 significant TWIST1-binding DNA peaks were obtained (Supplemental Table 1). This collection contained sequences from the promoter regions of known TWIST1 target genes, such as SNAI2 (Casas et al. 2011), and also new targets, such as α2-macroglobulin (A2M) and BMP4 (Supplemental Fig. S1A,B). We randomly selected five non-TWIST1-binding genomic regions and seven TWIST1-binding regions and used quantitative PCR (qPCR) to validate the ChIP-seq results. The fragments containing TWIST1-binding sites showed a significant enrichment compared with non-TWIST1-binding site fragments (Supplemental Fig. S1D). TWIST1-binding peaks are highly enriched at intergenic and intronic regions of the human genome (Supplemental Fig. S1E), which is consistent with the location of TWIST-occupied genomic regions previously observed in Drosophila.
To uncover novel DNA motifs bound by TWIST, a de novo motif enrichment analysis was performed using the HOMER algorithm (http://biowhat.ucsd.edu/homer) (Heinz et al. 2010). We evaluated sequences covering ±100 base pairs (bp) from the center of individual TWIST1-binding peaks. As expected, the most highly enriched motif (>75% of total TWIST1-binding peaks) identified by HOMER is the canonical E-box motif (CANNTG), which further validates our TWIST1 ChIP-seq data (Fig. 1A,B). Surprisingly, the second most highly enriched TWIST1-binding motif comprised two closely spaced E-boxes, which accounted for 41.4% of the TWIST-binding peaks (Fig. 1A,B). This motif shows 27-fold enrichment over the calculated random occurrence frequency (2.18%) in the human genome (P-value = 1 × 10−2097). This novel TWIST1-binding motif, which we designated as the “double E-box” motif, contains two canonical E-box sequences separated by exactly 5 nt (Fig. 1A). More importantly, computational permutation analysis revealed that the double E-box motifs containing exactly 5-nt spacing between the two canonical E-boxes are much more common than any other spacing arrangements ranging from 0 nt to 10 nt (Fig. 1C; Supplemental Fig. S2). Furthermore, there was no enrichment for motifs containing more than two canonical E-boxes, indicating that this enrichment is not due to an increase of E-box numbers. It is also worth noting that minimal additional nucleotide sequences surrounding the canonical E-box were strongly enriched, consistent with what was observed in Drosophila (Ozdemir et al. 2011). Together, these findings suggest that human TWIST1 frequently occupies a double E-box motif with a unique 5-nt spacing configuration.
Figure 1.
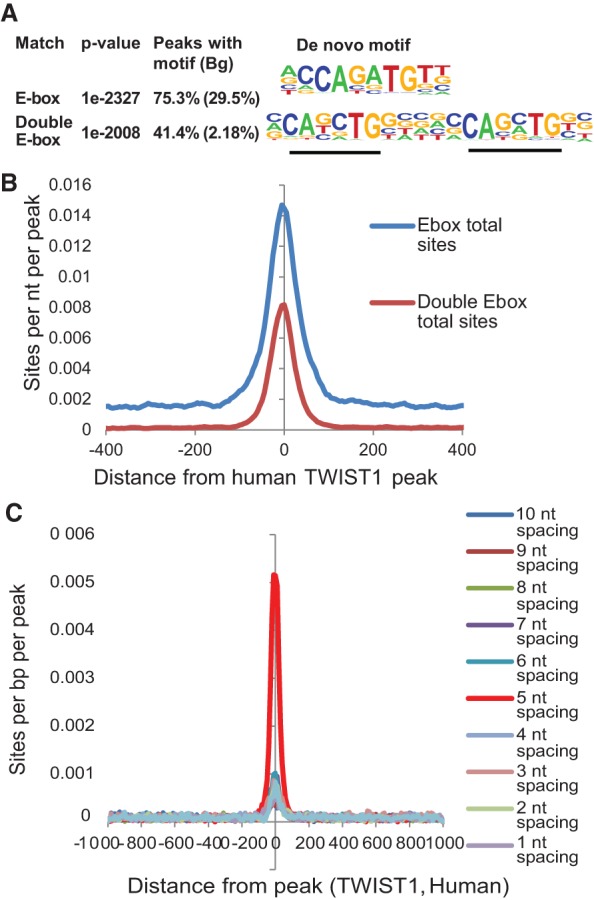
Human TWIST1 recognizes a double E-box motif with a unique spatial configuration. (A) A de novo motif enrichment analysis was performed on sequences covering ±100 bp from the center of individual human TWIST1-binding peaks by the HOMER algorithm. The top two most-enriched motifs representing an E-box and a double E-box are shown. Bg is the calculated random occurrence frequency of each motif in the genome. (B) The profile of motif occurrences for both the single E-box motif and the double E-box motif is shown relative to the center of TWIST1 ChIP-seq peaks. (C) Permutation analysis for the enrichment of the motifs containing two E-boxes separated by 1–10 nt among human TWIST1-binding peaks.
The double E-box motif is recognized by both Drosophila and human TWIST proteins but not by other bHLH proteins
To determine whether the double E-box motif is evolutionarily conserved across species, we performed the same HOMER analysis on the previously published Drosophila Twist ChIP-seq data (Ozdemir et al. 2011). Indeed, we found that Drosophila Twist also preferentially bound the double E-box motif where two E-boxes are separated by exactly 5-nt spacing with an 11-fold enrichment over the calculated random occurrence frequency (P-value = 1 × 10−35) (Fig. 2A). Permutation analysis also shows more enrichment for the 5-nt spacing double E-box motif (Fig. 2B; Supplemental Fig. S2), similar to human TWIST1. Furthermore, inspection of well-established and direct target genes for Drosophila Twist revealed that the promoter regions of Tinman (Yin et al. 1997) and rho (Ozdemir et al. 2011) all contain the double E-box motifs (Fig. 2C). These results strongly suggest that TWIST recognition of this double E-box motif is evolutionarily conserved between Drosophila and humans and occurs in its bona fide target genes.
Figure 2.

The double E-box TWIST-binding motif is conserved in Drosophila but is not bound by other bHLH transcription factors. (A) The profile of motif occurrences for both motifs is shown relative to the center of Twist ChIP-seq peaks from Drosophila. (B) Permutation analysis for the enrichment of the motifs containing two E-boxes separated by 0–10 nt among Drosophila Twist-binding peaks. (C) Two well-characterized Drosophila Twist target genes, tinman and rho, both contain the double E-box motif on their regulatory regions. (D) Permutation analysis for the enrichment of the motifs containing two E-boxes separated by 1–10 nt among individual DNA peaks bound by E2A in B cells, NEUROD1 in mouse islet cells, and c-MYC in embryonic stem (ES) cells.
To determine whether recognition of the double E-box motif is unique to the TWIST family of bHLH transcription factors, we analyzed the publicly available ChIP-seq data for NEUROD1 (murine Islet cells) (Tennant et al. 2013), E47 (murine B cells) (Lin et al. 2010), and c-MYC (murine embryonic stem [ES] cells) (Chen et al. 2008). Remarkably, the double E-box motif is not enriched in the ChIP-seq data with these bHLH proteins (Fig. 2D). Together, these data demonstrate that the novel double E-box TWIST-binding motif is not only highly conserved between Drosophila and humans but also specific to recognition of the E-box by the TWIST family bHLH transcription factor.
Molecular modeling suggests that heterodimeric TWIST complexes can co-occupy the double E-box motif
We next employed computational modeling to understand the molecular basis for TWIST complexes to preferentially bind the double E-box motif. Since there is no extant structure of TWIST, we first modeled the three-dimensional (3D) structure of the human TWIST1 bHLH domain using the published crystal structure of the NeuroD1/E47 bHLH domain heterodimer bound to a single E-box site (Protein Data Bank [PDB] 2ql2). A model for association of two TWIST–E47 heterodimers was then created by duplicating the NeuroD1/E47 bHLH domain heterodimer separated by five DNA bases. As shown in Figure 3A, the 5-nt spacing between two E-boxes allows a full turn of the DNA double helix (6 nt of the E-box motif plus 5-nt spacing) between the two E-boxes; therefore, the conserved base pairs in each E-box face the same spatial direction in DNA. Modeling of two TWIST/E47 heterodimers onto the double E-box site shows that this mode of occupancy is highly feasible, with no observable spatial constraints or steric hindrance among all proteins and DNA when the DNA gap between E-box sequences is 5 nt. Moreover, the model suggests that co-occupancy of two E-boxes with 5-nt spacing would spatially align the two heterodimers in the same orientation on DNA such that protein–protein interactions between the two heterodimers are in direct contact via hydrogen bonding and hydrophobic interaction (Fig. 3A; Supplemental Fig. S3A). In contrast, DNA sequences of <5 nt between the double E-boxes result in steric clashing of these loops, while gaps of >5 nt remove loop contacts (Fig. 3B). This model strongly suggests that the two TWIST/E47 heterodimers may bind to the double E-box together to form a stable ternary complex.
Figure 3.

Molecular modeling shows that two TWIST1/E47 heterodimers bind to the double E-box motif to allow high-affinity and specificity binding. (A) Molecular modeling of two TWIST1/E47 bHLH protein heterodimers bound to the double E-box motif. (B) Comparison of the configuration of two TWIST1/E47 bHLH protein heterodimers bound to two E-boxes separated by 3–7 nt. (C) Computational analysis of the double E-box motif at human TWIST1-binding peaks shows that the CANNTG sequences with zero or one mismatched nucleotides are significantly enriched above the calculated random occurrence frequency of each motif (P << 1 × 10−100).
In-depth analysis of the double E-box sites obtained from the genomic ChIP fragments indicated that human TWIST1 binds not only the double E-box motif containing two canonical E-boxes with perfectly matched CANNTG consensus sequence but also the double E-box motif in which one of the E-boxes contains one mismatched nucleotide among the CANNTG consensus sequence (Fig. 3C). In contrast, the single E-box motif with one mismatched nucleotide showed no enrichment over background (Supplemental Fig. S3B). These observations suggest that in the double E-box motif, a nonconsensus half site may be tolerated to a significant degree because of compensation by the presence of the other half-site in the double E-box motif.
TWIST binds the double E-box motif with high affinity and requires the 5-nt spacing
Next, we set out to understand why TWIST preferentially binds to the double E-box motif compared with the single E-box motif. We first analyzed peak scores for all single and double E-box-binding sites in both our human TWIST1 ChIP-seq data set and the published Drosophila data set. The peak scores correspond to the number of DNA tags obtained by sequencing for individual peaks, thus directly indicative of the binding affinity of TWIST to this peak. Among human TWIST1-binding peaks, peaks with scores >300 are 2.7-fold more likely to contain the double E-box motif than a single E-box, while peaks with scores <300 are equally likely to have either a single or double E-box (P-value < 1 × 10−16, Fisher exact test) (Fig. 4A). Similarly, in the Drosophila Twist ChIP-seq data set that contains much fewer peaks than the human data set, we still observed a significant enrichment for the double E-box motif than a single E-box in peaks with scores >300 versus peaks with scores <300 (P-value < 0.02) (Fig. 4B). Taken together, these data suggest that the double E-box motif provides a higher-affinity platform for recognition by TWIST.
Figure 4.

TWIST presents a higher binding affinity toward the double E-box motif compared with the single E-box motif. (A) Human TWIST1-binding peaks were grouped by their peak scores, and the percentage of the peaks containing a single E-box versus a double E-box within each group was calculated. (B) Drosophila Twist-binding peaks were analyzed as in A. (C) The domain structures of all proteins used in the EMSA study. (D) The wild-type and mutant double E-box sequences based on the human A2M promoter are used in the EMSA study. (E,F) EMSA analyses of human TWIST1/E12 or TWIST1/E47 bound to a 30-nt oligo containing the double E-box motif from the human A2M promoter or the same oligo with one of the E-boxes mutated. Antibodies against TWIST1, E12, or E47 were added into the corresponding reactions to either supershift (for E12) or block (for TWIST1 and E47) the protein/DNA complexes. Solid arrows point to the TWIST1/E12- or TWIST1/E47-containing complexes, and dashed arrows point to the complexes containing E12 or E47 alone.
To define and quantify the parameters of TWIST1/E–protein heterodimers binding to single versus double E-box sites, we first analyzed the complex configurations when human TWIST1 binds to the double E-box motif compared with the single E-box motif by electrophoresis mobility shift assays (EMSAs). Since the relative importance of E12 versus E47 as heterodimer partners for TWIST1 is unknown, we used both E47 and E12 (collectively termed “E proteins”) separately as binding partners for TWIST1. Human full-length TWIST1 protein (TWIST1 FL), the E12 bHLH domain (E12 bHLH), and the E47 bHLH domain (E47 bHLH) were produced separately by in vitro translation. S35-labeled proteins were used to normalize the amounts of protein used in EMSAs, which were produced in parallel translations using nonradioactive methionine (Fig. 4C; Supplemental Fig. S4A). TWIST1 and individual E proteins were preincubated in equal molar amounts to ensure heterodimer formation, and the ability of TWIST1 to bind to E47 was also confirmed in the coimmunoprecipitation experiment (Fig. 6E, below). The DNA fragments used for EMSAs were the 30-bp native double E-box-containing element from the human A2M gene promoter (wild type) described in Supplemental Figure S1A or the variants in which one of the E-boxes was mutated (MUT1) (Fig. 4D). All EMSA assays were performed in probe excess, as evidenced by the equal amounts of unbound probes in each lane (Supplemental Fig. S4B). As shown in Figure 4, E and F, TWIST1 alone displayed no binding activity toward the wild-type probe or the MUT1 probe (lane 2), while E12 or E47 alone bound to both probes as a single complex (lanes 4,9, dashed arrow), and these complexes can be completely supershifted by an E12 antibody (lane 5) or largely blocked by an E47 antibody (lane 10). TWIST1 and E12 proteins together formed two distinct complexes on the wild-type double E-box DNA probe: one with the faster mobility than the E12 complex alone and one with much slower mobility (Fig. 4E,F, lane 6, solid arrows). Importantly, both of these new complexes contain TWIST1 and E12 proteins, as both are abolished by a TWIST1-blocking antibody (Fig. 4E,F, lane 7) or supershifted by an E12 antibody (Fig. 4E,F, lane 8). The TWIST1+E47 complex behaved very similarly to the TWIST1+E12 complex, except that the E47 antibody showed both partial blocking and partial supershift activities. Furthermore, when one of the two E-boxes was mutated on this DNA oligo (A2M MUT1 probe), TWIST1+E12 or TWIST1+E47 only formed the single faster mobility complex on the MUT1 probe (Fig. 4F, lanes 6,11, solid arrows). This suggests that an interaction between TWIST1/E-protein heterodimers (but not E12 or E47 homodimers alone) on the double E-box DNA is responsible for forming this slower mobility complex. Together with data from our molecular modeling and ChIP-seq analyses, these results suggest that the double E-box motif is likely to be bound by two TWIST1/E-protein heterodimeric complexes.
Figure 6.

The WR domain of TWIST1 is required for the high-affinity binding of TWIST1 toward the double E-box motif. (A) The domain structures of TWIST1 and TWIST1ΔWR used in the EMSA analyses. (B,C) EMSA analysis of human TWIST1 versus TWIST1ΔWR bound to a 30-nt oligo containing the double E-box motif from the human A2M promoter or the same oligo with one of the E-boxes mutated. Antibodies against TWIST1, E12, or E47 were added into the corresponding reactions to either supershift (for E12) or block (for TWIST1 and E47) the protein/DNA complexes. Solid arrows point to the TWIST1/E12- or TWIST1/E47-containing complexes, and dashed arrows point to the TWIST1ΔWR/E12- or TWIST1ΔWR/E47-containing complexes. The asterisk points to a minor band in lanes 5, 6, 11, and 12, and this complex contains only E12 or E47, but not TWIST1, because only the E12 and E47 antibodies, but not the TWIST1-blocking antibody, affected this complex. (D) HA-TWIST1 wild type (WT), HA-TWIST1ΔWR, or a control vector was transfected with Myc-E47bHLH into 293T cells and processed for immunoprecipitation using the HA antibody. The resulting immunoprecipitation samples and input samples were analyzed by SDS-PAGE gel and probed with antibodies against Myc and HA. (E) HA-TWIST1 wild type, HA-TWIST1ΔWR, or a control vector was transfected together with Flag-TWIST1 wild type and Myc-E47 bHLH in 293T cells. The resulting cell lysates were immunoprecipitated with the Flag antibody in the presence or absence of the BMP4 wild-type DNA oligo. The resulting immunoprecipitation samples and input samples were analyzed by SDS-PAGE gel and probed with antibodies against HA, Flag, and Myc. An asterisk points to the antibody light chain (bottom band) on the blot for Flag-TWIST1.
Since EMSA is a rough measure of the overall stability of DNA–protein complexes during electrophoresis and does not readily yield kinetic data such as relative affinities and on–off rates, we sought to quantify such interaction by surface plasmon resonance (SPR)-based kinetic analyses. Purified human TWIST1 protein containing the bHLH domain and the WR domain (TWIST1 bHLH+WR) together with the E47 bHLH domain was used for SPR analyses (Fig. 5A; Supplemental Fig. S5A). The DNA probes were first biotinylated and then bound to SPR streptavidin chips and used for real-time, flow-based kinetic binding studies in a BiacoreT200 machine. Consistent with the EMSA results, TWIST1 alone did not bind DNA, and TWIST1/E47 complexes showed a higher affinity toward the double E-box motif than E47 alone (Supplemental Fig. S5B–D). In addition to the double E-box probe, we also tested probes that contained a 2-nt or 10-nt spacing between the two E-boxes for comparison (Fig. 5B). In the binding isotherms in Figure 5C, TWIST1/E47 protein showed characteristic transcription factor–DNA interactions with rapid on rates and slow dissociation from the wild-type double E-box, with a calculated dissociation rate kd of 0.00169 sec−1 (Fig. 5C,F). Interestingly, using the same analyte, the off rates were drastically increased in flow cells containing double E-box mutants with either 2-nt or 10-nt spacing (Fig. 5D,E), with kd values of 0.00501 sec−1 and 0.00941 sec−1, respectively (Fig. 5F). To further define the spacing requirement for the double E-box motif, we also performed SPR-based kinetic analyses using mutant DNAs in which the two E-boxes are separated by 4-nt or 6-nt spacing. The off rates for these two mutant probes were also increased compared with the wild-type double E-box motif (Fig. 5G–J), although the difference is less pronounced than probes with 2-nt and 10-nt spacing. This result suggests that the 4-nt and 6-nt spacing configurations may allow some binding affinity in the context of a DNA oligo and when using purified proteins in SPR assays in vitro. Since our ChIP-seq analyses identified the 5-nt spacing as the most enriched TWIST1-binding motif in cells, it suggests that in the context of the intact chromatin and the presence of additional factors in the TWIST1/E47 transcription complex in vivo, the 4-nt and 6-nt spacing configurations are less favored compared with the 5-nt spacing. Together, these data further support our model that the 5-nt spacing between the two E-boxes is critical for the apparent stable binding of the double E-box motif by two TWIST1/E47 heterodimers.
Figure 5.

Human TWIST1 presents a higher binding affinity toward the double E-box motif with 5-nt spacing but not other spacing. (A) The domain structures of all proteins used in the SPR-based kinetic analyses. (B) The double E-box sequences based on the human BMP4 promoter with 5-nt, 2-nt, and 10-nt spacing were used in the SPR-based kinetic analyses. (C–E) SPR-based kinetic analysis of the association and dissociation rates between TWIST1/E47 and the double E-box motif with 5-nt spacing (C), 2-nt spacing (D), or 10-nt spacing (E). (F) Summary of the binding parameters obtained by the SRP-based kinetic analysis shown in C–E. (G–I) SPR-based kinetic analysis of the association and dissociation rates between TWIST1/E47 and the double E-box motif with 5-nt spacing (G), 4-nt spacing (H), or 6-nt spacing (I). (J) Summary of the binding parameters obtained by the SRP-based kinetic analysis shown in G–I.
The WR domain of TWIST1 stabilizes two TWIST1/E-protein heterodimers on the double E-box motif
The molecular modeling studies suggest that the 5-nt spacing between two E-boxes allows two TWIST1/E-protein heterodimers to be aligned spatially on the same face of the DNA and that they might interact with each other to form a ternary complex of two dimers on the double E-box motif (Fig. 3A). We then asked whether any domain of TWIST might facilitate the formation of the ternary complex. Both human and Drosophila TWIST preferentially binds to the double E-box motif (Figs. 1B, 2A). In addition to the bHLH domain that is responsible for binding to DNA and E proteins, the only additional domain conserved between them is the C-terminal WR domain (Fig. 4C). Molecular models of the WR domain were created and found to be a well-folded and stable α helix. Thus, we performed molecular simulation to model the structure of TWIST1/E47 binding toward the double E-box motif and found that the helical WR domains from each TWIST1 could interact with one another in a ternary complex (Fig. 3A). Distances for the connection of two WR domains to the TWIST1 bHLH domain were highly favored with two E-boxes separated by 5 nt, as other nucleotide spacing between the two E-boxes did not allow for interaction of the WR domain dimers (Fig. 3B; Supplemental Fig. S2). Therefore, molecular modeling strongly suggests that each heterodimer could provide a WR domain from TWIST1 that interacts in a parallel association of helices to stabilize two heterodimers to the double E-box.
To test this model, we first deleted the WR domain from human TWIST1 (TWIST1ΔWR) (Fig. 6A; Supplemental Fig. S4B). Using coimmunoprecipitation, we found that the WR domain was not required for TWIST1 to heterodimerize with E47 (Fig. 6D), consistent with previous reports indicating that the HLH domain is responsible for interacting with E proteins (Laursen et al. 2007). Interestingly, in EMSA analyses, TWIST1ΔWR+E12 (or TWIST1ΔWR+E47) only formed a single fast migration complex on the double E-box DNA (Fig. 6B, lanes 5,11, dashed arrow) instead of the two complexes formed by TWIST1 FL (Fig. 6B, lanes 2,8, solid arrows). Importantly, TWIST1ΔWR+E12 formed a single complex on the MUT1 DNA with a single E-box (Fig. 6C, lanes 5,11, dashed arrow). Antibody addition experiments further confirmed that the single complex contains both TWIST1ΔWR and E proteins (Fig. 6B,C). Together with data presented in Figure 4, E and F, these results strongly indicate that TWIST1ΔWR together with E proteins is very competent to bind to a single E-box; however, the WR domain plays an essential role in mediating the binding of two TWIST1/E-protein heterodimers to the double E-box motif, which is observed on the EMSA gel as a stable slowly migrating complex.
To directly demonstrate that the WR domain is required for mediating TWIST1/TWIST1 interaction, we expressed HA-tagged TWIST1 wild type or TWIST1ΔWR together with Flag-tagged TWIST1 wild type and Myc-tagged E47 bHLH in 293T cells. We immunoprecipitated with the Flag antibody in the presence or absence of the double E-box DNA oligos to determine whether Flag-tagged TWIST1 wild type could pull down HA-tagged TWIST1 proteins. Interestingly, we found that only HA-TWIST1 wild type bound to Flag-TWIST1 wild type; in contrast, deletion of the WR domain (HA-TWIST1ΔWR) completely abolished its interaction with Flag-TWIST1 wild type (Fig. 6E), suggesting that the WR domain is required for TWIST1/TWIST1 interaction. It is worth noting that WR–WR domain interaction does not require binding to the double E-box DNA. In addition, TWIST1ΔWR was not able to form a complex with TWIST1 wild type via its bHLH domain (Fig. 6E), although TWIST1ΔWR was competent to use the same bHLH domain to bind to E47 (Fig. 6D). Together with our observation that human TWIST1 alone without E proteins could not bind to E-boxes (Fig. 4E,F), these data suggest that human TWIST1 cannot form homodimers via its bHLH domain to bind to E-boxes. Instead, these data indicate that the WR–WR domain interaction between two heterodimeric TWIST1/E47 complexes is specifically responsible for binding to the double E-box motif.
The WR domain of TWIST1 is essential for TWIST1-induced EMT and impacts TWIST1 target gene expression
To determine whether the WR domain is required for the ability of TWIST1 to induce EMT, we stably expressed either wild-type human TWIST1 or TWIST1ΔWR in HMLE cells and tested their abilities to induce EMT. The wild-type TWIST1 and TWIST1ΔWR mutant proteins were detected in the nucleus at similar levels in HMLE cells expressing them (Fig. 7B). The HMLE cells expressing wild-type TWIST1 became scattered, lost cell–cell junctions, and assumed a spindle-like morphology, indicative of EMT. In contrast, HMLE cells expressing the TWIST1ΔWR mutant maintained an epithelial phenotype and did not exhibit morphological EMT (Fig. 7A). Immunofluorescence staining of the cells showed strong E-cadherin signals at the cell–cell junctions in the HMLE-TWIST1ΔWR cells, while the E-cadherin signal was completely lost in the HMLE-Twist1 cells. In addition, HMLE-Twist1ΔWR cells express an intermediate level of N-cadherin compared with either the HMLE or HMLE-Twist1 cells (Fig. 7B). Western blotting analysis confirmed that HMLE-Twist1ΔWR cells still retained a high level of the epithelial cell marker E-cadherin and only expressed an intermediate level of mesenchymal markers vimentin and N-cadherin (Supplemental Fig. S6A). These data demonstrate that the WR domain of TWIST1 is required for induction of EMT.
Figure 7.

The WR domain of TWIST1 is required for TWIST1-induced EMT and TWIST1 target gene expression. (A) Bright-field images of HMLE cells expressing a control vector, Twist1, or Twist1ΔWR. (B) Immunofluorescence images of HMLE cells expressing a control vector, Twist1, or Twist1ΔWR that are stained for TWIST1 (green), DAPI (blue), E-cadherin (red), and N-cadherin (red). (C) qPCR analysis of a list of genes that contain the double E-box motif at their proximate promoter regions in HMLE cells expressing TWIST1 FL or TWIST1ΔWR. Their folds of mRNA induction were compared with their expression in HMLE cells and normalized to GAPDH. The error bar is the standard error of the mean from triplicate samples.
To further understand the impact of the WR domain on TWIST target gene expression, we selected a group of human TWIST1 target genes that contain the double E-box motif near their transcription start sites (Supplemental Fig. S6B) and analyzed their mRNA induction in HMLE cells expressing TWIST1 or TWIST1ΔWR compared with control HMLE cells. Significantly, among all genes analyzed, the fold induction in HMLE-TWIST1ΔWR cells was reduced by >50% compared with the levels in HMLE-TWIST1 cells (Fig. 7C). These functional data further support our conclusion that the WR domain-mediated stabilization of TWIST binding at the double E-box motif is critical for the induction of EMT by TWIST.
Discussion
Through a series of genomic, biochemical, and functional analyses, our results strongly support a new model to explain how the TWIST family bHLH transcription factors recognizes their target DNA sequences with high affinity and specificity. The salient pieces of data supporting this model can be summarized as follows: (1) TWIST recognizes a double E-box motif containing two canonical E-boxes spaced by 5 nt. (2) The double E-box element is bound by TWIST at its bona fide target genes in both humans and Drosophila but not by other bHLH-binding proteins. (3) Two TWIST1/E47 heterodimers bind the same face of the double E-box motif, and this high-affinity binding requires 5-nt spacing between E-boxes. (4) The WR domains of each TWIST1 can form a parallel association of α helices to stabilize a tetramer on DNA and are required for the induction of EMT by TWIST1. Together, these data strongly suggest that the double E-box motif is a composite, high-affinity, and TWIST-selective binding site for the TWIST family of bHLH proteins.
A major unanswered question in the E-box field has been to define how target gene specificity is achieved when upwards of 50 or so protein complexes, spanning bHLH and zinc finger protein subtypes, nominally recognize the same short 6-nt E-box consensus that occurs frequently in the genome. While there is significant but subtle specificity within the E-box sequence itself for binding by some bHLH proteins, most notably at the “NN” nucleotides in the CANNTG sequence and possibly extending to nucleotides just outside of the consensus, these variations are not enough to account for the significant specificity in target gene regulation shown by the vast set of bHLH proteins. Thus, it was remarkable that upon surveying published ChIP-seq studies of bHLH transcription factors, including NEUROD1 (Tennant et al. 2013), E2A (Lin et al. 2010), and c-MYC (Chen et al. 2008), we did not detect the double E-box motif, suggesting that the novel double E-box motif is unique to the TWIST proteins and confers its target specificity. Unlike many known bHLH transcription factors that recognize specific nucleotide sequences surrounding the canonical E-box motif, the TWIST subfamily of bHLH proteins has evolved a unique strategy to recognize the spatial architecture of the double E-box motif to achieve specificity.
One important finding from this study is that the unique double E-box TWIST-binding motif is highly conserved between human and Drosophila Twist proteins. Although human and mouse TWIST1 share an ∼95% sequence identity, the overall homology between mammalian and Drosophila Twist proteins is dramatically reduced to only ∼35%. Even with this difference, it is noteworthy that data from Drosophila Twist ChIP-seq studies (Ozdemir et al. 2011) presented the same preference for the double E-box motif as human TWIST1. Another ChIP-seq study found that the Twist-binding landscape is highly conserved across six Drosophila species (He et al. 2011). Indeed, upon reanalyzing the Drosophila melanogaster ChIP-seq data set from this study, we also observed the specific enrichment of the double E-box motif (data not shown). Furthermore, as shown in Figure 4D, human TWIST1 does not contain any canonical transcription activation domain and heterodimerizes with E proteins to bind to E-boxes, while Drosophila Twist can homodimerize to activate transcription due to three CAX domains on the N terminus of Twist. Therefore, the preference for the double E-box motif can be achieved by a TWIST1/E-protein heterodimer in humans or a Twist/Twist homodimer in Drosophila. The human TWIST family contains two family members—TWIST1 and TWIST2—that are 100% identical in their WR domains and 95% identical in their bHLH domains. Therefore, we believe that the double E-box motif uncovered in this study is a highly specific binding characteristic of the TWIST family bHLH factors that has been conserved across multiple species and emphasizes its importance for TWIST-mediated gene regulation.
We showed that the highly conserved WR domain of human TWIST1 is required for two TWIST1/E-protein heterodimers to bind the double E-box motif and for the ability of TWIST1 to induce a complete EMT. The role of the TWIST WR domain in regulating gene expression and TWIST function has also been examined in several previous studies. Consistent with our results, a recent study showed that the WR domain of TWIST1 is required for TWIST1-mediated prostate cancer metastasis (Gajula et al. 2013). Laursen et al. (2007) showed that truncation of the WR domain from TWIST1 reduced transactivation of gene expression using a luciferase reporter assay. Interestingly, the promoter region used in this study is from a well-known target of Drosophila Twist, Tinman, whose promoter contains two sets of the double E-box motif (Laursen et al. 2007). This study proposed that the WR domain serves a transcriptional activation domain; however, this proposed model is inconsistent with the fact that Drosophila Twist already contains three classical transcription activation domains, and the mammalian TWIST1 homodimer does not present transactivation activity. Instead, the double E-box-binding model described here could possibly be the mechanistic explanation for the requirement of the WR domain in TWIST-mediated transcription regulation. The WR domain was also reported to bind to RUNX2 (Bialek et al. 2004), SOX9 (Gu et al. 2012), the PPA E3 ligase (Lander et al. 2011), p53 (Piccinin et al. 2012), and RELA (Li et al. 2012) in certain biological settings. It is possible that the WR domain has pleiotropic functions as a homotypic or heterotypic protein–protein interaction module in different cells or species or during different developmental programs, which could greatly expand the flexibility of target gene regulation by TWIST complexes. Regardless of the exact mechanism used, the conservation in the WR domain and the double E-box TWIST-binding motif between Drosophila and humans argues that the role of the WR domain in mediating TWIST binding to the double E-box motif is likely to be one of its original functions during early evolution.
TWIST1 heterozygous insufficiency causes Saethre-Chotzen syndrome with defective fusion of skull bones. Recent case studies described a similar clinical presentation of Saethre-Chotzen syndrome in patients with mutations in the TWIST1 WR domain (Seto et al. 2007; Pena et al. 2010). The milder phenotype seen in these patients can possibly be explained by the blunted, but not abolished, ability of TWIST WR domain mutants to bind to the double E-box motif with high affinity. Consistent with these data, our functional analysis showed that even overexpression of the WR domain deletion mutant of TWIST1 was not able to induce a full EMT, further highlighting the essential role of the WR domain in TWIST-mediated gene regulation. Together, our genomic, biochemical, and functional studies uncovered a novel strategy that the TWIST family of bHLH transcription factors uses to recognize a unique DNA architecture to achieve target gene specificity.
Materials and methods
Cell culture and media
HMLE cells immortalized with large T and telomerase containing the Twist1-ER fusion protein (HMLE T-ER) were grown in a 1:1 mixture of Lonza MEGM and DMEM-F/12 media supplemented with 5 ng/mL EGF, 5 µg/mL insulin, 0.5 µg/mL hydrocortisone, L-glutamine, and 0.5% penicillin/streptomycin.
ChIP-seq
HMLE/Twist1-ER cells were treated with 20 nM 4-hydroxy tamoxifen for 4 d and then fixed and cross-linked with paraformaldehyde at a final concentration of 1% for 15 min. Cross-linking was quenched with 0.4 M glycine for 10 min. The cells were harvested on ice and washed sequentially with PBS, buffer 1 (0.25% Triton X-100, 10 mM EDTA, 500 μM EGTA, 10 mM HEPES), and buffer 2 (200 mM NaCl, 1 mM EDTA, 500 μM EGTA, 10 mM HEPES). The cells were then resuspended in 900 μL of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-Cl, protease inhibitor) and sonicated to shear the chromatin to be between 0.5 and 1.0 kb in size. Dilution buffer (1% Triton-X, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-Cl with protease inhibitor) was added at 1:10 to dilute SDS concentration. One milliliter of diluted lysate was incubated with 3 μg of mouse anti-estrogen receptor (clone TE111.5D11, Thermo Scientific) or 3 μg of mouse IgG control antibody overnight at 4°C. Protein G Dynabeads (Life Technology) were washed with PBS–0.1% BSA, added to the lysates, and incubated for 2 h at 4°C. The beads were then sequentially washed in the following buffers: TSE1 (0.1% SDS, 1% Triton-X, 2 mM EDTA, 20 mM Tris-Cl, 150 mM NaCl), TSE2 (0.1% SDS, 1% Triton-X, 2 mM EDTA, 20 mM NaCl, 500 mM NaCl), buffer 3 (250 mM LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-Cl), and TE buffer. Fragments were eluted from beads with 300 μL of 1% SDS and 0.1 M NaHCO3 and then underwent reverse cross-linking by incubating overnight at 65°C. Chromatin was purified by phenol:chloroform extraction and resuspended in 50 μL of TE buffer.
Chromatin samples were prepared for sequencing using the Illumina ChIP-seq sample preparation kit and sequenced in the Illumina GAII system according to the manufacturer's instructions. The first 25 bp for each sequence tag returned by the Illumina Pipeline was aligned to the hg18 assembly (National Center for Biotechnology Information). Only the tags uniquely mapped to the genome were used for further analysis. ChIP-seq results were mapped and visualized using the University of California at Santa Cruz Genome Browser.
ChIP-seq peak and DNA sequence analysis
ChIP-seq reads were first mapped to the hg18/NCBI 36.1 version of the human genome using Eland, allowing up to two mismatches per read. ChIP-seq enriched peaks were called using MACS 1.3.7 with the following parameters: mfold = 20, bw = 300, and P-value = 1 × 10−5 (Zhang et al. 2008). The peak location data are included in Supplemental Table 1. DNA motif analysis was performed using HOMER (http://homer.salk.edu/homer; Heinz et al. 2010). De novo motif discovery was performed using 200-bp regions centered on the TWIST1 ChIP-seq peaks for motif lengths between 6 and 20 bp. Motif enrichment histograms were generated using the annotatePeaks.pl program in HOMER. Individual 2× E-box configuration motifs with different spacing and allowable mismatches were generated using the seq2profile.pl script in HOMER.
Molecular modeling
A dimer model of the WR domain was created by I-TASSER (Roy et al. 2010) model generation using a 14xGly linker placed between two TWIST1 segments of amino acids 166–202. Following removal of the 14xGly linker and addition of water with a pH of 7.4, the model was energy-minimized using the AMBER03 force field (Duan et al. 2003). A double E-box DNA model was created by manually joining the DNA of two models of PDB structure 2ql2 (E-box DNA with E47/NeuroD1 complex bound) with five DNA bases separating the two E-box sequences. Amino acids missing in the loop of E47 and also replacing one of the E47 sequences with that from TWIST1 (amino acids 109–161 of the bHLH domain) were modeled using YASARA. The WR dimer was added between the two E47/TWIST1 dimers, and the amino acids were added to fill in the loops of the TWIST1 molecule and energy-minimized using the YASARA2 force field (Krieger et al. 2002). For the TWIST1-only dimer of dimer models, a second TWIST1 complex (dimerized through the WR domain) was aligned to the E47 structures using MUSTANG (Konagurthu et al. 2006), and the E47 was removed.
Twist1 gene design, cloning, recombinant protein expression, and purification
To improve protein production in Escherichia coli, a fully synthetic human Twist1 coding sequence was designed using codon optimization strategies and inserted into the pQE30 vector (Qiagen) at the Bam HI and Hind III sites. A SacI site 5′ of the bHLH region was created during gene design to allow further subcloning of the TWIST1 bHLH+WR region into the pQE30 vector at the SacI and HindIII sites. The SacI and PstI sites were used to release the TWIST1 bHLH ΔWR fragments from the pQE30-TWIST1 bHLH+WR plasmid, and the fragment was cloned into the pQE30 vector at corresponding sites to result in a pQE30-TWIST1 bHLH ΔWR vector.
The primers 5′-CATCACGGATCCGACCACTCGGAGGAGGAGAAGAAGGAGCTG-3′ and 5′-CTAATTAAGCTTGTTTTGTTTACTTTCACATGTGCCCGGCGGG-3′ were used to amplify the coding region of the E47 bHLH region, including its C terminus, using the pHBAneo-E47 plasmid as a PCR template (gift from Dr. Cornelis Murre, University of California at San Diego). The DNA fragment was cloned into the pQE30 vector at the Bam HI and HindIII sites. The E12 cDNA cloned in VICTR gene trapping vector was used to amplify the E12 bHLH region (a gift from Dr. Robert Benezra, Memorial Sloan-Kettering Cancer Center).
The protein expression vectors were transformed into E. coli SG13009 cells (Qiagen), and the cells were propagated with aeration at 37°C in 0.8 L of Luria broth in the presence of 100 μg/mL ampicillin and 25 μg/mL kanamycin to an A600 of ∼0.6 followed by addition of 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) to induce protein expression overnight at 25°C. The His-tagged TWIST1 FL, TWIST1 bHLH+WR, TWIST1 bHLH ΔWR, and E47 bHLH proteins expressed from bacteria were purified under denaturing conditions as recommended by the manufacturer (Qiagen). Briefly, the bacterial pellet was resuspended in 0.1 M Na-phosphate, 0.1 M Tris-HCl, and 6 M guanidine-HCl (pH 8.0) with 1 mM fresh PMSF and stirred for 2 h at 4°C to lyse the cells and solubilize the proteins under denaturing condition. The cell extract was centrifuged at 12,000g for 20 min. The supernatant fraction containing soluble protein was incubated in a batch with Ni-NTA resin for 1 h. The resin was washed once with 0.1 M Na-phosphate, 0.1 M Tris-HCl, and 6 M guanidine-HCl (pH 6.3) and twice with 0.1 M Na-phosphate, 0.1 M Tris-HCl, and 6 M guanidine-HCl (pH 6.3) with an additional 20 mM imidazole. The resin was loaded into a column, and the protein was eluted with 0.1 M Na-phosphate, 0.1 M Tris-HCl, 6 M guanidine-HCl (pH 4.6), and 300 mM imidazole with 1 mM PMSF. The eluted protein was dialyzed against three changes of 1× PBS and 1 mM DTT with 0.1 mM PMSF. The soluble proteins were concentrated with Amicon Ultra 0.5-mL centrifugal filters (3-kDa cutoff, Merck Millipore Ltd.). Bio-Rad Bradford protein assay and Coomassie staining were used to measure the concentration and purity of the proteins. In some cases, the soluble proteins were subjected to an additional purification step using HIS-Select spin columns (Sigma-Aldrich, Inc.) under native conditions following the manufacturer's protocol. The purified protein was dialyzed against two changes of 1× PBS and 1 mM DTT with 0.1 mM PMSF.
In vitro transcription and translation
The following primer pairs were used in PCR to amplify regions coding for E12 bHLH, E47 bHLH, TWIST1 FL, or TWIST1▵WR proteins. The forward primers (FOR) contain an in-built T7 promoter followed by Kozak consensus, while the reverse complementary (RC) primers contain an in-frame stop codon followed by a stretch of “A” residues to generate oligo dT sequences at the 3′ ends of in vitro transcribed RNAs: E12/E47 FOR T7 Quick, GGATCCTAATACGACTCACTATAGGGAACAGCCACCATGGACCACTCGGAGGAGGAG; E12/E47 RC T7 Quick, AAAAAAAAAAAAAAAAAAAAAAAAAAAAAATCACATGTGCCCGGCGGG; Twist1 FL/dWR FOR T7 Quick, GGATCCTAATACGACTCACTATAGGGAACAGCCACCATGATGCAGGACGTGTCCAG; Twist1 FL RC T7 Quick, AAAAAAAAAAAAAAAAAAAAAAAAAAAAAATTAGTGGGACGCGGACATGG; and Twist1 dWR RC_T7 Quick, AAAAAAAAAAAAAAAAAAAAAAAAAAAAAACTACTTGGAGTCCAGCTCG.
One microgram of either plasmid DNA or PCR products was used as a template, and in vitro transcription and translation reactions were carried out using a T7 Quick TNT kit for plasmid DNA or T7 Quick TNT for PCR DNA (Promega), respectively. The 35S-labeled proteins were used to determine the amounts of protein to be used in EMSAs, which were produced in parallel translations using cold methionine.
EMSAs
The promoter fragment (−2507 to −2537) of the human A2M gene that contains the native double E-box binding sites was used. Synthetic oligonucleotide duplex DNAs for A2M wild type and A2M MUT1 were 32P-end-labeled with T4 polynucleotide kinase (Roche Applied Science) and used as probes. In vitro translated Flag TWIST1 FL, TWIST1ΔWR, E12bHLH, and E47 bHLH proteins used in EMSAs were first mixed in equal molar ratios and incubated for 30 min at 30°C to allow formation of the heterodimers. Next, the protein mixture was incubated with 32P-radiolabeled probes in 20 μL of 20 mM HEPES (pH 7.6), 100 mM KCl, 10% glycerol, 1 mM DTT, 0.5 mg/mL BSA, and 0.05 mg/mL poly (dI–dC) for 30 min at 30°C. The nucleoprotein complexes were separated by native 5% PAGE at 300 V in buffer containing 50 mM Tris, 45 mM boric acid, and 1% glycerol for 2 h at 4°C. The gel was fixed in 10% acetic acid plus 10% methanol and dried, and signals were visualized by autoradiography.
The A2M double E-box oligonucleotides used for EMSA were as follows (E-boxes in the sequences are underlined): A2M wild type, 5′-CTCGAATCAGATGGAAGCCATGTGTTAAGG-3′ and 3′-GAGCTTAGTCTACCTTCGGTACACAATTCC-5′; and A2M MUT1, 5′-CTCGAATAATTTAGAAGCCATGTGTTAAGG-3′ and 3′-GAGCTTATTAAATCTTCGGTACACAATTCC-5′.
SPR-based kinetic analysis
The Biacore system T200 (GE Healthcare) was used in this analysis. The running buffer used for all experiments was 1× PBS, 1 mM DTT, and 0.01% Tween-20.
The BMP4 double E-box DNAs for use on the chip were generated by annealing one 5′ biotinylated synthetic oligonucleotide with its complementary strand of nonbiotinylated oligonucleotide using standard conditions. The biotinylated dsDNAs were immobilized on the series S sensor chip SA as the ligand after the chip was conditioned with 1 M NaCl and 50 mM NaOH per the manufacturer's instructions. The annealed DNAs were diluted to 0.1 ng/μL in running buffer and applied to the chips in individual flow cells at a flow rate of 10 μL/min followed by washing. A flow cell with no ligand was used in all runs as the reference well in the analysis. TWIST1 bHLH+WR/E47bHLH heterodimers diluted in running buffer at 4 μM were used as analytes to detect the individual RL in each flow cell.
The primers used were as follows (E-boxes in the sequences are underlined and/or in bold): biotinylated BMP4 wild type, 5′-biotin-GAAGCGGCTGGGGCTCACCTGGGGACCACGTGCGGAGGTACTAGAAA-3′ and 3′-CTTCGCCGACCCCGAGTGGACCCCTGGTGCACGCCTCCATGATCTTT-5′; biotinylated BMP4 2 nt 5′-biotin-GAAGCGGCTGGGGCTCACCTGGACACGTGCGGAGGTACTAGAAA-3′ and 3′-CTTCGCCGACCCCGAGTGGACCTGTGCACGCCTCCATGATCTTT-5′; biotinylated BMP4 10 nt, 5′-biotin-GAAGCGGCTGGGGCTCACCTGGGGACGGGACCACGTGCGGAGGTACTAGAAA-3′ and 3′-CTTCGCCGACCCCGAGTGGACCCCTGCCCTGGTGCACGCCTCCATGATCTTT-5′; biotinylated BMP4 4 nt, 5′-biotin- GAAGCGGCTGGGGCTCACCTGGGGACACGTGCGGAGGTACTAGAAA-3′ and 3′-CTTCGCCGACCCCGAGTGGACCCTGGTGCACGCCTCCATGATCTTT-5′; and biotinylated BMP4 6 nt, 5′-biotin-GAAGCGGCTGGGGCTCACCTGGGGACGCACGTGCGGAGGTACTAGAAA-3′ and 3′-CTTCGCCGACCCCGAGTGGACCCCTGCGTGCACGCCTCCATGATCTTT-5′.
Serial dilutions of 4 μM, 2 μM, 1 μM, 0.5 μM (in duplicates), 0.25 μM, and 0.125 μM were used for each of the kinetics/affinity runs. The wizard method was used with a flow rate of 30 μL/min, a contact time of 180 sec, and a dissociation time of 400 sec. A regeneration buffer (1× PBS, 850 mM NaCl, 1 mM DTT) with a flow rate 30 μL/min for 60 sec was used to wash off the analyte.
Viral production and infection
Stable Twist1- and Twist1ΔWR-overexpressing HMLE cell lines were generated with retroviral infection using the pWZL-Blast vector and selected with 10 μg/mL blasticidin, as described previously (Eckert et al. 2011).
Immunoprecipitation, immunoblotting, and immunofluorescence
All DNA constructs were transfected into 293T cells using Fugene 6 (Promega) and harvested 48 h later. An anti-Flag M2 affinity gel (Sigma, A2220) and an HA clone 16B12 monoclonal antibody (Covance, MMS-101R) were used for immunoprecipitation. The following primary antibodies were used for immunoblotting: mouse anti-TWIST1 2c1a (Santa Cruz Biotechnology), mouse anti-E-cadherin (BD, 610182), mouse anti-vimentin V9 (Thermo Scientific, MS-129-P), chicken anti-actin (Abcam, ab13822), anti-Flag (Sigma, F3165), anti-HA (Genetex, GTX115044), and anti-Myc hybridoma supernatant (clone 9E10).
Immunofluorescence was performed in eight-chamber slides by seeding 10,000 cells per well. Cells were fixed with 4% paraformaldehyde for 30 min and blocked in 5% goat serum/PBS-Tween for 1 h. Both primary and secondary antibodies were incubated in 5% goat serum/PBS-Tween overnight and for 1 h, respectively. The primary antibodies used were mouse anti-TWIST1 2c1a (Santa Cruz Biotechnology), rabbit anti-E-cadherin (Cell Signaling, 3195S), mouse anti-vimentin V9 (Thermo Scientific, MS-129-P), and nuclear DAPI staining (VectorShield). All images were imaged with an Olympus FV-1000 confocal microscope.
qPCR
All qPCR reactions used an 8-μL mixture of the Applied Biosystems SYBR Green PCR master mix, 0.2 μM forward and reverse primers, and water added to 2 μL of DNA sample. PCR and analysis were done with the Applied Biosystems Fast 7500 system. For validation of ChIP-seq data, primers were designed to flank regions with and without TWIST1-binding sites based on the ChIP-seq analysis. ChIP chromatin from noninduced and 4-d induced HMLE-Twist1ER cells were used, and mRNA levels were compared between the two samples to detect the level of enrichment.
For gene expression qPCR, mRNA was harvested from 70% confluent 60-mm plates using the QIAshredder and RNeasy minikits (Qiagen). Two micrograms of RNA was reverse-transcribed using the high-capacity cDNA reverse transcription kit (Life Technology). Primers used for PCR are listed in the Supplemental Material. All qPCR analyses were performed in triplicates and repeated in three biological replicates that showed consistent results.
Supplementary Material
Acknowledgments
We thank Dr. Wei Wang, Dr. Tao Wang, and Dr. Sarah Kinnings for their advice. We thank Dr. Cornelis Murre for his suggestions and the E47 cDNA. We thank members of the Yang laboratory, especially Spencer Wei, for technical help and discussions. We thank David Schultz for help with the Biacore assays. This work was supported by National Institutes of Health (NIH) grants (DP2OD002420, 1RO1CA168689, and 1R01CA174869), the Sidney Kimmel Foundation for Cancer Research, the Hartwell Foundation, and the Mary Kay Ash Charitable Foundation to J.Y.; NIH grants (P30-CA010815, R01-CA129883, R01-CA163761, R01-CA167151, and R01-CA175691), the Department of Defence Breast Cancer Research Program (W81XWH-11-1-0494), The Samuel Waxman Cancer Research Foundation, Susan G. Komen for the Cure (KG-110708), The Noreen O'Neill Foundation for Melanoma Research, and the Ovarian Cancer Research Foundation (291009) to F.J.R.; an NIH grant (1RO1HG004659) to X.-D.F.; an NIH predoctoral training grant (5T32GM007752) and an NIH National Research Science Award (5F31GM090678) to A.T.C.; an NIH training grant (5T32NCI09171) to Y.L.; and an NIH Institutional Research and Academic Career Development Award training grant (5K12GM068524) to H.P.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.242842.114.
References
- Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, et al. 2004. A TWIST code determines the onset of osteoblast differentiation. Dev Cell 6: 423–435. [DOI] [PubMed] [Google Scholar]
- Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. 2011. Snail2 is an essential mediator of TWIST1-induced epithelial mesenchymal transition and metastasis. Cancer Res 71: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanon I, Baylies MK. 2002. A TWIST in fate: evolutionary comparison of TWIST structure and function. Gene 287: 11–22. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117. [DOI] [PubMed] [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. 2003. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 24: 1999–2012. [DOI] [PubMed] [Google Scholar]
- Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. 2011. TWIST1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19: 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajula RP, Chettiar ST, Williams RD, Thiyagarajan S, Kato Y, Aziz K, Wang R, Gandhi N, Wild AT, Vesuna F, et al. 2013. The TWIST box domain is required for TWIST1-induced prostate cancer metastasis. Mol Cancer Res 11: 1387–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Boyer TG, Naski MC. 2012. Basic helix–loop–helix transcription factor TWIST1 inhibits transactivator function of master chondrogenic regulator Sox9. J Biol Chem 287: 21082–21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Bardet AF, Patton B, Purvis J, Johnston J, Paulson A, Gogol M, Stark A, Zeitlinger J. 2011. High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nat Genet 43: 414–420. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. 2004. An overview of the basic helix–loop–helix proteins. Genome Biol 5: 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. 2006. MUSTANG: a multiple structural alignment algorithm. Proteins 64: 559–574. [DOI] [PubMed] [Google Scholar]
- Krieger E, Koraimann G, Vriend G. 2002. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins 47: 393–402. [DOI] [PubMed] [Google Scholar]
- Lander R, Nordin K, LaBonne C. 2011. The F-box protein Ppa is a common regulator of core EMT factors TWIST, Snail, Slug, and Sip1. J Cell Biol 194: 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen KB, Mielke E, Iannaccone P, Fuchtbauer EM. 2007. Mechanism of transcriptional activation by the proto-oncogene TWIST1. J Biol Chem 282: 34623–34633. [DOI] [PubMed] [Google Scholar]
- Leptin M. 1991. TWIST and snail as positive and negative regulators during Drosophila mesoderm development. Genes Dev 5: 1568–1576. [DOI] [PubMed] [Google Scholar]
- Leptin M, Grunewald B. 1990. Cell shape changes during gastrulation in Drosophila. Development 110: 73–84. [DOI] [PubMed] [Google Scholar]
- Li S, Kendall SE, Raices R, Finlay J, Covarrubias M, Liu Z, Lowe G, Lin YH, Teh YH, Leigh V, et al. 2012. TWIST1 associates with NF-κB subunit RELA via carboxyl-terminal WR domain to promote cell autonomous invasion through IL8 production. BMC Biol 10: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. 2010. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari ME, Murre C. 2000. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 20: 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir A, Fisher-Aylor KI, Pepke S, Samanta M, Dunipace L, McCue K, Zeng L, Ogawa N, Wold BJ, Stathopoulos A. 2011. High resolution mapping of TWIST to DNA in Drosophila embryos: efficient functional analysis and evolutionary conservation. Genome Res 21: 566–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. 2007. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7: 415–428. [DOI] [PubMed] [Google Scholar]
- Pena WA, Slavotinek A, Oberoi S. 2010. Saethre-Chotzen syndrome: a case report. Cleft Palate Craniofac J 47: 318–321. [DOI] [PubMed] [Google Scholar]
- Piccinin S, Tonin E, Sessa S, Demontis S, Rossi S, Pecciarini L, Zanatta L, Pivetta F, Grizzo A, Sonego M, et al. 2012. A ‘TWIST box’ code of p53 inactivation: TWIST box: p53 interaction promotes p53 degradation. Cancer Cell 22: 404–415. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann T, Girardot C, Brehme M, Tongprasit W, Stolc V, Furlong EE. 2007. A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes Dev 21: 436–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto ML, Hing AV, Chang J, Hu M, Kapp-Simon KA, Patel PK, Burton BK, Kane AA, Smyth MD, Hopper R, et al. 2007. Isolated sagittal and coronal craniosynostosis associated with TWIST box mutations. Am J Med Genet A 143: 678–686. [DOI] [PubMed] [Google Scholar]
- Tennant BR, Robertson AG, Kramer M, Li L, Zhang X, Beach M, Thiessen N, Chiu R, Mungall K, Whiting CJ, et al. 2013. Identification and analysis of murine pancreatic islet enhancers. Diabetologia 56: 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Morgan M. 2004. Breast cancer progression with a TWIST. Nat Med 10: 777–778. [DOI] [PubMed] [Google Scholar]
- Thisse B, el Messal M, Perrin-Schmitt F. 1987. The TWIST gene: isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res 15: 3439–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Yang J. 2013. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 27: 2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. 2012. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. 2004. TWIST, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939. [DOI] [PubMed] [Google Scholar]
- Yin Z, Xu XL, Frasch M. 1997. Regulation of the TWIST target gene tinman by modular cis-regulatory elements during early mesoderm development. Development 124: 4971–4982. [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Zinzen RP, Stark A, Kellis M, Zhang H, Young RA, Levine M. 2007. Whole-genome ChIP-chip analysis of Dorsal, TWIST, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev 21: 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.