

The tumor suppressor ING4 (inhibitor of growth protein 4) is involved in various cellular processes by virtue of its epigenetic regulatory capability and through its positive regulation of p53 and negative regulation of NFκB. Yan et al. find that the F-box protein JFK targets ING4 for ubiquitination and degradation through assembly of an Skp1-Cul1-F-box (SCF) complex. JFK-mediated ING4 destabilization leads to the hyperactivation of the canonical NFκB pathway and promotes angiogenesis and metastasis of breast cancer.
Keywords: angiogenesis, metastasis, breast cancer, E3 ligase, ING4, JFK
Abstract
Loss of function/dysregulation of inhibitor of growth 4 (ING4) and hyperactivation of NF-κB are frequent events in many types of human malignancies. However, the molecular mechanisms underlying these remarkable aberrations are not understood. Here, we report that ING4 is physically associated with JFK. We demonstrated that JFK targets ING4 for ubiquitination and degradation through assembly of an Skp1–Cul1–F-box (SCF) complex. We showed that JFK-mediated ING4 destabilization leads to the hyperactivation of the canonical NF-κB pathway and promotes angiogenesis and metastasis of breast cancer. Significantly, the expression of JFK is markedly up-regulated in breast cancer, and the level of JFK is negatively correlated with that of ING4 and positively correlated with an aggressive clinical behavior of breast carcinomas. Our study identified SCFJFK as a bona fide E3 ligase for ING4 and unraveled the JFK–ING4–NF-κB axis as an important player in the development and progression of breast cancer, supporting the pursuit of JFK as a potential target for breast cancer intervention.
ING4 is a member of the inhibitor of growth (ING) family, which consists of five evolutionarily conserved proteins (ING1–5) (He et al. 2005). These proteins have been defined as type II tumor suppressors and implicated in various critical cellular processes, such as cell proliferation, apoptosis, DNA repair, senescence, angiogenesis, and drug resistance (Garkavtsev et al. 2004; Unoki et al. 2009). Dysregulation of ING4 has been reported in various malignancies, and down-regulation of ING4 is correlated with high-grade tumors and poor outcome of patients in several human cancers (Coles and Jones 2009). However, how ING4 is regulated under normal conditions and dysregulated in malignancies is poorly understood.
The ING proteins are characterized by a high homology in their C-terminal domain containing a nuclear localization sequence and a plant homeodomain (PHD) (He et al. 2005). These proteins have been reported to act as both “readers” and “writers” of the epigenetic histone code (Chi et al. 2010). Specifically, ING4 reads chromatin through the recognition of mono-, di-, and trimethyl histone H3 Lys4 (H3K4me1/2/3) (Hung et al. 2009) and associates with the HBO1 histone acetyltransferase (HAT) complex, which is responsible for chromatin remodeling and transcriptional regulation (Lalonde et al. 2013). ING4 also directly interacts with p53 and promotes the transactivation of p53 (Shiseki et al. 2003). In addition, by positive regulation of IκB activity (Coles et al. 2010) or negative regulation of the level of the RelA subunit (p65) of NF-κB (Hou et al. 2014), ING4 negatively regulates NF-κB-responsive gene transcription (Nozell et al. 2008; Byron et al. 2012). Interestingly, consistent with the down-regulation of ING4, NF-κB is continually hyperactivated in many types of human cancers and directs the expression of an array of target genes, including anti-apoptotic genes BCL2L1 and XIAP and angiogenesis-related genes IL-6, IL-8, and cyclooxygenase-2 (COX-2) (Basseres and Baldwin 2006). NF-κB also directly activates the transcription of the epithelial–mesenchymal transition (EMT) inducers Snail, Twist, and matrix metalloproteinase-9 (MMP9) and indirectly down-regulates the expression of the metastasis suppressor genes RKIP and E-cadherin (Karin 2006). Clearly, the negative regulation of NF-κB activity by ING4 is critically implemented in the control of apoptosis, angiogenesis, and cell migration. Therefore, understanding how the ING4–NF-κB pathway is dysregulated and NF-κB is constitutively activated in malignancies is of great importance to the understanding of tumor development and progression.
The protein homeostasis in cells is mainly controlled by the ubiquitin–proteasome system (Hershko and Ciechanover 1998). This system is involved in the regulation of cell proliferation, differentiation, and survival, and, similarly, dysregulation in this system is associated with various pathological states, including cancers (Hoeller et al. 2006). The ubiquitination cascade involves a ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s). E3 ubiquitin ligases are classified into four groups: HECT type, RING finger type, U-box type, and PHD finger type (Pickart 2004). SCF (Skp1–Cul1–F-box) ubiquitin ligase is the best-characterized multisubunit RING finger complex composed of four subunits: intrinsic constituents Skp1, Cul1, and Rbx1 and variable component F-box protein (FBP) (Petroski and Deshaies 2005). Featuring an F-box domain, FBPs act as receptors for substrate recognition and specification (Cardozo and Pagano 2004). However, of the 69 FBPs identified in humans, the substrates for the majority remain unidentified (Hermand 2006). In addition to the F-box domain, FBPs contain additional C-terminal motifs capable of protein–protein interaction, with WD repeats and leucine-rich repeats being the most common motifs in yeast and human FBPs. Although Kelch domain-containing FBPs make up the majority of the FBPs in Arabidopsis (Lechner et al. 2006), our previous studies reported that JFK is the only Kelch domain-containing FBP in humans (Sun et al. 2009, 2011).
Here we report that ING4 is physically associated with JFK in vivo. We demonstrated that JFK targets ING4 for ubiquitination and degradation through assembly of an SCF ubiquitin ligase. We showed that SCFJFK-mediated ING4 destabilization potentiates NF-κB signaling and promotes the angiogenesis and metastasis of breast cancer in vitro and in vivo. We found that the expression of JFK is markedly up-regulated in breast cancers and that JFK protein level is negatively correlated with that of ING4 and positively correlated with an aggressive clinical behavior of breast carcinomas.
Results
ING4 is physically associated with JFK in the context of an SCF complex
In an effort to better understand the mechanistic role of ING4 in malignant transformation, we employed affinity purification and mass spectrometry to screen the proteins that are associated with ING4 in vivo. In these experiments, MCF-7 cells were transfected with Flag-tagged ING4 (Flag-ING4). Whole-cell extracts were prepared and subjected to affinity purification using an anti-Flag affinity column. After extensive washing, the bound proteins were eluted with excess Flag peptides, resolved on SDS-PAGE, and then visualized by silver staining. The protein bands on the gel were recovered and analyzed by mass spectrometry. The results indicate that ING4 was copurified with a number of proteins, including CLIP1, LATS2, Jade-1, p65, EEF1A1, and WDR77. Among these proteins, Jade-1 (Doyon et al. 2006) and p65 (Hou et al. 2014) are known to interact with ING4. Interestingly, JFK, the only Kelch domain-containing FBP in humans (Sun et al. 2009, 2011), and Skp1, an integral component of the SCF complex (Petroski and Deshaies 2005), were also identified in the ING4-containing protein complex (Fig. 1A; Supplemental Table S1).
Figure 1.

ING4 is physically associated with JFK in the context of an SCF complex. (A) Mass spectrometry analysis of ING4-associated proteins. Cellular extracts from MCF-7 cells stably expressing Flag-ING4 were immunopurified with anti-Flag affinity column and eluted with Flag peptides. The eluates were resolved by SDS-PAGE and silver-stained. The protein bands were retrieved and analyzed by mass spectrometry. (B) Fractionation of ING4-associated proteins by fast protein liquid chromatography (FPLC). Cellular extracts from MCF-7 cells transfected with Flag-ING4 were fractionated on Superose 6 size exclusion columns. Chromatographic elution profiles, silver staining, and Western blot analysis of the chromatographic fractions are shown. The elution positions of calibration proteins with known molecular masses are indicated, and an equal volume from each fraction was analyzed. (C) ING4 interacts with JFK in the context of the SCF complex. Whole-cell lysates from MCF-10A cells, MCF-7 cells, T47D cells, and MDA-MB-231 cells were immunoprecipitated with antibodies against ING4 followed by immunoblotting with the antibodies against the indicated proteins. (D) ING4 interacts with JFK in vitro. GST pull-down experiments were performed with bacterially expressed GST-ING4 and in vitro transcribed/translated JFK, Cul1, Skp1, or Rbx1. (E) In vivo molecular interaction of JFK with ING4. MCF-7 cells were cotransfected with Myc-ING4 and Flag-JFK or JFK mutants. Cellular lysates were immunoprecipitated with anti-Flag followed by immunoblotting with anti-Myc. Schematic diagrams of wild-type JFK and JFK deletion mutants are shown.
To further support the observation that ING4 is physically associated with JFK in vivo, protein fractionation experiments were carried out by fast protein liquid chromatography (FPLC) with Superose 6 columns and a high-salt extraction and size exclusion approach. The result indicates that ING4 from MCF-7 cells was eluted with an apparent molecular mass much greater than that of the monomeric protein; ING4 immunoreactivity was detected in chromatographic fractions from the Superose 6 column with a relatively symmetrical peak centered between ∼669 and ∼2000 kDa (Fig. 1B). Significantly, the chromatographic profile of ING4 largely overlapped with that of JFK as well as that of Cul1 and Skp1, two constituents of the SCF complex (Fig. 1B, fractions 17–21). To validate our experimentation, Jade-1, which is known to interact with ING4 in vivo (Doyon et al. 2006), was also identified in the ING4-purified protein complex in our FPLC experiments. These results support the observation that ING4 is physically associated with JFK in vivo and suggest that ING4 interacts with JFK in the context of an SCF complex.
To support the proposition that ING4 is associated with JFK in the context of the SCF complex in vivo, total proteins from MCF-7 cells were extracted, and coimmunoprecipitation experiments were performed. Immunoprecipitation with antibodies against ING4 and immunoblotting with antibodies against JFK, Cul1, Skp1, or Rbx1 showed that JFK as well as the integral components of the SCF complex—Cul1, Skp1, and Rbx1—were indeed efficiently coimmunoprecipitated with ING4 (Fig. 1C). The association between ING4 and JFK or Skp1 was also detected in MCF-10A cells, T47D cells, and MDA-MB-231 cells (Fig. 1C). Glutathione S-transferase (GST) pull-down assays with bacterially expressed GST-ING4 and in vitro transcribed/translated JFK, Cul1, Skp1, or Rbx1 showed that ING4 interacts with JFK but not Cul1, Skp1, and Rbx1 (Fig. 1D), consistent with the feature of the SCF ubiquitin ligase in which only the FBP directly interacts with the substrate.
Further analysis by coimmunoprecipitation experiments in MCF-7 cells that were cotransfected with wild-type JFK, F-box-deleted JFK (JFKΔF-box), or Kelch domain-deleted JFK (JFKΔKelch) together with Myc-tagged ING4 (Myc-ING4) demonstrated that the Kelch domain of JFK is responsible for its interaction with ING4, since although the JFKΔF-box still retained the capacity to interact with ING4, JFKΔKelch failed to do so (Fig. 1E). Moreover, GST pull-down assays with bacterially expressed GST-JFK, GST-JFKΔF-box, or GST-JFKΔKelch and in vitro transcribed/translated ING4 showed that while both wild-type JFK and JFKΔF-box were capable of interacting with ING4 in vitro, JFKΔKelch was not (Supplemental Fig. S1A). These results are consistent with the feature of an SCF ubiquitin ligase in which the FBP recognizes and binds to corresponding substrates through its variable protein–protein interaction domain. Analogously, GST pull-down with bacterially expressed JFK and in vitro transcribed/translated Myc-tagged ING4 or ING4 deletion mutants showed that the novel conserved domain (NCR; 61–120 amino acids) (He et al. 2005) of ING4 is responsible for its interaction with JFK (Supplemental Fig. S1B). Together, these data support a notion that ING4, through the substrate receptor JFK, interacts with the SCF complex in vivo.
JFK promotes ING4 degradation through an SCF-dependent pathway
Our previous studies showed that JFK promotes the ubiquitination and degradation of p53 through the assembly of an SCF complex (Sun et al. 2009, 2011). The physical interaction of ING4 with JFK in the context of the SCF complex prompted us to investigate whether ING4 is also destabilized by JFK via an SCF-dependent mechanism. To this end, MCF-7, MDA-MB-231, MCF-10A, and T47D cells were transfected with empty vector or JFK or treated with control siRNA or JFK siRNA, and Western blotting analysis of the cellular lysates revealed that JFK overexpression resulted in a decrease and JFK depletion led to an increase in the level of ING4 protein (Fig. 2A; Supplemental Fig. S2A), whereas overexpression of either JFKΔF-box or JFKΔKelch in MCF-7 cells did not result in evident changes in the ING4 level (Fig. 2B). In addition, there were no detectable changes in ING4 protein steady-state level when cells were transfected with β-Trcp (the FBP for p100) (Nakayama et al. 2003), Fbw7 (the FBP for cyclin E and p100) (Koepp et al. 2001; Fukushima et al. 2012), or Skp2 (the FBP for cyclin E) (Supplemental Fig. S2B; Nakayama et al. 2000), suggesting that ING4 degradation is JFK-specific. In support of this notion, Western blotting analysis revealed a JFK dose-dependent decay of ING4 in MCF-7 cells (Fig. 2C). Moreover, JFK-dependent ING4 destabilization could be effectively blocked by MG132 (Fig. 2C), suggesting that this process is probably mediated by the ubiquitin–proteasome pathway.
Figure 2.
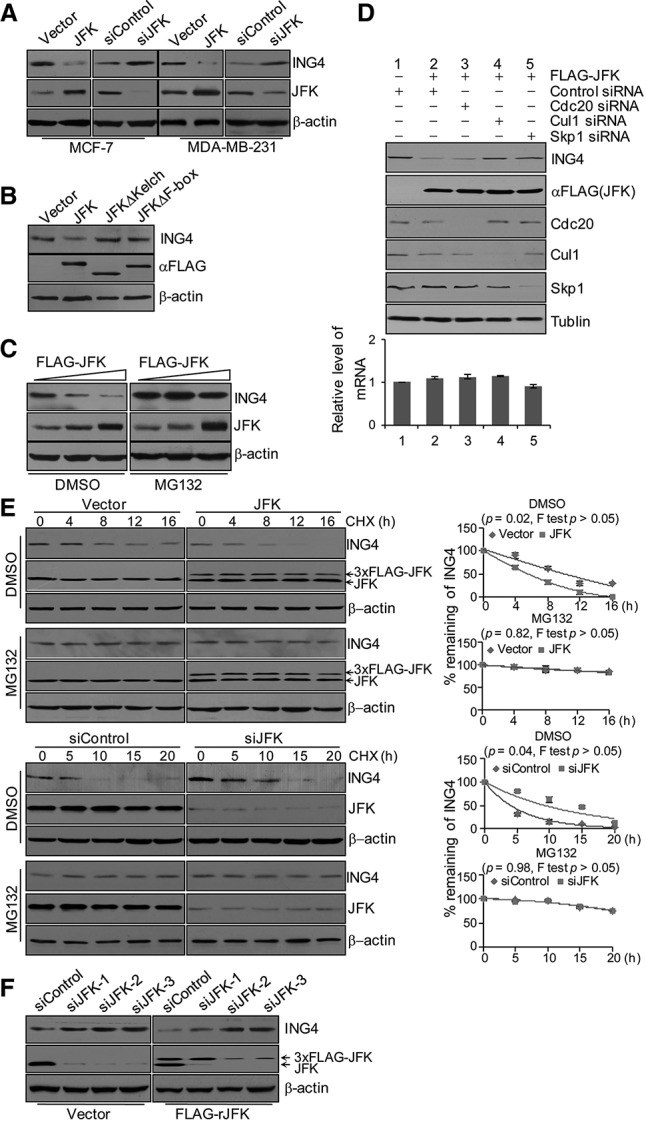
JFK promotes ING4 degradation through an SCF-dependent pathway. (A) Wild-type JFK promotes ING4 degradation in breast cancer cell lines. MCF-7 and MDA-MB-231 cells were transfected with empty vector or Flag-tagged JFK or treated with control siRNA or JFK siRNA. Cellular extracts were prepared, and Western blotting was performed with the antibodies against the indicated proteins. (B) Wild-type JFK, but not JFK mutants, promotes ING4 degradation. MCF-7 cells were transfected with Flag-tagged JFK, JFKΔF-box, or JFKΔKelch. Cellular extracts were prepared, and Western blotting was performed with the antibodies against the indicated proteins. (C) JFK negatively affects the steady-state level of the ING4 protein. MCF-7 cells were transfected with increasing amounts of Flag-JFK. Forty-eight hours after transfection, cells were treated with DMSO or MG132 for 6 h before cells were collected for Western blotting analysis. (D) The destruction of ING4 by JFK is mediated by an SCF complex. MCF-7 cells were transfected with Flag-JFK and treated with siRNAs for Cul1, Skp1, or Cdc20 as indicated. Cellular proteins were extracted for Western blotting analysis with antibodies against the indicated proteins, or total RNAs were extracted and analyzed for ING4 expression by real-time RT–PCR. Bars represent the mean ± SD for triplicate experiments. (E) JFK negatively regulates ING4 half-life. MCF-7 cells were transfected with empty vector or Flag-JFK or treated with control siRNA or JFK siRNA. Forty-eight hours after transfection, cells were treated with 50 μg/mL cycloheximide (CHX) for the indicated times in the presence or absence of MG132 before cellular proteins were extracted for Western blotting analysis. Quantitation was done by densitometry and expressed as signals of ING4/β-actin. Bars represent the mean ± SD for triplicate experiments. (F) JFK siRNA is specific. MCF-7 cells were transfected with vector or a JFK siRNA-1-resistant form (rJFK) together with control siRNA or JFK siRNA. Cellular proteins were extracted for Western blotting analysis with antibodies against the indicated proteins.
Next, MCF-7 cells were treated with specific siRNAs against Cdc20, Cul1, or Skp1 to knock down the expression of these proteins. Western blotting analysis indicates that, in both Cul1- and Skp1-depleted cells, overexpression of JFK was no longer associated with an increased turnover of ING4, whereas depletion of Cdc20, the activator subunit of the mitotic APC (anaphase-promoting complex) (Peters 2006), had no effect on JFK-mediated ING4 destruction (Fig. 2D), suggesting that JFK promotes ING4 protein degradation through an SCF-dependent pathway. The decreased ING4 protein expression under JFK overexpression was not due to transcriptional regulation of ING4, as real-time RT–PCR measurements indicated that JFK overexpression did not affect ING4 mRNA level (Fig. 2D).
Cycloheximide (CHX) chase assays were then performed in MCF-7 cells that were either transfected with JFK or treated with JFK-specific siRNAs. Western blotting analysis revealed that JFK overexpression was associated with a decrease and JFK knockdown resulted in an increase in the half-life of ING4, effects that only occurred in the absence of MG132 (Fig. 2E).
As stated before, one of the molecular activities of ING4 is to interact with another tumor suppressor, p53 (Shiseki et al. 2003). In light of our previous observations that JFK is also physically and functionally associated with p53 (Sun et al. 2009, 2011), we asked whether the JFK-mediated ING4 degradation is in some way connected to the function of p53. In order to address this, gain of function and loss of function of JFK were performed in HCT116 p53+/+ and HCT116 p53−/− cells. Western blotting analysis showed that JFK overexpression resulted in a decrease and JFK depletion led to an increase in the level of ING4 protein regardless of p53 status (Supplemental Fig. S2C). Thus, we concluded that p53 is not involved in JFK-mediated ING4 degradation. The silencing specificity of JFK siRNA was validated using a JFK siRNA-1-resistant form (rJFK) that was generated by synonymous mutations (Fig. 2F). Collectively, these results support the argument that JFK promotes ING4 destruction through the assembly of an SCF complex (SCFJFK) and is a novel negative regulator of ING4.
JFK promotes polyubiquitination of ING4
To further strengthen the receptor–substrate relationship between JFK and ING4 and investigate the proteasome-dependent mechanism of JFK-mediated ING4 degradation, we next determined whether JFK-promoted ING4 destabilization is a consequence of ING4 ubiquitination. To this end, MCF-7 cells were cotransfected with Flag-tagged JFK, JFKΔF-box, or JFKΔKelch together with Myc-ING4 and HA-tagged wild-type ubiquitin or ubiquitin mutants (UbKO and UbK48R) defective in polyubiquitin chain assembly (Thrower et al. 2000) in the presence of MG132. Immunoprecipitation of the cellular lysates with an antibody against Myc followed by immunoblotting with an antibody against HA indicated that JFK, but not JFKΔF-box and JFKΔKelch, promoted ING4 polyubiquitination in vivo (Fig. 3A). Consistently, when wild-type ubiquitin used in the ubiquitination assay was replaced with ubiquitin mutant UbKO or UbK48R, JFK-promoted ING4 polyubiquitination was no longer detected (Fig. 3A). Moreover, in vitro ubiquitination assays with bacterially expressed GST-JFK, GST-JFKΔF-box, or GST-JFKΔKelch and in vitro transcribed/translated ING4 support the observation that only wild-type JFK promotes ING4 polyubiquitination (Fig. 3B).
Figure 3.
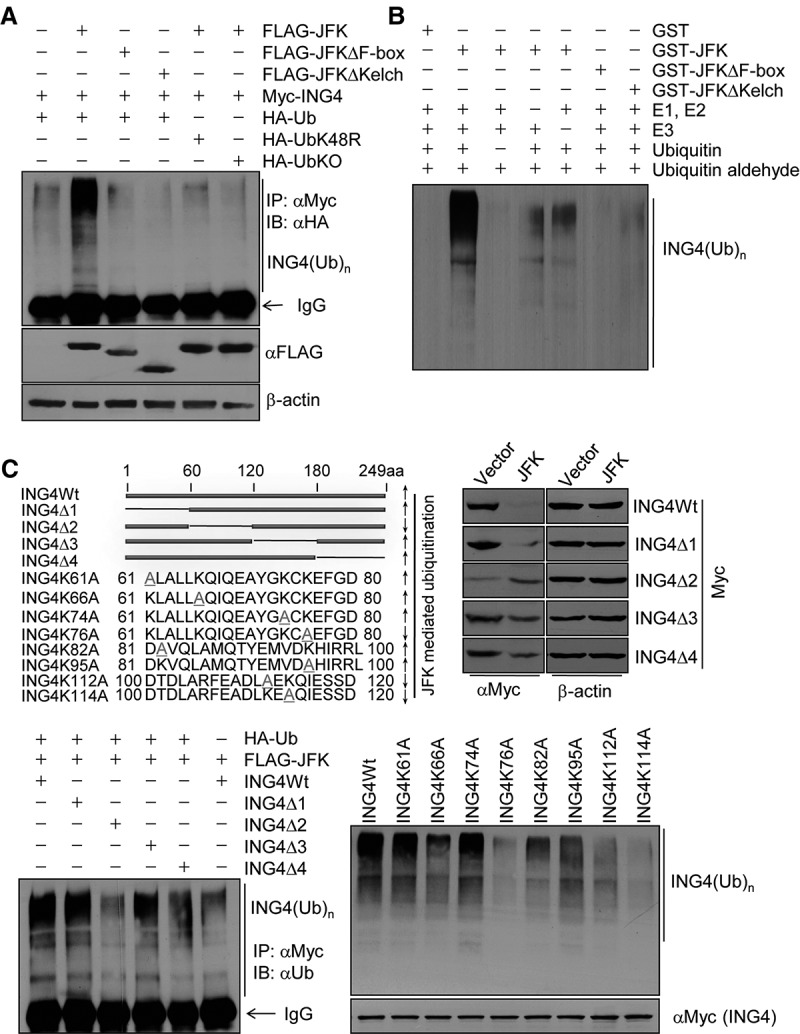
JFK promotes polyubiquitination of ING4. (A) JFK promotes ING4 ubiquitination in vivo. MCF-7 cells were cotransfected with the indicated plasmids. Forty-eight hours after transfection, cells were treated with MG132 for 12 h before cellular extracts were prepared for immunoprecipitation (IP) with anti-Myc followed by immunoblotting (IB) with anti-HA. (B) JFK promotes ING4 ubiquitination in vitro. Bacterially expressed GST-JFK or JFK mutants were incubated with in vitro transcribed/translated ING4 for ubiquitination assays. The reaction mixture was resolved on SDS-PAGE. (C) Molecular insight into JFK-promoted ING4 polyubiquitination. (Top left) Schematic representation of wild-type ING4 and ING4 mutants. (Top right) MCF-7 cells were cotransfected with Flag-JFK and wild-type ING4 or ING4 deletion mutants. Cellular proteins were extracted for Western blotting analysis. (Bottom left) MCF-7 cells were cotransfected with Flag-JFK, HA-ubiquitin, and Myc-tagged wild-type ING4 or ING4 deletion mutants. Cells were treated with MG132 for 12 h before cellular extracts were prepared for immunoprecipitation assays with anti-Myc followed by immunoblotting with anti-ubiquitin. (Bottom right) Bacterially expressed GST-JFK was incubated with in vitro transcribed/translated wild-type ING4 or ING4 point mutants for ubiquitination assays. The reaction mixture was resolved on SDS-PAGE.
MCF-7 cells were then cotransfected with Flag-JFK and Myc-ING4 or Myc-tagged serial deletions of ING4 (ING4Δ1–Δ4). Western blotting analysis of the cellular lysates showed that JFK overexpression was associated with decreases in the protein level of ING4Δ1, ING4Δ3, and ING4Δ4 but not of ING4Δ2 (Fig. 3C, top right), suggesting that ING4 polyubiquitination occurs in residues 61–120, which are absent in ING4Δ2. Indeed, in vivo ubiquitination assays with Myc-tagged ING4 or ING4Δ1–Δ4 showed that while wild-type ING4 and ING4Δ1, ING4Δ3, and ING4Δ4 were polyubiquitinated by JFK, ING4Δ2 was not (Fig. 3C, bottom left). In support of this observation, eight lysines in amino acid residues 61–120 of ING4 were individually replaced with alanine (Fig. 3C, top left). In vitro ubiquitination assays with bacterially expressed GST-JFK and in vitro transcribed/translated wild-type ING4 or ING4 mutants showed evident decreases in the polyubiquitination of ING4K76A, ING4K112A, and ING4K114A (Fig. 3C, bottom right), suggesting that these three lysines are the sites targeted by SCFJFK for polyubiquitination. Taken together, these data indicate that JFK targets ING4 for polyubiquitination and degradation and that SCFJFK is a bona fide E3 ubiquitin ligase for ING4.
The JFK–ING4 pathway intersects with NF-κB signaling
In order to explore the biological significance of JFK-mediated ING4 degradation, we analyzed the cellular signaling transduction pathways that are potentially influenced by JFK using the Human Signal Transduction PathwayFinder PCR array (SABioscience) (Hager-Theodorides et al. 2009; Olson et al. 2011). This array profiles the expression of ∼100 key genes representing 10 prominent cellular signal transduction pathways, including TGF-β, WNT, NF-κB, JAK/STAT, p53, Notch, Hedgehog, PPAR, oxidative stress, and hypoxia pathways. In these experiments, total mRNAs from MCF-7 cells overexpressing JFK were extracted. Corresponding cDNAs were synthesized, amplified under nonbiased conditions, and hybridized to the Human Signal Transduction PathwayFinder PCR array to analyze relative mRNA expression. The results reported a total of 38 genes whose expression was affected by JFK overexpression (Fig. 4A, top). Five of these genes—including CCL5, CSF1, ICAM1, STAT1, and TNF, whose expression was significantly up-regulated (Fig. 4A, bottom)—are cataloged to the NF-κB signaling pathway, a pathway that has been reported to be negatively regulated by ING4 (Supplemental Fig. S3A; Nozell et al. 2008; Coles et al. 2010; Byron et al. 2012; Hou et al. 2014). Based on these results, we hypothesized that the JFK–ING4 pathway intersects with and thus impacts on NF-κB signaling. Several lines of evidence support this hypothesis: First, confocal microscopic analysis of MCF-7 cells overexpressing JFK showed an increase in p65 (a core subunit of NF-κB) translocation from cytoplasm to nucleus (Fig. 4B). Second, gain of function of JFK was associated with increases in mRNA expression of NF-κB target genes CCL5, ICAM1, IL-6, IL-8, MMP9, STAT1, and VCAM1 (Basseres and Baldwin 2006) in MCF-7 cells, as measured by real-time RT–PCR, whereas loss of function of JFK was accompanied by dampened NF-κB signaling, as evidenced by decreased expression of these target genes (Fig. 4C). Third, in ING4-depleted MCF-7 cells, overexpression of JFK no longer led to increased expression of CCL5, IL-8, MMP9, and VCAM1, and knockdown of JFK no longer resulted in decreased expression of the NF-κB target genes, while overexpression of ING4 in JFK-overexpressing MCF-7 cells offset JFK-enhanced expression of NF-κB target genes (Fig. 4D). The silencing specificity of ING4 siRNA was validated using an ING4 siRNA-1-resistant form (rING4) that was generated by synonymous mutations (Supplemental Fig. S3B). Collectively, these results indicate that JFK, through destabilizing ING4, potentiates NF-κB transcriptional activity, supporting the notion that the JFK–ING4 pathway intersects with and thus influences NF-κB signaling.
Figure 4.
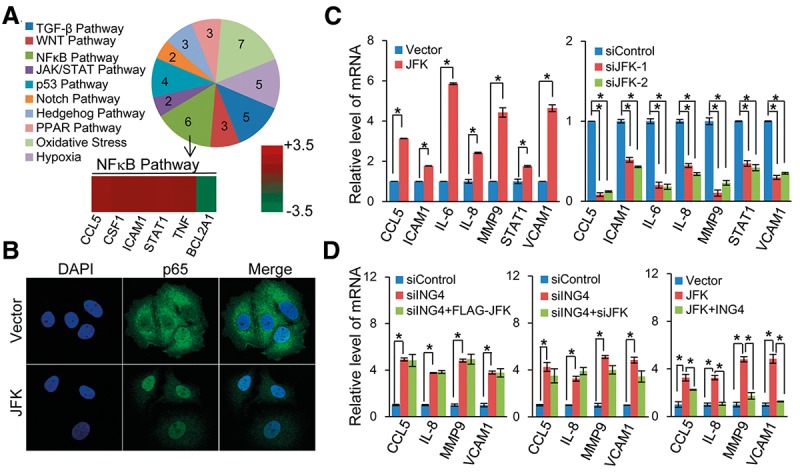
The JFK–ING4 pathway intersects with NF-κB signaling. (A) JFK potentiates NF-κB signaling. MCF-7 cells were transfected with empty vector or Flag-JFK. Total cellular RNAs were prepared, and PCR array was performed. The expression of the indicated genes in different signal pathways was analyzed, and the pie chart represents the number of changed genes in each pathway. (B) JFK facilitates p65 cellular translocation. MCF-7 cells were transfected with empty vector or Flag-JFK, stained with anti-p65, and analyzed by confocal microscopy. (C) The effect of JFK on the expression of NF-κB target genes. MCF-7 cells were transfected with the indicated plasmids or treated with the indicated siRNA. Total cellular RNAs were prepared for real-time RT–PCR analysis. Bars represent the mean ± SD for triplicate experiments. (*) P < 0.05. (D) Simultaneous overexpression of ING4 offsets JFK-enhanced expression of NF-κB target genes. MCF-7 cell were transfected with the indicated plasmids or treated with the indicated siRNA. Total cellular RNAs were prepared for real-time RT–PCR analysis. Bars represent the mean ± SD for triplicate experiments. (*) P < 0.05.
JFK promotes the angiogenesis of breast cancer cells
In order to further explore the biological significance of JFK-mediated ING4 degradation, we next investigated the effect of the JFK–ING4 pathway on NF-κB-regulated cellular events. In this regard, it is interesting to note that the NF-κB pathway regulates the expression of several prominent proangiogenic factors, including IL-6, IL-8, CCL5, and COX-2 (Karin and Greten 2005). Thus, it is logical to postulate that the JFK–ING4 pathway is implicated in the regulation of the angiogenesis through modulation of NF-κB activity. To test this hypothesis, we first performed an in vitro endothelial tube formation assay, which is based on the ability of endothelial cells to form three-dimensional capillary-like tubular structures when cultured on extracellular matrix gels prepared from Engelbreth-Holm-Swarm tumor cells (Grant et al. 1985). In the experiments, human umbilical vein endothelial cells (HUVECs) were infected with retroviruses carrying vector or JFK or lentiviruses carrying control siRNA or JFK siRNA. Conditioned media (CM) from cultures of MCF-7 cells that were also infected with retroviruses carrying vector or JFK or lentiviruses carrying control siRNA or JFK siRNA were added onto solidified extracellular matrix gels. After incubation, the formation of endothelial cell tubes was examined under light microscopy, and the number of tubes was counted. It was noted that JFK-overexpressing HUVECs or HUVECs cultured in the CM from JFK-overexpressing MCF-7 cells formed significantly more tubes than control groups, whereas JFK-depleted HUVECs or HUVECs cultured in the CM from JFK-deficient MCF-7 cells generated fewer tubes than controls (Fig. 5A), suggesting that JFK promotes the angiogenic potential of HUVECs. JFK-mediated ING4 degradation in this system was validated by Western blotting measurement of the ING4 expression in HUVECs (Fig. 5A), and the effect of JFK-mediated ING4 degradation on the NF-κB signaling was verified by real-time RT–PCR measurement of the expression of NF-κB target genes CCL5 and IL-8 in HUVECs (Fig. 5A).
Figure 5.
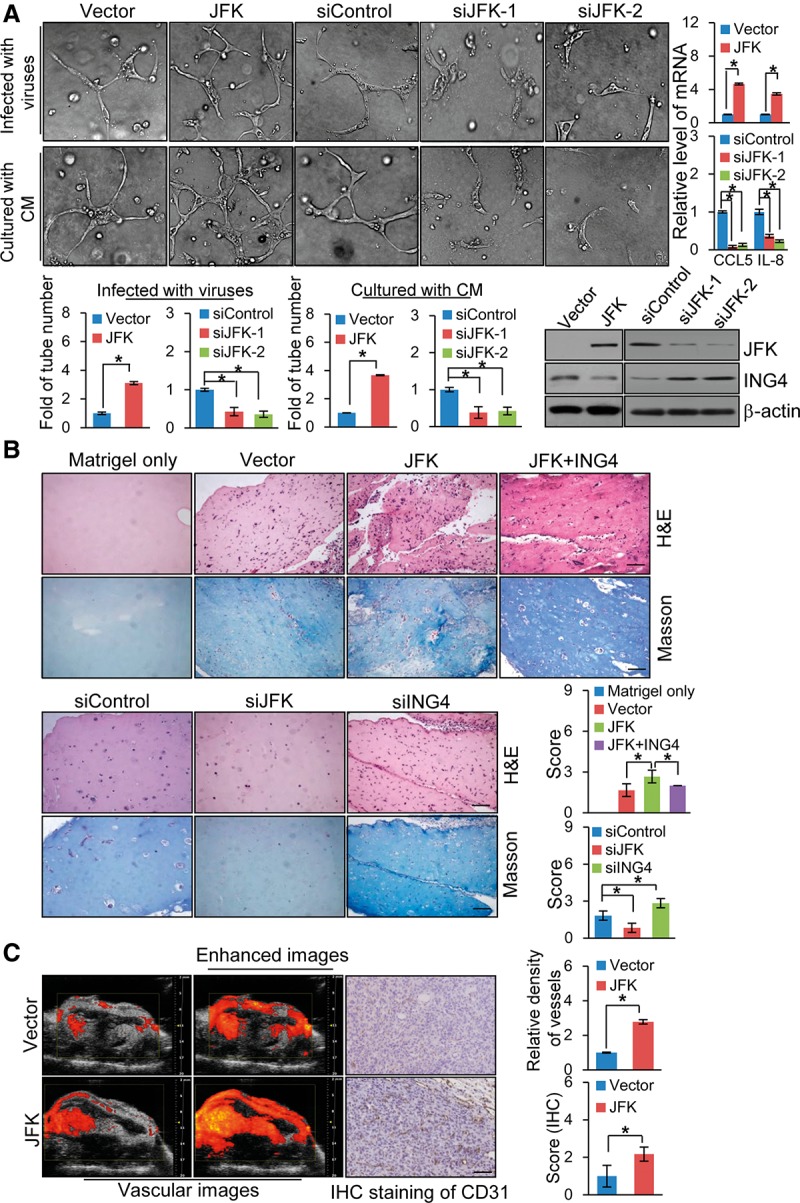
JFK promotes the angiogenic potential of breast cancer cells. (A) JFK promotes the angiogenic potential of breast cancer cells in vitro. HUVECs were infected with retroviruses carrying vector or JFK or lentiviruses carrying control siRNA or JFK siRNA or were cultured with CM from MCF-7 cells that were infected with retroviruses carrying vector or JFK or lentiviruses carrying control siRNA or JFK siRNA and were added onto a solidified extracellular matrix. After incubation, endothelial cell tube formation was assessed under light microscopy, and the tube number was counted. Representative images from each group are shown. Bars represent the mean ± SD for triplicate measurements. The expression of JFK and ING4 in HUVECs was detected by Western blotting, and the expression of NF-κB target genes in HUVECs was examined by real-time RT–PCR. (B) JFK promotes the angiogenic potential of breast cancer cells in vivo. Matrigels only or Matrigels mixed with MCF-7 cells infected with retroviruses carrying JFK and/or ING4 or lentiviruses carrying JFK siRNA or ING4 siRNA were injected subcutaneously into 6-wk-old BALB/c female mice (n = 6). Seven days after injection, the mice were sacrificed, and the Matrigel plugs were removed for H&E and Masson trichrome staining. Bar, 73 μm. (C) MDA-MB-231-Luc-D3H2LN cells were infected with retroviruses carrying empty vector or JFK and were implanted into the left abdominal mammary fat pad of immunocompromised 6-wk-old female SCID beige mice (n = 6). The vascular density was assessed with the Vevo 2100 imaging platform, and immunohistochemical staining was performed with antibodies against CD31. Bar, 73 μm.
Next, in vivo chicken yolk sac membrane (YSM) assays were carried out in which gelatin sponges that had adsorbed suspensions of MCF-7 cells that were transfected with JFK and/or ING4 or with control siRNA or JFK siRNA were placed on top of the YSM. Microscopic counting of the number of blood vessels entering the sponge showed that eggs treated with JFK-overexpressing MCF-7 cells developed enriched blood vessels toward the implants in a “spoked wheel” pattern and that the positive effect of JFK on blood vessel formation was offset by overexpression of ING4, whereas eggs treated with JFK-depleted MCF-7 cells formed significantly fewer blood vessels compared with controls (Supplemental Fig. S4A).
We then employed a well-established in vivo angiogenesis model—the mouse Matrigel plug assay (Angiolillo et al. 1995)—to investigate the effect of JFK on angiogenesis in vivo. In this assay, BALB/c female mice were randomly divided into seven groups, and the animals in each group (n = 6) were injected subcutaneously with either Matrigels only or Matrigels that were mixed with MCF-7 cells infected with retroviruses carrying JFK and/or ING4 or lentiviruses carrying control siRNA, JFK siRNA, or ING4 siRNA. Seven days after injection, the mice were sacrificed, and the Matrigel plugs were processed and stained with H&E (hematoxylin and eosin) and Masson trichrome. Microscopic examination of Matrigel plugs revealed that endothelial cells, often organized into blood vessels containing red blood cells, were enriched in the JFK-overexpressing group and that the positive effect of JFK on blood vessel formation was offset by simultaneous overexpression of ING4 (Fig. 5B, top). In contrast, only a few endothelial cells had invaded the plugs of JFK siRNA-treated Matrigels, while depletion of ING4 mimicked the enhancing effect of JFK overexpression on blood vessel formation (Fig. 5B, bottom).
To explore the role of JFK in breast cancer angiogenesis in vivo, MDA-MB-231-Luc-D3H2LN cells were infected with retroviruses carrying vector or JFK and were implanted into the left abdominal mammary fat pad of immunocompromised 6-wk-old female SCID beige mice (n = 6). Vascular density was assessed with the Vevo 2100 imaging platform in power Doppler mode 5 wk after tumor onset. The results showed that JFK overexpression led to a more than twofold increase in vascular density compared with the control group (Fig. 5C). Consistently, the expression of CD31, a marker for angiogenesis (Ozdemir et al. 2014), was also higher in the JFK group. Taken together, these results indicate that JFK promotes the angiogenic potential of breast cancer cells.
JFK promotes EMT and the invasive potential of breast cancer cells in vitro
One of the hallmarks of cancer is the ability of tumor cells to invade and metastasize (Hanahan and Weinberg 2011). At the very beginning of metastasis, cancer cells reprogram by turning on embryonic morphogenesis regulators to undergo EMT and turning off differentiation programs, allowing cancer cells to phenotypically transform, a process in which nonmotile, polarized epithelial cells embedded via cell–cell junctions in a cell collective dissolve their cell–cell junctions and convert into individual, nonpolarized, motile, and invasive mesenchymal cells (Mani et al. 2008). Based on the important role of the NF-κB pathway in cancer invasion and metastasis (Karin 2006), we next asked what roles, if any, the JFK–ING4 pathway plays in breast cancer metastasis. To this end, MCF-7 cells were transfected with JFK and/or ING4. The morphological alterations of the cells were examined by phase-contrast microscopy. Notably, JFK-overexpressing MCF-7 cells displayed loss of cell–cell contacts, scattering, and a spindle-like, fibroblastic morphology compared with the highly organized cell–cell adhesion, cell polarity, and cobblestone-like appearance of control MCF-7 cells (Fig. 6A). In agreement with these observations, immunofluorescent microscopy showed a reduction/loss of epithelial marker E-cadherin and γ-catenin staining and an increase in mesenchymal marker vimentin and fibronectin staining in JFK-overexpressing MCF-7 cells—effects that were partially attenuated by simultaneous overexpression of ING4 (Fig. 6A; Supplemental Fig. S4B). Consistently, Western blotting showed a reduction of the epithelial markers E-cadherin, α-catenin, β-catenin, and γ-catenin and induction of the mesenchymal markers N-cadherin, fibronectin, vimentin, and SM-actin in JFK-overexpressing MCF-7 cells—effects that were partially attenuated by simultaneous overexpression of ING4 (Fig. 6B). Significantly, JFK overexpression was associated with an enhanced NF-κB activity, as evidenced by increased expression of NF-κB target genes MMP9 and VCAM1, which was partially attenuated by simultaneous overexpression of ING4, as measured by real-time RT–PCR (Fig. 6B). In contrast, JFK depletion was associated with an elevation of epithelial markers and a decrease of the mesenchymal markers in MDA-MB-231 cells and was accompanied by weakened NF-κB activity, as evidenced by decreased expression of NF-κB target genes MMP9 and VCAM1 (Fig. 6B). Collectively, these data indicate that JFK, through destabilization of ING4 protein and derepression of NF-κB activity, promotes EMT in breast cancer cells.
Figure 6.
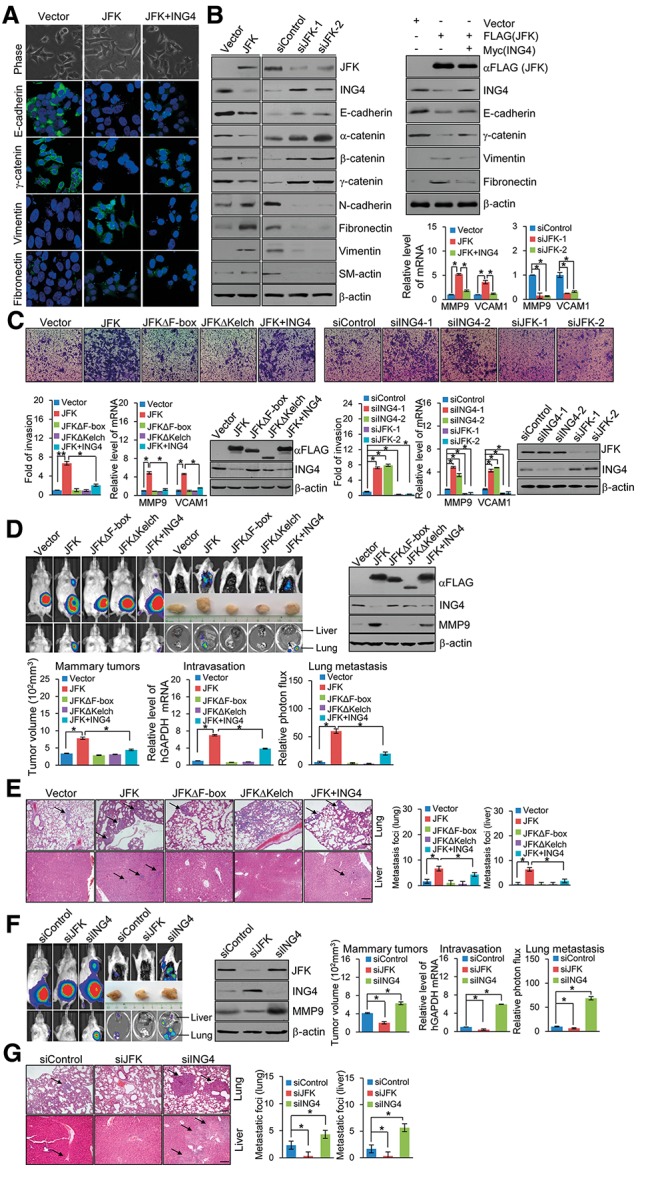
JFK promotes invasion and metastasis of breast cancer. (A) JFK-induced morphological changes in MCF-7 cells. MCF-7 cells were transfected with Flag-JFK and/or Myc-ING4. (Top) The morphological alterations of the cells were observed by phase-contrast microscopy. MCF-7 cells were transfected with Flag-JFK and/or Myc-ING4, and immunofluorescence staining of epithelial (E-cadherin and γ-catenin) and mesenchymal (vimentin and fibronectin) markers was visualized by confocal microscopy (green). (Bottom) DAPI staining was included to visualize the cell nucleus (blue). (B) Western blotting analysis of the epithelial and mesenchymal markers in MCF-7 cells transfected with Flag-JFK and/or Myc-ING4 or MDA-MB-231 cells treated with control siRNA or JFK siRNA. Expression of NF-κB target genes was measured by real-time RT–PCR. (C) MDA-MB-231 cells were infected with retroviruses carrying Flag-tagged JFK, JFKΔF-box, JFKΔKelch, and/or ING4 or lentiviruses carrying JFK siRNA or ING4 siRNA. Forty-eight hours later, cells were starved for 18 h before cell invasion assays were performed using Matrigel transwell filters. The invaded cells were stained and counted. The images represent one field under microscopy in each group. Bars indicate mean ± SD of a representative experiment performed in triplicate. P-values were determined by Student's t-test ([*] P < 0.05; [**] P < 0.01). Expression of NF-κB target genes was measured by real-time RT–PCR. The efficiency of retrovirus- or lentivirus-mediated gene expression or depletion in MDA-MB-231 cells was verified by Western blotting. (D) MDA-MB-231-Luc-D3H2LN cells were infected with retroviruses carrying Flag-tagged JFK, JFKΔF-box, JFKΔKelch, and/or ING4. These cells were inoculated into the left abdominal mammary fat pad (2 × 106 cells) of 6-wk-old immunocompromised female SCID beige mice. Tumor size was measured on day 42 (mammary tumors, n = 6). The presence of circulating tumor cells (intravasation, n = 6) was assessed by real-time RT–PCR of human GAPDH expression relative to murine β2-microglobulin in 1 mL of mouse blood perfusate. Lung and liver metastases were quantified using bioluminescence imaging after 6 wk of initial implantation, and representative in vivo bioluminescent images are shown. Bars represent mean ± SD. (n = 6). P-values were determined by Student's t-test ([*] P < 0.05). (E) Representative images of lung or liver sections stained with H&E are shown. Bar, 145 μm. (F) MDA-MB-231-Luc-D3H2LN cells were infected with lentiviruses carrying JFK siRNA or ING4 siRNA. These cells were inoculated into the left abdominal mammary fat pad (2 × 106 cells) of 6-wk-old immunocompromised female SCID beige mice. Tumor size was measured on day 42 (mammary tumors, n = 6). The presence of circulating tumor cells (intravasation, n = 6) was assessed by real-time RT–PCR of human GAPDH expression relative to murine β2-microglobulin in 1 mL of mouse blood perfusate. Lung and liver metastases were quantified using bioluminescence imaging after 6 wk of initial implantation, and representative in vivo bioluminescent images are shown. Bars represent mean ± SD. (n = 6). P-values were determined by Student's t-test ([*] P < 0.05). (G) Representative images of lung or liver sections stained with H&E are shown. Bar, 145 μm.
Western blotting analysis of the expression of endogenous ING4 and JFK in different human normal or breast cancer cell lines showed that ING4 expression was higher in the less-invasive cell lines MCF-10A, MCF-7, and T47D than in the highly invasive cell lines MDA-MB-231 and SUM1315, whereas the expression of JFK exhibited a reversed trend (Supplemental Fig. S4C), consistent with our proposition that JFK promotes the invasive potential of breast cancer cells through destabilizing ING4. To substantiate this proposition, transwell invasion assays were then performed in MDA-MB-231 stable clones infected with retroviruses carrying JFK, JFKΔF-box, JFKΔKelch, and/or ING4 or lentiviruses carrying JFK siRNA or ING4 siRNA. These experiments showed that stable expression of JFK, but not JFKΔF-box and JFKΔKelch, resulted in a more than sixfold increase in cell invasion (Fig. 6C, left), whereas JFK depletion led to a threefold decrease in cell invasion (Fig. 6C, right). Moreover, the effect of JFK overexpression on the invasive potential of MDA-MB-231 cells was probably through its negative regulation of ING4, as simultaneous overexpression of ING4 (JFK+ING4) resulted in an attenuated effect (Fig. 6C, left), and depletion of ING4 alone mimicked the effect of JFK overexpression (Fig. 6C, right). Furthermore, real-time RT–PCR measurements indicated that JFK overexpression was associated with an elevated expression of NF-κB target genes MMP9 and VCAM1, whereas JFK depletion was accompanied by a decreased expression of NF-κB target genes MMP9 and VCAM1 in MDA-MB-231 cells. The efficiency of retrovirus- or lentivirus-mediated gene expression or depletion was verified by Western blotting (Fig. 6C). Collectively, these data indicate that JFK promotes the invasive potential of breast cancer cells and does so through destabilization of ING4 protein and derepression of NF-κB activity.
JFK promotes breast cancer metastasis in vivo
To explore the role of JFK in breast cancer metastasis in vivo, bioluminescence imaging was performed using MDA-MB-231-Luc-D3H2LN cells that were engineered to stably express firefly luciferase. Cells were infected with retroviruses carrying JFK, JFKΔF-box, JFKΔKelch, and/or ING4 or lentiviruses carrying control siRNA, JFK siRNA, or ING4 siRNA and were implanted into the left abdominal mammary fat pad of immunocompromised 6-wk-old female SCID beige mice (n = 6). The growth/dissemination of tumors was monitored weekly by bioluminescence imaging with an IVIS imaging system (Xenogen Corporation). Tumor metastasis was defined as any detectable luciferase signal above background and away from the primary tumor site and was measured by quantitative bioluminescence imaging after 6 wk for orthotopically implanted groups. Breast cancer cell intravasations were also assessed by real-time RT–PCR analysis of the level of human GAPDH expression relative to that of murine β2-microglobulin in blood samples or were quantified by bioluminescence imaging. The results showed that, in mice implanted with MDA-MB-231-Luc-D3H2LN tumors overexpressing JFK, a larger size of primary tumors and enhanced cell intravasation were detected (Fig. 6D; Supplemental Fig. S5A). In these animals, spontaneous lung or liver metastases were evident (Fig. 6D,E). However, in mice implanted with MDA-MB-231-Luc-D3H2LN tumors overexpressing either JFKΔF-box or JFKΔKelch, no significant differences in primary tumor size, cell intravasation, and metastasis were detected, similar to control mice implanted with cells infected with viruses carrying empty vectors (Fig. 6D,E). In mice implanted with MDA-MB-231-Luc-D3H2LN tumors overexpressing JFK+ING4, the primary tumor size, cell intravasation, and metastasis were significantly alleviated compared with mice implanted with cells infected with viruses carrying JFK (Fig. 6D,E). Consistently, bioluminescence imaging detected smaller primary tumor sizes, reduced cell intravasations, and decreased lung or liver metastases in mice carrying JFK-depleted MDA-MB-231-Luc-D3H2LN tumors (Fig. 6F,G). The expression of JFK, ING4, and the NF-κB target gene MMP9 in primary tumors was measured and verified by Western blotting (Fig. 6D,F). In agreement with our in vitro results, a higher JFK level was associated with a lower ING4 level and an elevated MMP9 level in tumors from animals bearing JFK-overexpressing implants (Fig. 6D), whereas in tumors from animals bearing JFK-depleted implants, a higher level of ING4 and lower level of MMP9 were detected compared with tumors from control mice implanted with cells infected with viruses carrying control siRNA (Fig. 6F). In addition, MDA-MB-231-Luc-D3H2LN cells infected with retroviruses carrying vector or JFK were injected into the left ventricle of immunocompromised 6-wk-old female SCID beige mice (n = 6). Bioluminescence imaging showed no significant difference in metastasis burdens between JFK-overexpressing and control implants (Supplemental Fig. S5B). These data indicate that JFK promotes the metastasis of breast cancer and does so through destabilization of ING4 protein and derepression of NF-κB activity.
The expression of JFK is up-regulated in breast carcinomas and positively correlated with the aggressive clinical behaviors of breast cancer
To further support the role of the JFK–ING4 pathway in the invasion and metastasis of human cancers and explore the clinical significance of this pathway, a series of carcinoma samples from breast, liver, lung, renal, pancreatic, colon, esophageal, rectal, and gastric cancer patients were collected, with each type of carcinoma having at least six samples paired with the adjacent normal tissues. Tissue microarray analysis by immunohistochemical staining showed that the expression of JFK is up-regulated in breast, renal, and pancreatic carcinomas, with the highest JFK in breast ductal carcinomas (Fig. 7A). To further support the role of the JFK–ING4 axis in the progression and metastasis of breast cancer in a clinicopathologically relevant context, we collected the samples of distant organ metastasis (DOM; lung and liver) from breast cancer patients and performed immunohistochemical staining with antibodies against JFK or ING4. The intensity of the JFK or ING4 immunopositivity was scored as follows: 0 for negative, 1 for weak, 2 for moderate, and 3 for strong. Statistical analysis showed that JFK expression is negatively correlated with ING4 in DOM (Fig. 7B). In addition, in 15 paired samples of breast carcinomas and adjacent tissues, the protein levels of JFK, IL-8, and MMP9 were found to be higher in tumor tissues than in adjacent tissues, and the level of ING4 generally showed a trend inverse with that of JFK (Fig. 7C, top). Quantification revealed that the relative JFK protein level was negatively correlated with that of ING4 and positively correlated with that of IL-8 and MMP9 (Fig. 7C, bottom).
Figure 7.
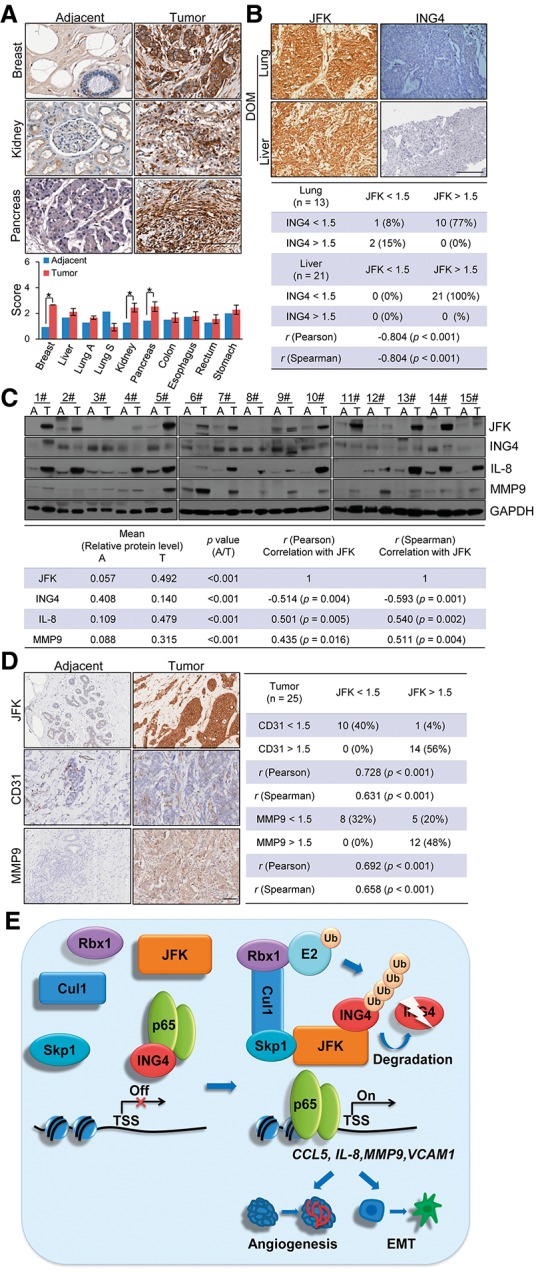
The expression of JFK is negatively correlated with that of ING4 and positively correlated with the aggressive clinical behaviors of breast cancer. (A) Immunohistochemical staining of JFK in paired samples of breast ductal carcinoma, hepatocellular carcinoma, lung adenocarcinoma (lung A), lung squamous carcinoma (lung S), suprarenal epithelioma, pancreatic ductal carcinoma, colon adenocarcinoma, esophageal squamous carcinoma, rectal adenocarcinoma, and gastric adenocarcinoma. Representative sections from tumors and adjacent normal tissues stained with JFK antibodies are shown. Bar, 200 μm. (B) The expression of ING4 and JFK in DOM samples was measured by immunohistochemical analysis. Representative images are shown. Bar, 200 μm. Statistical analysis of the intensity of the JFK or ING4 immunopositivity in DOM is shown. The staining intensity was scored as follows: (0) negative; (1) weak; (2) moderate; (3) strong. (C) Total proteins from 15 paired samples of breast carcinomas versus adjacent normal breast tissues were extracted for Western blotting analysis with antibodies against the indicated proteins. Quantitation was done by densitometry and expressed as signals of the indicated proteins to GAPDH, and the correlation between JFK and ING4, IL-8, or MMP9 was analyzed by Pearson or Spearman correlation. (D) Immunohistochemical staining of JFK, CD31, and MMP9 in 25 paired samples of normal breast tissues and breast carcinomas. Bar, 73 μm. (E) Proposed model of the JFK–ING4–NF-κB pathway in breast cancer angiogenesis and metastasis. In normal mammary cells or noninvasive breast cancer cells, ING4 is associated with p65 (the core subunit of NF-κB) to repress the canonical NF-κB pathway. During the development and progression of breast adenocarcinomas, ING4 is specifically recognized by JFK and degraded by SCFJFK complex, leading to the derepression of NF-κB signaling and promotion of angiogenesis and metastasis of breast cancer.
To support the role of the JFK–ING4 axis in the angiogenesis and metastasis of breast cancer in a clinicopathologically relevant context, we collected another 25 paired samples of breast carcinomas and adjacent tissues and performed immunohistochemical staining with antibodies against JFK, CD31 (a marker for angiogenesis) (Ozdemir et al. 2014), or MMP9. The intensity of immunopositivity was scored as follows: 0 for negative, 1 for weak, 2 for moderate, and 3 for strong. Statistical analysis revealed that high immunopositivity of JFK is associated with high levels of CD31 (Fig. 7D)—indicative of elevated vessel density (Ozdemir et al. 2014)—and high expression of MMP9 (Fig. 7D).
To support the importance of the JFK–ING4–NF-κB axis in breast cancer progression clinicopathologically, we analyzed the breast cancer data sets downloaded from Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo; GSE1456 and GSE31519). The results showed that higher JFK expression was associated with poorer overall survival in luminal A and basal-like breast cancer patients (Supplemental Fig. S6A), and higher JFK expression was also associated with poorer disease-free survival of basal-like breast cancer patients (Supplemental Fig. S6B). Moreover, the correlation analysis from the data sets also showed that the level of JFK mRNA was positively correlated with that of CCL5 and VCAM1 (Supplemental Fig. S6C), targets of the NF-κB pathway. In addition, Kaplan-Meier survival analysis of renal or pancreatic cancer data sets downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo; GSE22541 and GSE28735) also showed that a higher expression of JFK is associated with poorer disease-free survival of renal cancer but not with overall survival of pancreatic cancer (Supplemental Fig. S6D). Collectively, our observations indicate that JFK plays an important role in promoting breast cancer progression.
Discussion
The tumor suppressor ING4 is implicated in a variety of cellular processes, including cell cycle regulation, apoptosis, senescence, DNA repair, angiogenesis, and cell mobility (Unoki et al. 2009), and dysregulation of ING4 has been reported in a number of malignancies such as breast carcinoma, glioma, melanoma, and head and neck squamous cell carcinoma (Cai et al. 2009; Klironomos et al. 2010; Zhang et al. 2012). Especially in breast cancer and melanoma, down-regulation of ING4 is correlated with high-grade tumors and poor outcome of patients, implying that ING4 plays an important role in tumor initiation and metastasis (Raho et al. 2007; Kim et al. 2010). However, how the ING4 protein is regulated in a normal or tumorigenic environment is currently unknown. In this study, we report that ING4 is physically associated with JFK, the only Kelch domain-containing FBP in humans (Sun et al. 2009, 2011). We demonstrated that JFK targets ING4 for ubiquitination and degradation through the assembly of an SCF ubiquitin ligase and propose that SCFJFK is a bona fide E3 ubiquitin ligase for ING4.
The ubiquitin–proteasome system has emerged as a major regulatory principle that influences many cellular processes (Micel et al. 2013), and dysregulation in this system is also associated with various pathological outcomes, including cancers (Hoeller et al. 2006). As the best-characterized multisubunit ubiquitin ligase, SCF ubiquitin ligase-regulated protein turnover is critically involved in cell cycle progression, apoptosis, gene transcription, signal transduction, DNA replication, maintenance of genome integrity, and tumorigenesis (Ang and Wade Harper 2005; Mani and Gelmann 2005). Containing three static subunits (Skp1, Cul1, and Rbx1/2), the SCF ubiquitin ligase features a variable FBP functioning for substrate recognition. As mentioned before, of the 69 FBPs that have been found in humans, the majority are still without a substrate and a defined function (Pickart 2004; Hermand 2006). Thus, our identification of ING4 as a substrate for JFK adds to the understanding of the substrate and function of the F-box family of proteins.
Previously, we reported that SCFJFK also catalyzes ubiquitination and degradation of another tumor suppressor, p53, leading to the inhibition of cell apoptosis and promotion of cell proliferation (Sun et al. 2009, 2011). Based on the report that ING4 also directly interacts with p53 and promotes the transactivation of p53 (Shiseki et al. 2003), it is interesting to know whether JFK-directed ING4 degradation is in some way connected to p53. Our experiments in HCT116 p53+/+ and HCT116 p53−/− cells indicated that JFK-mediated ING4 degradation is not dependent on the presence of p53. In light of the positive functional relationship between ING4 and p53, it is intriguing to speculate that destabilization of both ING4 and p53 by the same SCFJFK E3 ubiquitin ligase acts as a double assurance, allowing efficient proliferation of somatic or stem cells. Alternatively, ING4 and p53 are degraded in response to different environmental stimuli and/or under different cellular micromilieus. It is also possible that JFK-directed degradation of p53 and ING4 is physiologically implemented in cell context-dependent or spatiotemporally dependent manners. Clearly, future investigations are warranted to delineate these scenarios.
Initially recognized for its essential roles in inflammation and innate immunity, NFκB is increasingly recognized as a crucial player in many steps of cancer development, including tumorigenesis, EMT, and metastasis (Karin and Greten 2005). It has been reported that NF-κB cooperates with multiple signaling pathways and forms several prominent cross-talk nodes that are mediated by other transcription factors, such as STAT3, p53, and the ETS-related gene ERG (Baeuerle and Baltimore 1996; Karin 2006). These transcription factors either directly interact with NFκB subunits or affect NFκB target genes. In addition, different signaling molecules, such as ING4, GSK3-β, p38, and PI3K, can modulate NFκB transcriptional activity or affect its upstream signaling (Hayden and Ghosh 2004; Coles et al. 2010). ING4 has been linked to the down-regulation of NF-κB signaling by acting as either an activator for IκB transcriptional activation (Coles et al. 2010) or an E3 ligase to target the RelA subunit (p65) of NF-κB for ubiquitination and degradation (Hou et al. 2014). Consistent with the negative functional relationship between ING4 and the NF-κB pathway, down-regulation of the tumor suppressor ING4 has been reported in various malignancies (Coles and Jones 2009), and hyperactivation of NFκB signaling has been observed in many types of human cancers (Basseres and Baldwin 2006). However, the molecular mechanism underlying these aberrations and the relative significance of these abnormalities in cancer development and progression are not clear. In the present study, we report that JFK-mediated ING4 destabilization leads to the up-regulation of the canonical NF-κB pathway. Through a series of in vitro and in vivo experiments, we demonstrated that JFK promotes the angiogenesis and metastasis of breast cancer and showed that it does so through destabilization of the ING4 protein and derepression of NF-κB activity. These findings place the FBP JFK upstream of the ING4–NF-κB axis, pointing to a predominant and causative role for JFK, through degradation of ING4 (possibly also p53) and hyperactivation of NF-κB, in carcinogenesis, angiogenesis, and metastasis of breast cancer. Indeed, we found that the expression of JFK is markedly up-regulated in various types of human cancer, especially in breast cancer, and JFK protein level is negatively correlated with that of ING4 and positively correlated with an aggressive clinical behavior of breast carcinomas. Furthermore, since the tumor suppressor ING4—being a native subunit of the HBO1 complex to drive H3 acetylation through binding to H3K4me3 at ING4 target promoters—has also been reported to be an epigenetic modifier (Doyon et al. 2006; Hung et al. 2009), the turnover of ING4 protein by JFK links this FBP to epigenetic regulation, and it is now abundantly clear that dysregulation of epigenetic modifiers actively contributes to carcinogenesis (Chi et al. 2010). Although the precise cellular activity of ING4 is out of the scope of our present investigation, the destabilization of the tumor suppressor ING4 establishes JFK as a potent promoter of angiogenesis and metastasis of breast cancer. In support of this notion, a recent study proposed that JFK (FBXO42) is a potential target for therapeutic intervention of high-risk triple-negative breast cancers (Lee et al. 2013).
The functional relevance and significance of the proteins that were also copurified with ING4, including protein kinase LATS2 and WDR77, remain to be investigated. It is also important to investigate the scope and variety of JFK function in terms of both substrate and cell context. Perhaps more relevant to our current work, the proteasome inhibitor bortezomib has been approved by the Food and Drug Administration and effectively used in the treatment of multiple myeloma (Lawasut et al. 2012). Meanwhile, a small molecule inhibitor of the NEDD8-activating enzyme (MLN4924) has been developed as a more specific drug to inactivate the cullin-based SCF ubiquitin ligases (Tanaka et al. 2012). Molecules that specifically inhibit or block the activity of JFK or the interaction of JFK with its substrates, such as JFKΔF-box, will potentially be more effective and safer for breast cancer intervention.
In conclusion, our study identified SCFJFK as a bona fide E3 ubiquitin ligase for ING4 and demonstrated the JFK–ING4–NF-κB axis as a potent promoter in the angiogenesis and metastasis of breast cancer, supporting the pursuit of JFK as a potential target for breast cancer intervention.
Materials and methods
Patients and specimens
The samples of carcinomas and the adjacent normal tissues were obtained from surgical specimens from patients with breast, liver, lung, renal, pancreatic, colon, esophageal, rectal, or gastric cancer. DOM (lung and liver) was procured from surgical specimens from patients with breast cancer. Samples were selected from patients for whom complete information on clinicopathological characteristics was available. All studies were approved by the Ethics Committee of the Peking University Health Science Center, and informed consent was obtained from all patients.
Statistical analysis
Results are reported as mean ± SD unless otherwise noted. SPSS version 13.0 was used for statistical analysis. Comparisons between cancers and adjacent normal tissues were performed using paired samples t-test based on a bidirectional hypothesis for continuous variables or using Pearson or Spearman correlation. Breast, renal, and pancreatic data sets were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo): GSE1456, GSE31519, GSE28735, and GSE22541.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (91219201 and 81130048 to Y.S., and 81372223 and 81422034 to L.S.) and the Ministry of Science and Technology of China (973 Program 2011CB504204 to Y.S., and 2014CB542004 to J.L.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.254292.114.
References
- Ang XL, Wade Harper J. 2005. SCF-mediated protein degradation and cell cycle control. Oncogene 24: 2860–2870. [DOI] [PubMed] [Google Scholar]
- Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, Kleinman HK, Reaman GH, Tosato G. 1995. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med 182: 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. 1996. NF-κB: ten years after. Cell 87: 13–20. [DOI] [PubMed] [Google Scholar]
- Basseres DS, Baldwin AS. 2006. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 25: 6817–6830. [DOI] [PubMed] [Google Scholar]
- Byron SA, Min E, Thal TS, Hostetter G, Watanabe AT, Azorsa DO, Little TH, Tapia C, Kim S. 2012. Negative regulation of NF-κB by the ING4 tumor suppressor in breast cancer. PLoS One 7: e46823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Li X, Zheng S, Wang Y, Li H, Yang J, Sun J. 2009. Inhibitor of growth 4 is involved in melanomagenesis and induces growth suppression and apoptosis in melanoma cell line M14. Melanoma Res 19: 1–7. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Pagano M. 2004. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 5: 739–751. [DOI] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. 2010. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10: 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles AH, Jones SN. 2009. The ING gene family in the regulation of cell growth and tumorigenesis. J Cell Physiol 218: 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles AH, Gannon H, Cerny A, Kurt-Jones E, Jones SN. 2010. Inhibitor of growth-4 promotes IκB promoter activation to suppress NF-κB signaling and innate immunity. Proc Natl Acad Sci 107: 11423–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, Lane WS, Tan S, Yang XJ, Cote J. 2006. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 21: 51–64. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Matsumoto A, Inuzuka H, Zhai B, Lau AW, Wan L, Gao D, Shaik S, Yuan M, Gygi SP, et al. 2012. SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep 1: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, Barnett GH, Jain RK. 2004. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature 428: 328–332. [DOI] [PubMed] [Google Scholar]
- Grant DS, Kleinman HK, Leblond CP, Inoue S, Chung AE, Martin GR. 1985. The basement-membrane-like matrix of the mouse EHS tumor: II. Immunohistochemical quantitation of six of its components. Am J Anat 174: 387–398. [DOI] [PubMed] [Google Scholar]
- Hager-Theodorides AL, Furmanski AL, Ross SE, Outram SV, Rowbotham NJ, Crompton T. 2009. The Gli3 transcription factor expressed in the thymus stroma controls thymocyte negative selection via Hedgehog-dependent and -independent mechanisms. J Immunol 183: 3023–3032. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. 2004. Signaling to NF-κB. Genes Dev 18: 2195–2224. [DOI] [PubMed] [Google Scholar]
- He GH, Helbing CC, Wagner MJ, Sensen CW, Riabowol K. 2005. Phylogenetic analysis of the ING family of PHD finger proteins. Mol Biol Evol 22: 104–116. [DOI] [PubMed] [Google Scholar]
- Hermand D. 2006. F-box proteins: more than baits for the SCF? Cell Div 1: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu Rev Biochem 67: 425–479. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Hecker CM, Dikic I. 2006. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer 6: 776–788. [DOI] [PubMed] [Google Scholar]
- Hou Y, Zhang Z, Xu Q, Wang H, Xu Y, Chen K. 2014. Inhibitor of growth 4 induces NFκB/p65 ubiquitin-dependent degradation. Oncogene 33: 1997–2003. [DOI] [PubMed] [Google Scholar]
- Hung T, Binda O, Champagne KS, Kuo AJ, Johnson K, Chang HY, Simon MD, Kutateladze TG, Gozani O. 2009. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol Cell 33: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. 2006. Nuclear factor-κB in cancer development and progression. Nature 441: 431–436. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. 2005. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5: 749–759. [DOI] [PubMed] [Google Scholar]
- Kim S, Welm AL, Bishop JM. 2010. A dominant mutant allele of the ING4 tumor suppressor found in human cancer cells exacerbates MYC-initiated mouse mammary tumorigenesis. Cancer Res 70: 5155–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klironomos G, Bravou V, Papachristou DJ, Gatzounis G, Varakis J, Parassi E, Repanti M, Papadaki H. 2010. Loss of inhibitor of growth (ING-4) is implicated in the pathogenesis and progression of human astrocytomas. Brain Pathol 20: 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. 2001. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294: 173–177. [DOI] [PubMed] [Google Scholar]
- Lalonde ME, Avvakumov N, Glass KC, Joncas FH, Saksouk N, Holliday M, Paquet E, Yan K, Tong Q, Klein BJ, et al. 2013. Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev 27: 2009–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawasut P, Chauhan D, Laubach J, Hayes C, Fabre C, Maglio M, Mitsiades C, Hideshima T, Anderson KC, Richardson PG. 2012. New proteasome inhibitors in myeloma. Curr Hematol Malig Rep 7: 258–266. [DOI] [PubMed] [Google Scholar]
- Lechner E, Achard P, Vansiri A, Potuschak T, Genschik P. 2006. F-box proteins everywhere. Curr Opin Plant Biol 9: 631–638. [DOI] [PubMed] [Google Scholar]
- Lee U, Frankenberger C, Yun J, Bevilacqua E, Caldas C, Chin SF, Rueda OM, Reinitz J, Rosner MR. 2013. A prognostic gene signature for metastasis-free survival of triple negative breast cancer patients. PLoS One 8: e82125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani A, Gelmann EP. 2005. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol 23: 4776–4789. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. 2008. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 133: 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micel LN, Tentler JJ, Smith PG, Eckhardt GS. 2013. Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J Clin Oncol 31: 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, et al. 2000. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 19: 2069–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Hatakeyama S, Maruyama S, Kikuchi A, Onoe K, Good RA, Nakayama KI. 2003. Impaired degradation of inhibitory subunit of NF-κB (IκB) and β-catenin as a result of targeted disruption of the β-TrCP1 gene. Proc Natl Acad Sci 100: 8752–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozell S, Laver T, Moseley D, Nowoslawski L, De Vos M, Atkinson GP, Harrison K, Nabors LB, Benveniste EN. 2008. The ING4 tumor suppressor attenuates NF-κB activity at the promoters of target genes. Mol Cell Biol 28: 6632–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson N, Kasahara DI, Hristova M, Bernstein R, Janssen-Heininger Y, van der Vliet A. 2011. Modulation of NF-κB and hypoxia-inducible factor-1 by S-nitrosoglutathione does not alter allergic airway inflammation in mice. Am J Respir Cell Mol Biol 44: 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. 2014. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. 2006. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7: 644–656. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. 2005. Function and regulation of cullin–RING ubiquitin ligases. Nat Rev Mol Cell Biol 6: 9–20. [DOI] [PubMed] [Google Scholar]
- Pickart CM. 2004. Back to the future with ubiquitin. Cell 116: 181–190. [DOI] [PubMed] [Google Scholar]
- Raho G, Miranda C, Tamborini E, Pierotti MA, Greco A. 2007. Detection of novel mRNA splice variants of human ING4 tumor suppressor gene. Oncogene 26: 5247–5257. [DOI] [PubMed] [Google Scholar]
- Shiseki M, Nagashima M, Pedeux RM, Kitahama-Shiseki M, Miura K, Okamura S, Onogi H, Higashimoto Y, Appella E, Yokota J, et al. 2003. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res 63: 2373–2378. [PubMed] [Google Scholar]
- Sun L, Shi L, Li W, Yu W, Liang J, Zhang H, Yang X, Wang Y, Li R, Yao X, et al. 2009. JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proc Natl Acad Sci 106: 10195–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Shi L, Wang F, Huangyang P, Si W, Yang J, Yao Z, Shang Y. 2011. Substrate phosphorylation and feedback regulation in JFK-promoted p53 destabilization. J Biol Chem 286: 4226–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Nakatani T, Kamitani T. 2012. Negative regulation of NEDD8 conjugation pathway by novel molecules and agents for anticancer therapy. Curr Pharm Des 19: 4131–4139. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. 2000. Recognition of the polyubiquitin proteolytic signal. EMBO J 19: 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki M, Kumamoto K, Takenoshita S, Harris CC. 2009. Reviewing the current classification of inhibitor of growth family proteins. Cancer Sci 100: 1173–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang Y, Zhang F, Zhang Q. 2012. Correlation between tumor suppressor inhibitor of growth family member 4 expression and microvessel density in breast cancer. Hum Pathol 43: 1611–1617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.