

1. Introduction
In 1963, Chambon et al. reported the detection of a nicotinamide mononucleotide-activated, DNA-dependent enzymatic activity in rat liver extracts that catalyzed the synthesis of a polyadenylic acid.1 The product of this reaction was later identified as poly(ADP-ribose) or PAR, a polymer of ADP-ribose (ADPR) monomers derived from the oxidized form of nicotinamide adenine dinucleotide (NAD+).2 These initial studies have led to half a century of research on the chemistry, enzymology, structure, function, biology, physiology, and pathology of ADPR, PAR, and their derivatives, as well as the enzymes that catalyze their synthesis and degradation, and the effector proteins that interact with or are posttranslationally modified by them. In this Review, we describe the biological chemistry of PAR and its associated enzymes, effector proteins, and targets, with a particular emphasis on their roles in gene regulation, from chromatin to RNA biology.
1.1. The PARP Family
The synthesis of PAR from NAD+ is catalyzed by poly(ADP-ribose) polymerase (PARP) enzymes belonging to the PARP family (EC 2.4.2.30), which contains at least 17 distinct proteins (Table 1).3 Not all PARP family members are enzymatically active, and some may function as mono(ADP-ribosyl)transferases rather than PARPs.4 As a consequence, a new nomenclature describing PARPs more accurately as ADP-ribosyltransferases (ARTs) has been proposed.5 The 17 PARP family members can be subdivided into four subfamilies based on their domain architectures (Table 1).3 These include: (1) DNA-dependent PARPs (PARP-1, PARP-2, and PARP-3), which are activated by discontinuous DNA structures (for PARPs 1 and 2, through their amino-terminal DNA binding domains) (Figure 1); (2) tankyrases, including PARP5a (tankyrase-1) and PARP-5b (tankyrase-2), which contain large ankyrin domain repeats that mediate protein–protein interactions; (3) CCCH PARPs, including PARP-7 (tiPARP), PARP-12, PARP13.1, and PARP13.2, which contain Cys-Cys-Cys-His zinc fingers that bind to RNA, as well as WWE domains, which can exhibit PAR binding activity; and (4) macroPARPs, including PARP-9 (BAL1), PARP14 (BAL2, CoaSt6), and PARP-15 (BAL3), which contain macrodomain folds that can bind ADPR and derivatives. As these examples illustrate, nature through the course of evolution has modified the PARP catalytic domain and functionalized it with a variety of other protein domains to create a set of proteins with varied activities, subcellular locations, and functions.
Table 1. List of PARP Family Members, Highlighting Those PARPs Discussed in This Review.
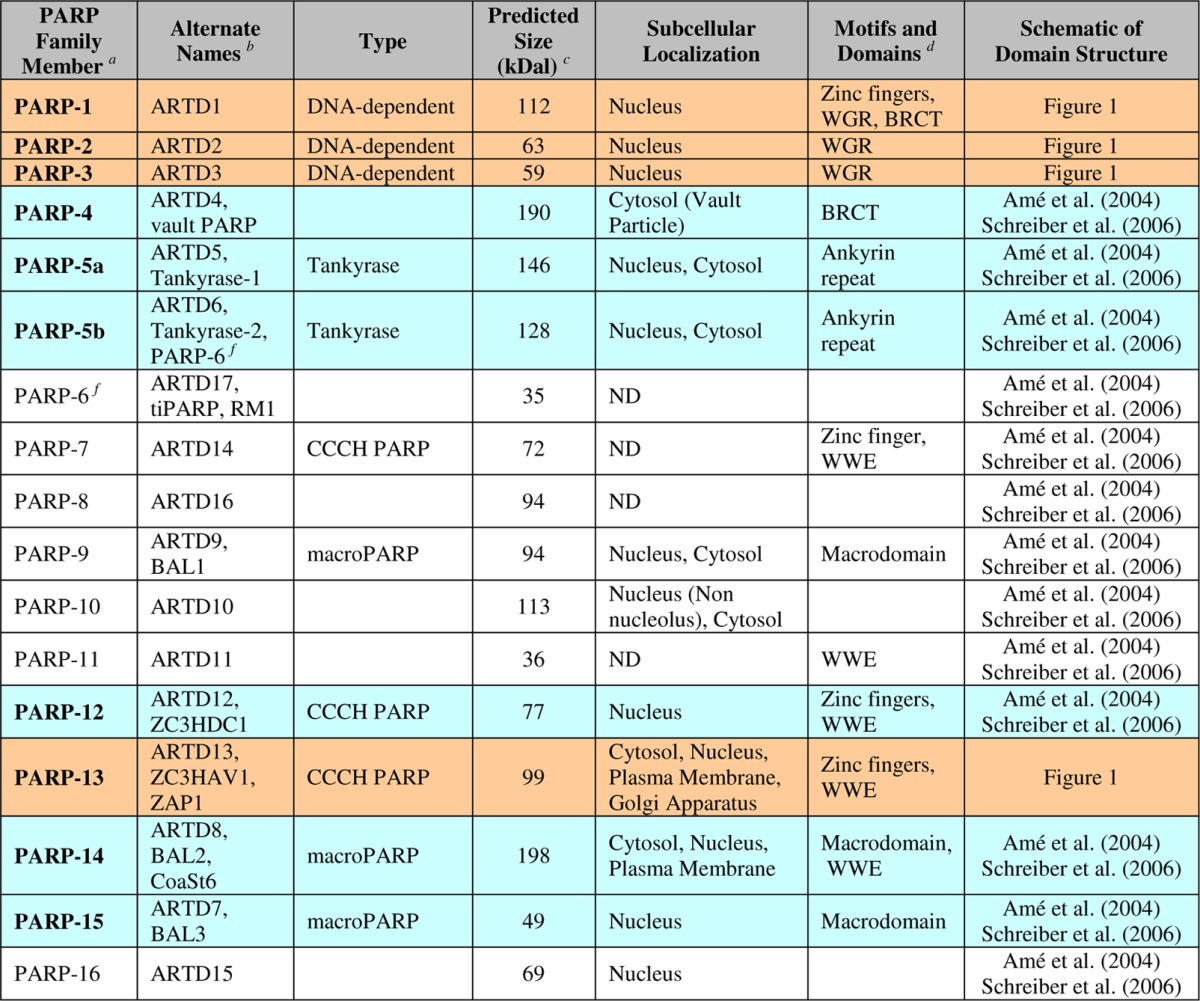
PARP family members listed in bold are discussed in this Review. The four PARPs highlighted in orange are the primary focus, while the six PARPs highlighted in blue are the secondary focus.
The “ARTD” names are based on the revised nomenclature of Hottiger et al., 2010.5
Calculated on the basis of the number of amino acids in each protein.
All PARP family members contain a PARP domain and the PARP signature motif.
Readers are directed to Amé et al., 2004,3a and Schreiber et al., 2006,3b for schematics of the PARP family members not highlighted in this Review.
PARP-6 refers to two different proteins in the literature: PARP-5b/ARTD6/Tankyrase 2 and ARTD17.
Figure 1.

Structural and functional organization of nuclear DNA-dependent PARPs, as well as PARP-13. PARPs 1, 2, and 3 comprise a subset of nuclear PARPs whose catalytic activity is stimulated by discontinuous DNA structures. In the case of PARPs 1 and 2, this activation by DNA occurs through their N-terminal DNA binding domains. Unlike PARPs 1 and 2, PARP-3 does not have a well-defined DNA-binding domain, but it can interact with chromatin and bind to DNA in vitro.210,215 PARPs 1 and 2 are poly(ADP-ribosyl) transferases, while PARP-3 is a mono(ADP-ribosyl) transferase. PARP-13.1 and PARP-13.2 contain Cys-Cys-Cys-His zinc fingers that bind to RNA, as well as WWE domains, which can bind to PAR. PARP-13.1 contains a PARP domain with an H–Y–V motif instead of the H–Y–E catalytic triad motif found in many enzymatically active PARPs, such as PARPs 1, 2, and 3, and is thus catalytically inactive. PARP-13.2 is a truncated version of PARP-13.1 lacking the PARP homology region. Abbreviations as are follows: Zn = zinc binding domains; NLS = nuclear localization signal; NES = nuclear export signal; LZ = leucine zipper motif (thought to function as a protein–protein interaction motif); BRCT = BRCA1 C-terminal motif (thought to function as a phosphopeptide binding motif); WGR = tryptophan-glycine-arginine-containing motif (may function as a nucleic acid binding motif); WWE = tryptophan-tryptophan-glutamate-containing motif (functions as a PAR binding motif); H–Y–E and H–Y–V, H = histidine, Y = tyrosine, E = glutamate, V = valine.
PARP-1 (ARDT1) is the prototypical and founding member of the PARP family (Table 1). It is a 116 kDa protein containing a set of well-characterized structural and functional domains (Figure 1, top).3 These include (from the amino to carboxyl termini of the protein): (1) an amino-terminal DNA binding domain containing two zinc finger motifs, a zinc binding domain, and a nuclear localization signal (NLS); (2) an automodification domain containing a BRCA1 C-terminus (BRCT) motif (although sites of automodification outside of the historical automodification domain have now been identified6); (3) a WGR (Trp-Gly-Arg) motif; and (4) a carboxyl-terminal catalytic domain containing the highly conserved PARP signature motif, which forms the NAD+ binding site and defines the PARP family of proteins. This collection of structural and functional domains comprises a ubiquitous and abundant protein that is ideally suited to carry out a wide variety of functions in the nucleus.7 Many of the functional studies of PARP family members to date have been done with PARP-1. As such, many of the examples herein are derived from the PARP-1 literature.
The catalytic activities of the DNA-dependent PARPs (PARPs 1, 2, and 3) are activated by discontinuous DNA structures. Although PARPs 1, 2, and 3 have in common carboxyl-terminal WGR and catalytic domains, their amino-termini are quite different, with PARP-1 containing a large zinc finger-containing extension (Figure 1). These structural differences suggest potential differences in function, including differences in specificity for DNA-dependent activation. Indeed, Langelier et al. have shown that PARP-2 and PARP-3 are preferentially activated by DNA breaks harboring a 5′ phosphate, suggesting that different DNA repair intermediates may drive the activities of these enzymes as compared to PARP-1.8 Unlike PARP-1, the amino-termini of PARP-2 and PARP-3 are not required for DNA binding or DNA-dependent activation, yet all three PARPs share an allosteric regulatory mechanism of DNA-dependent catalytic activation through a local destabilization of the catalytic domain.8
1.2. Overview of the Molecular, Cellular, and Biological Functions of PAR and the PARP Family
PARP family members exhibit a wide array of subcellular distributions and expression patterns, suggesting a broad and varied biology for this family.4,9 Although some PARP-mediated cellular responses may be independent of their catalytic activity, many of the best-characterized actions of PARP family members require PAR production and may involve distinct PAR-binding modules present in key regulatory proteins (see below). The functions of PARPs can be understood at (1) the molecular level, relating to the chemical biology of PAR, (2) the cellular level, relating to the cellular processes that they control, and (3) the biological level, relating to the physiological and pathological processes in which they play key roles (Figure 2). With respect to the first, three general types of regulatory mechanisms have been ascribed to PAR: inhibition of protein–protein interactions, formation of interaction scaffolds, and regulation of ubiquitylation.10 With respect to the second, PARPs have been shown to function as regulatory proteins in a wide array of cellular processes, from transcription and DNA repair (Figure 3), to mitochondrial function and the formation of suborganellar bodies.4,10,11 With respect to the third, PARPs have been shown to function as key components of stress responses, as well as other critical homeostatic mechanisms (Figure 2).7a,12 Given the dependence of PARP catalytic activity on NAD+, the functions of the PARP family members may be physically and functionally linked to cellular metabolic processes and the enzymes that control them.13 For the purposes of this Review, we have focused on the role of PARPs in gene regulation, with a particular emphasis on the newest area of study: the role of PARPs in RNA biology.
Figure 2.
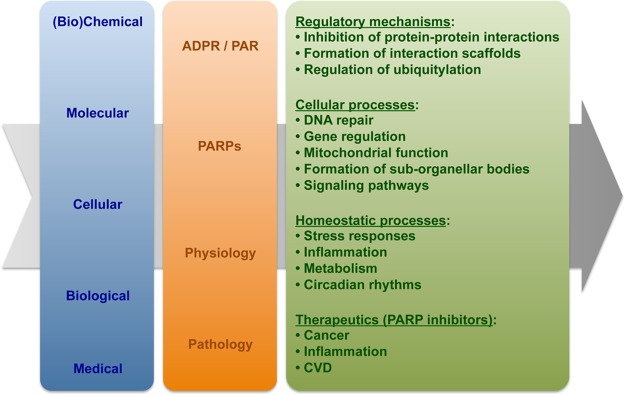
Overview of the molecular, cellular, and biological functions of PAR and the PARP family. This schematic relates the functions of PAR and PARPs to biological outcomes. Details are provided in the text.
Figure 3.
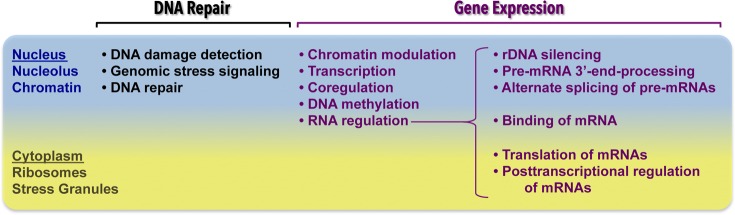
Overview of key molecular functions of PARP-1 (DNA repair and gene regulation), with an emphasis on the new biology of PARPs in the regulation of RNA. PARP family members function in the nucleus to control DNA repair and gene expression, as well as the nucleus and cytoplasm to regulate RNA. Details are provided in the text.
2. NAD+ Metabolism and PARP-1
2.1. NAD+ Biosynthesis Pathways
The cellular NAD+ biosynthetic pathways supply NAD+ for use by PARP-1 and other PARP family members. Thus, the means by which NAD+ is synthesized and consumed is relevant to our understanding of PARP function and biology.
2.1.1. NAD+ as a Signaling Molecule
More than a century ago, NAD+ was first discovered as a cofactor in fermentation.14 Subsequent years of study have revealed it to be a universal energy-carrying molecule that acts as a cofactor in multiple cellular redox reactions. In these reactions, this pyridine nucleotide is reversibly oxidized (NAD+) or reduced (NADH) by various oxidoreductases, yet the total pool remains unaltered.
A novel aspect of NAD+ as a signaling molecule has emerged more recently, with the identification of NAD+-dependent enzymes, such as PARPs and sirtuins (SIRTs). These enzymes use NAD+ as a substrate to catalyze their respective enzymatic reactions. However, unlike oxidoreductases, NAD+-dependent enzymes irreversibly degrade NAD+, which can lead to the depletion of cellular NAD+ contents.15 Thus, the regeneration and maintenance of nuclear NAD+ is crucial for maintaining cellular signaling function.
2.1.2. NAD+ Biosynthesis Pathways
NAD+ can be synthesized from L-tryptophan via the de novo pathway or from other nucleotides via the salvage pathway (Figure 4).15 In the de novo pathway, tryptophan is converted to quinolinic acid, which is subsequently processed by quinolinate phosphoribosyltransferase (QPRT) to form nicotinic acid mononucleotide (NaMN), a pyridine mononucleotide precursor for NAD+.16 Alternatively, the salvage pathway utilizes the nucleobases, nicotinic acid (NA) and nicotinamide (NAM), and nucleosides, nicotinamide riboside (NR) and nicotinic acid riboside (NAR), to generate a pyridine mononucleotide.15 Nicotinic acid phosphoribosyltransferase (NAPRT) converts NA to NaMN, while nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide riboside kinases (NRKs) use NAM and NR, respectively, to generate nicotinamide mononucleotide (NMN), another pyridine mononucleotide precursor for NAD+.15
Figure 4.

NAD+ biosynthetic pathways. In mammals, NAD+ can be synthesized de novo from L-tryptophan (the de novo pathway) or from other nucleotides such as nucleobases or nucleosides (the salvage pathway).
The only enzyme shared by both NAD+ synthesis pathways is nicotinamide mononucleotide adenylyl transferase (NMNAT), which condenses ATP and a pyridine mononucleotide, NMN or NaMN, to generate pyridine dinucleotide NAD+ or NaAD+, respectively, and NaAD+ is further catalyzed by NAD synthetase (NADS) to produce NAD+.17 Given that NMNAT is the only enzyme known to catalyze such reactions and is the only common enzyme between both the de novo and salvage pathways, NMNAT is considered an indispensible enzyme for NAD+ biosynthesis. NAMPT acts in direct support of the NMNATs in the salvage pathway and may catalyze the rate-limiting step in NAD+ biosynthesis.18
2.1.3. NMNATs
The enzymatic activity of NMNATs was first discovered in the 1950s by Arthur Kornberg, who detected an enzymatic activity in yeast extracts that catalyzes the synthesis of NAD+ from NMN and ATP.19 Additional studies revealed that NMNAT transfers the adenylyl moiety from ATP to NMN or NaMN and releases pyrophosphate and also catalyzes a reversible reaction through its NAD+ pyrophosphorylase activity.20 In vitro, equilibrium favors the reverse reaction,21 but under physiological conditions, NAD+ synthesis is preferred, possibly due to abundant ATP levels and limited pyrophosphate concentrations.
Human NMNAT (hNMNAT) belongs to the nucleotidyl transferase superfamily and exists as three isoforms (NMNAT-1, NMNAT-2, and NMNAT-3), which are numbered according to the order in which they were cloned.22 These NMNAT isoforms exhibit different tissue expression patterns and, more interestingly, unique subcellular localizations: NMNAT-1 in the nucleus,21,23 NMNAT-2 in the cytosol and Golgi,21,22c and NMNAT-3 in the cytosol and mitochondria.21,22c Although evidence to support the contention that cells have a compartmentalized pool of NAD+ is limited, mainly due to the lack of in vivo NAD+ detection methods, the unique subcellular localization of NMNATs suggests that NAD+ biosynthesis is also compartmentalized within the cell. Indeed, overexpression of hNMNAT-1 in MCF-7 cells increases NAD(P)H levels in the nucleus, while having little effect on the cytosolic and mitochondrial pools of NAD(P)H.13b Additionally, depletion of NMNAT-1, but not NMNAT-2 or NMNAT-3, in primary myoblasts reduces the expression of mitochondrially encoded OXPHOS genes, mitochondrial DNA content, and ATP levels.24 Moreover, Nmnat1 null mice have been reported to be embryonic lethal,25 indicating that the cytosolic or mitochondrial pool of NAD+ cannot compensate for the loss of nuclear NAD+ production during embryonic development. These data support the possibility that NAD+ is compartmentalized; however, additional studies are needed, and a key step in determining the compartmentalization of NAD+ will be to establish technologies that allow direct detection of NAD+.
Structural analyses have revealed clearer insight into how hNMNAT utilizes pyridine mononucleotides and ATP to generate NAD+. Three independent groups solved crystal structures of hNMNAT-1 and found the enzyme to be a homohexameric protein (Figure 5A).26 Each monomer contains six parallel β-sheets flanked by α-helices containing the mononucleotide-binding motif.26 Ligand binding of hNMNAT-1 is mediated by a number of conserved amino acids. Trp169 (conserved in all NMNAT sequences) stacks against the pyridine ring of nicotinamide, and Trp92 (conserved among the human NMNATs) interacts with the pyridine ring in face-to-edge fashion on the other side.26a,26c Trp92, together with Glu94, also interacts with the ribose oxygen, while Ser16 and Lys57 contact the ribose phosphate.26c An interesting feature of the hNMNAT-1 active site as compared to bacterial and archaeal NMNAT is the presence of structural water molecules (ω), which together with Asp173 can subtly change the electrostatic distribution within the substrate-binding site, allowing hNMNAT-1 to bind NMN and NaMN without conformational changes.26a
Figure 5.

Mechanisms of NAD+ synthesis and utilization. (A) NAD+ synthesis by NMNAT-1. NMN and ATP bind to the mononucleotide-binding motif of NMNAT-1, which catalyzes the generation of NAD+. The crystal structure of hNMNAT-1 bound by NAD+ reveals conserved amino acid residues (W169, W92, and E94 in cyan, S16 and K57 in purple) that mediate its catalytic reaction (PDB: 1KQN).26a (B) Structure of hPARP-1 bound to DNA (PDB: 4DQY).216 PARP-1 utilizes NAD+ to generate PAR polymers. NAD+ (left, shown in light brown) was positioned into the catalytic domain of hPARP-1 bound by carba-NAD (cNAD; PDB: 1A26)30 by homology modeling with NAD+ bound diphtheria toxin (PDB: 1TOX). The triad of conserved residues, H–Y–E (H862, Y896, and E988, shown in green), is required for NAD+ binding. ADP from cNAD (right, shown in blue) represents the terminal region of the PAR chain. Conserved residues from acceptor sites (purple) interact with ADP from the terminal ADP-ribose.
2.2. NAD+ Utilization and Synthesis of Poly(ADP-ribose) (PAR) Polymer by PARP-1
2.2.1. NAD+ Binding and PAR Synthesis
PARP proteins utilize NAD+ as a donor of ADP-ribose units and transfer these units to their target proteins. ADP-ribose transfer occurs at the catalytic domain of PARPs, which contains a donor site with a PARP signature motif that binds NAD+ and an acceptor site where ADP-ribose chains are extended.27 While the crystal structure of PARP-1 bound to NAD+ has yet to be determined, structural homology modeling of the PARP-1 catalytic domain with NAD+ bound diphtheria toxin,28 as well as in silico characterization of PARPs,29 suggest that the conserved His, Tyr, and Glu residues are important for ligand binding (Figure 5B). This conserved “H–Y–E” triad has been predicted to be critical for the positioning of NAD+ during ADP-ribosylation by stacking with the nicotinamide ring of NAD+ (Tyr896) and forming hydrogen bonds with the hydroxyl group of adenine ribose and nicotinamide ribose (His862 and Glu988, respectively).27 Glu988 also forms hydrogen bonds with the hydroxyl group of the acceptor ribose (at the end of the PAR chain), which is a critical residue for adding new ADP-ribose units onto the acceptor ribose.10 Extension of ADP-ribose is further mediated by conserved residues residing in the acceptor site of the catalytic domain of PARPs, such as His826, Lys903, Tyr907, Met890. Site-directed mutagenesis of these conserved residues significantly reduces PARP-1 poly(ADP-ribosyl)ation (PARylation) activity while maintaining mono-ADP-ribosylation activity.30
During ADP-ribose transfer reactions, one molecule of NAD+ is catabolized to generate ADP-ribose and nicotinamide. The ADP-ribose unit is then transferred to the Lys, Glu, and Asp residues of target proteins, releasing nicotinamide as a byproduct of the reaction. The ADP-ribose chain can grow by up to 200 units by repeated attachment of an ADP-ribose unit to the adenine-proximal ribose unit through an α (1–2) O-glycosidic bond at the end of the PAR chain (elongation) (Figure 6A). In addition to linear extension, the PAR polymer can also form branches every 20–50 ADP-ribose units by forming an α (1–2) glycosidic bond between two nicotinamide-proximal riboses (Figure 6A).31 Each PAR residue contains an adenine moiety with two phosphate groups that carry negative charges. Because of diverse elongation and branching processes, PAR forms strongly negatively charged heterogeneous polymers both in vitro and in vivo, although the significance of this heterogeneity has remained elusive.
Figure 6.

The structure of PAR and the mechanism of PAR action. (A) Chemical structure of PAR illustrating elongation of the PAR chain by an α(1–2) O-glycosidic bond between riboses (elongation, orange line) and branching of the PAR chain by an α(1–2) glycosidic bond between two nicotinamide-proximal riboses (branching, blue line). (B) PARylation of a protein inhibits protein–protein or protein–nucleic acid interactions by masking interaction sites or introducing charge repulsion with strongly negatively charged polymers.10 (C) PAR recruits PAR-binding protein to its sites of action, serving as an interaction scaffold.10 (D) PARylation of PARP-1 triggers recruitment of the E3 ligase RNF146, which contains a PAR-binding WWE domain and brings about subsequent ubiquitylation and proteasome-mediated degradation of the target protein.51a,53
Despite our understanding of how PAR is synthesized, many questions remain about the direct proteins targets of PARylation (the “PARylome”), versus those that bind to PAR. Proteomics can be an effective tool for resolving these issues.6a,6c,32 Nonetheless, determining the specific protein targets of each individual PARP remains challenging because all enzymatically active PARPs (1) use the same substrate, NAD+, and (2) produce the same product, mono(ADP-ribose) or chains of ADP-ribose, which are indistinguishable by mass spectrometry. A breakthrough approach to identifying PARP-1 targets proteome-wide has been recently reported by Carter-O’Connell et al. using PARP-1 and PARP-2 mutants that bind a “clickable” NAD+ analog, followed by copper-catalyzed conjugation to an azidoalkyl reporter (“click” chemistry) and tandem mass spectrometry (Figure 7).33 In this study, a conserved lysine residue in the catalytic domain of PARP-1 (K903), or a homologous residue in PARP-2, was mutated to alanine, and 5-ethyl-6-alkyne-NAD+ was used as a substrate. Only the PARP mutants, but not their wild-type counterparts or other wild-type PARPs, can use the NAD+ analog, leading to the identification of specific targets for specific PARPs (i.e., PARP-1 and PARP-2).33 However, the PARP-1 K903A mutant catalyzes only mono-ADP ribosylation, rather than PARylation, which may affect the faithful identification of PARP-1 targets. Therefore, improved methodologies will be required to overcome this limitation.
Figure 7.

Strategy for defining proteome-wide PARP-1-specific targets. NAD+ analogue (5-Et-6-a-NAD+) and PARP-1 mutant (K903A) were used to identify specific targets for PARP-1.33 Ethyl group and alkyne tags were added to the C-5 position of the nicotinamide ring and the N-6 position of adenosine ring, respectively, to generate the NAD+ analogue, which can only be utilized by the K903A PARP-1 mutant and not by wild-type PARP-1 or other PARP family members. Following conjugation with biotin azide and subsequent enrichment of biotinylated proteins, samples were subjected to LC–MS/MS to identify the proteome-wide targets of PARP-1. (A) NAD+ analog with wild-type PARP-1 (WT). (B) NAD+ analog with analog-sensitive mutant PARP-1 (K903A).
2.2.2. Mechanisms of PAR Action
The synthesis of a long, negatively charged polymer affects a wide array of biological processes through various mechanisms. PARylation of the protein alters its interaction with other binding partners, including proteins and nucleic acids (Figure 6B). For example, PARP-1 PARylates the chromatin remodeling factor FACT upon DNA damage and disrupts FACT–nucleosome interactions as well as FACT-mediated H2A/H2AX exchange.34 PARP-1 also modifies the ATP-dependent chromatin remodeler ISWI and histone demethylases KDM5B and KDM4D by reducing their binding to the nucleosome.35 PARP-1-dependent PARylation has also been shown to regulate many transcription factors in a similar manner, including, but not limited to, Sp1,36 NFAT,37 Sox2,38 Smad,39 and CLOCK40 as well as nuclear receptors such as farnesoid X receptors (FXR)41 and estrogen receptor (ER)42 (see below). A plausible explanation for these effects could be that PARP-1-dependent PARylation masks protein–protein interaction sites or introduces charge repulsion with strongly negatively charged polymers.
Many proteins have also been shown to bind noncovalently to PAR through PAR-binding domains or motifs.43 In this regard, PAR may act as a scaffold to recruit regulatory proteins (Figure 6C). The mechanism of scaffold function is well studied in the DNA damage response process. Upon laser-induced DNA damage, PARP-1 is rapidly activated, and PAR is accumulated at the site of damage (within seconds), followed by recruitment of scaffold proteins such as XRCC1,44 which preferentially binds to PARylated PARP-1,44a and chemical inhibition of PAR synthesis or genetic depletion of PARP-1 abolishes XRCC1 recruitment.44 Additionally, PAR polymers direct polycomb complexes and the nucleosome remodeling and histone deacetylase (NuRD) complex to DNA damage sites. Recruitment of these complexes deposits repressive histone marks on the chromatin, allowing a transient repressive chromatin structure at the site of DNA damage that blocks transcription and facilitates DNA repair.45
Recent studies have led to the further identification of PAR-dependent recruitment of proteins to their sites of action, mediated by PAR-binding domains (Figure 8). CHFR (checkpoint protein with FHA and RING domains) and APLF (aprataxin PNK-like factor) both contain the PAR-binding C2H2 zinc finger motif (PBZ) and are recruited in a PAR-dependent manner for checkpoint regulation and DNA damage responses, respectively (Figure 6C).46 MacroH2A.1 and ALC1 (amplified in liver cancer 1) also require PAR to be recruited to their target.47 However, these proteins contain a macrodomain, an ancient and highly conserved domain that recognizes PAR polymers in submicromolar affinities.10,48 MacroH2A.1 and ALC1 interact with PAR chains through their macrodomains, and inhibition of PAR synthesis or mutation in the macrodomain fails to recruit these proteins to laser-induced DNA damage sites.47 XRCC1 and BARD1 bind to ADP-ribose through their BRCA1 C-terminus (BRCT) motifs,49 while APTX and PNKP bind to iso-ADP-ribose through Forkhead-associated (FHA) domains.49a The interactions between PAR and the BRCT or FHA domains mediate the relocation of the proteins containing these domains to DNA damage sites.49 Finally, human ssDNA-binding protein 1 (hSSB1) binds to PAR and is recruited to sites of DNA damage via its oligonucleotide/oligosaccharide-binding (OB) fold, a ssDNA or RNA binding motif found in prokaryote and eukaryotes.50
Figure 8.

Recognition of PAR chains by PAR-binding modules. (A) PAR-binding proteins utilize various PAR-binding modules to recognize PAR. The PBZ domain (blue) uses a zinc-coordinated fold that recognizes the α(1 → 2) O-glycosidic bond between two ribose units.10 Solution structure of the first PBZ domain in a complex with ribofuranosyladenosine (upper right panel, PDB: 2KQD)217 and CHFR bound to P(1)P(2)-diadenosine 5′-pyrophosphate (lower right panel, PDB: 2XOY)218 are shown as examples. The macrodomain binds to the terminal ADP-ribose residue of PAR (red, upper left panel, PDB: 2BFQ)78a or mono-ADP ribosylated protein,219 and the WWE domains recognize the iso-ADP-ribose residue (green). Human RNF146 WWE domain in complex with iso-ADP-ribose is shown as an example (lower left, PDB: 3V3L).52 (B) A table summarizing different PAR-binding modules. PAR-binding motifs (PBM) are short amino acid sequences found in PAR-binding proteins such as XRCC1.10 The OB fold is a ssDNA- or RNA-binding motif in prokaryotes and eukaryotes; however, the OB fold of human SSB1 recognizes iso-ADP-ribose.50 BRCT and FHA domain also interact with PAR by recognizing ADP-ribose or iso-ADP-ribose unit from PAR chain, respectively.49
Another layer of biological process that PAR modulates is protein degradation through ubiquitylation. One mechanism of PAR-dependent ubiquitylation is through RNF146, in which the E3 ligase RNF146 binds to PAR through its WWE domain and subsequently ubiquitylates the Lys residue of the PARylated protein (Figures 6D and 8).51 Like other PAR-regulated proteins, RNF146 binds PAR, but is not covalently modified through PARylation.32a,51a,52 RNF146 protects against DNA damage-induced cell death by ubiquitylating PARP-1 in a PAR-dependent manner, leading to proteasomal degradation of PARP-1.51a,53 Interestingly, RNF146 has also been shown to regulate the Wnt signaling pathway and downstream gene expression. In this case, axin is PARylated by tankyrase (PARP-5), and RNF146 interacts with PARylated axin and controls its degradation.54 Regulation of the cellular signaling pathway through PAR-dependent protein degradation can be another interesting mechanism that PARP-1 might apply to regulate transcription, likely through controlling the stability of PARylated transcription factors or chromatin-modifying enzymes. However, whether PARP-1 adopts a similar mechanism in transcription regulation has yet to be determined.
2.3. NAD+-Dependent Regulation of PARP-1 and Crosstalk with SIRT1
2.3.1. Functional Interplay with NMNAT-1
An interesting aspect of the enzymatic reaction of NAD+-dependent enzymes is the consumption of NAD+ and the generation of NAM as a byproduct of the reaction. As described previously, NAM is a substrate for the NAD+ salvage pathway but also a potent inhibitor of NAD+-dependent enzymes, such as PARPs and SIRTs.55 This dual role indicates the possibility of a functional interplay between NAD+ synthesis and consumption. In the nucleus, PARP-1 activity is a major NAD+-consuming process. Upon activation, PARP-1 can rapidly use NAD+, and when hyper-activated, PARP-1 can deplete the cellular NAD+ pool.3b Therefore, appropriate synthesis of nuclear NAD+ is required for the cells to maintain their enzymatic activity.
Among the three isoforms of NMNATs, NMNAT-1 is the only enzyme that resides exclusively in the nucleus.21 Its unique subcellular localization suggests that it may be responsible for the regulation of nuclear NAD+-dependent enzymes, such as PARP-1 or SIRT1. Functional interplay between NMNAT-1 and NAD+-dependent enzymes was first suggested by the Wallerian degeneration slow (Wlds) mouse model, a dominant mouse mutation that can significantly delay axon degeneration.56 The protein responsible for this phenotype was found to be a chimeric NMNAT-1 that consists of the N-terminal 70 amino acids of the Ub24B (ubiquitylation assembly factor 4B) and the full coding sequence of NMNAT-1.57 It was proposed that this chimeric protein protects neuronal degeneration by increasing NAD+, leading to the subsequent activation of SIRT1.58 Although the clear mechanism of this neuroprotective effect still remains elusive, overexpression of NMNAT-1 or supplying NMN, NaMN, or NR supports a protective role for NAD+ synthesis during the axonal degeneration process.58b,59
Until recently, it was unclear how enzymes involved in the NAD+ synthesis pathway regulate PARP-1. The first biochemical evidence of the link between nuclear NAD+ synthesis and PARP-1 was proposed by Berger and colleagues in relation to DNA damage, where NMNAT-1 interacts with PARP-1 in a PAR-dependent manner.60 Upon binding, NMNAT-1 stimulates PARP-1 enzymatic activity. The NMNAT-1–PARP-1 interaction is regulated through phosphorylation of NMNAT-1 by protein kinase C (PKC), which reduces NMNAT-1 binding to PARP-1.60 Moreover, Zhang and colleagues suggested that there is functional interplay between NMNAT-1 and PARP-1 in the context of transcription regulation (Figure 9A).13b In MCF-7 cells, NMNAT-1 is recruited to promoters of target genes via PARP-1. This study also revealed that NMNAT-1 could enhance PARP-1 enzymatic activity upon binding, although the interaction was rather direct, instead of through the PAR polymer. Moreover, the enzymatic activity of NMNAT-1 was required for PARP-1-dependent PARylation at the promoters, indicating that NMNAT-1 regulates PARP-1 through protein–protein interactions as well as providing the PARP-1 substrate, NAD+.13b Although the functional link between NMNAT-1 and PARP-1 has been established, how this interplay affects biological processes requires further study.
Figure 9.

NAD+-dependent regulation of PARP-1 and PARP-1–SIRT1 crosstalk. (A) NMNAT-1 regulates PARP-1 or SIRT1-dependent transcription. (B) Competition between PARP-1 and SIRT1 for the common substrate NAD+. (C) Deacetylation of PARP-1 by SIRT1.
2.3.2. Crosstalk between PARP-1 and SIRT1
As mentioned previously, PARPs and SIRTs require a common substrate, NAD+, for their enzymatic reactions, which indicates that NAD+ utilization by one enzyme can affect the enzymatic activity of the other (Figure 9B). In the nucleus, two major NAD+-dependent enzymes, PARP-1 and SIRT1, have been suggested to compete for NAD+. Many studies have reported that chemical inhibition or genetic depletion of PARP-1 can increase total cellular NAD+ content and induce SIRT1 enzymatic activity (see below). However, intracellular NAD+ concentration has been reported to fall within the 200–500 μM range, which is significantly higher than the Km value of PARP-1 (20–60 μM) or SIRT1 (150–200 μM) for binding of NAD+. A reasonable explanation for this discrepancy is a subcellular difference in NAD+ concentration or localized NAD+ production. Indeed, the nuclear NAD+ concentration, estimated by using two-photon microscopy, is 70–90 μM,61 a concentration that is likely to rate-limit SIRT1. Thus, regulating PARP-1 enzymatic activity may result in alteration of SIRT1-dependent deacetylation events, although the molecular mechanisms remain to be clarified.
Another possible mechanism that PARP-1 and SIRT1 might share is the interaction with NAD+-synthesizing enzymes. Like PARP-1, SIRT1 has also been shown to interact with NMNAT-1 and recruit NMNAT-1 to its target gene promoter (Figure 9A).62 Depletion of NMNAT-1 results in the alteration of SIRT1 histone deacetylase activity and downstream gene expression, suggesting that nuclear NAD+ synthesis by NMNAT-1 is required for SIRT1 to regulate target gene expression.62 Moreover, NMNAT-1 also interacts with the nucleolar protein nucleomethylin (NML), which forms a complex with SIRT1 to regulate rRNA (rRNA) synthesis.63 Taking into account the role of PARP-1 in rRNA transcription (see below), accessibility toward NMNAT-1 could be a mechanism by which cells coordinate NAD+-dependent enzymes for the regulation of both mRNA and rRNA transcription.
While the competition for NAD+ or NMNAT-1 might influence PARP-1 and SIRT1 activity, direct crosstalk between these enzymes has been proposed as another layer of regulatory mechanism. So far, there is no clear evidence that PARP-1 PARylates SIRT1; however, deacetylation of PARP-1 by SIRT1 has been demonstrated (Figure 9C). Acetylation of PARP-1 was reported in macrophages and cardiomyocytes in the context of NF-kB-dependent immune and stress responses, respectively.64 Acetylation activates PARP-1 independent of DNA damage, and SIRT1 inhibits PARP-1 activation by deacetylation.64b Interestingly, increasing NAD+ concentration inhibits the PARP-1–SIRT1 interaction in vitro, suggesting that SIRT1-dependent PARP-1 regulation might occur when NAD+ is the limiting factor.64b Altogether, these studies indicate that NAD+ signaling in the cell is tightly regulated by functional interplay between NAD+ synthesis and consumption as well as by crosstalk between the enzymes involved in these biological processes.
3. Overview of the Roles of PARPs in the Regulation of Gene Expression
Over the first four decades of studies into the functions of PAR and PARPs, the major emphasis by far has been on their role in DNA damage detection and repair processes.11a,65 While these studies have been fruitful and have revealed many interesting aspects regarding the functions of PAR and PARPs, they have missed some important aspects of the biology. Over the past two decades, a growing literature has revealed an important role for PARP-1 family members, with an emphasis on PARP-1, in the regulation of chromatin, transcription, and gene expression.7b,9,11b,11d,66 Most recently, an emerging literature has implicated PARPs in another aspect of gene regulation, RNA biology.4,67 Thus, the three major molecular roles of PARP family members characterized to date are (1) DNA damage detection and repair, (2) transcriptional regulation, and (3) RNA biology. In the sections below, we discuss the role of PARPs in gene regulation, again with a focus on PARP-1 and with an emphasis on newly discovered roles in RNA biology, beginning first with a brief overview.
3.1. Overview of the Mechanisms of PARP-1-Dependent Gene Expression
Although originally overlooked as an important aspect of PARP biology, gene regulatory and transcriptional roles for PARP-1 and other PARP family members are, by now, well established in the literature.7b,9,11b,11d,66 A survey of the literature reveals at least four distinct, but interrelated, ways in which PARP-1 acts to control how genes are expressed and how the levels of gene products are maintained. They are (1) modulation of chromatin, (2) transcriptional coregulation, (3) modulation of DNA methylation, and (4) regulation of RNA. These are discussed briefly below, followed by a detailed review of the literature on the role of PARPs in RNA biology in the next section. (Note that the first three topics have been reviewed extensively elsewhere7b,9,11b,11d,66 for readers interested in more detail than we have presented here.)
3.1.1. PARP-1 as a Modulator of Chromatin
Chromatin, a repeating array of nucleosomes, is a protein–DNA complex that comprises genomic DNA, core histones (i.e., H2A, H2B, H3, and H4, or perhaps core histone variants), linker histones (e.g., H1), and other chromatin-associated proteins.68 Many early studies on the nuclear functions of PARP-1 and PAR showed that they can modulate chromatin structure, promoting the decompaction of chromatin by reducing interactions between nucleosomes and reducing nucleosome-dependent higher-order structures.11a,66a For example, in Drosophila, activation of dPARP (the PARP-1 homologue) promotes decondensation of chromatin in response to heat shock or other cellular signaling pathways.69 Furthermore, PARP-1-dependent PARylation of native polynucleosomes promotes decondensation, mimicking the effects of linker histone H1 depletion.70 These effects may be mediated by PARylation of H1 by PARP-170 or competition between PARP-1 and H1 for binding to nucleosomes.71 PAR-dependent effects on the compaction state of chromatin are highly dynamic66a,69b due, in part, to enzymes that can hydrolyze PAR, such as poly(ADP-ribose) glycohydrolase (PARG)72 and ARH3,73 or remove the terminal ADP-ribose, such as macrodomain-containing hydrolases, MacroD1, MacroD2, and C6orf130/TARG1,6b,74 to remove PAR chains and proximal mono(ADP-ribose) moieties from proteins.
Other effects of PARP-1 on chromatin are mediated, in part, by its effects on core histones or core histone variants. PARP-1 has been shown to PARylate histones, as well as nonhistone, chromatin-associated proteins.11a,66a Presumably, histone ADP-ribosylation (either mono- or poly-) affects the biochemical properties of the histones, thus altering nucleosome structure, or promotes interactions with chromatin-modulating proteins that contain ADPR-binding modules.10,48 Emerging evidence supports the conclusion that damaged DNA is not required to stimulate the enzymatic activity of chromatin-associated PARP-1. In fact, PARP-1’s catalytic activity can be stimulated by histones, nucleosomes, cellular signaling pathways, and protein-binding partners.66,71a,75 For example, proinflammatory signaling induces PARP-1 enzymatic activity and histone ADP-ribosylation at transcriptionally active and accessible chromatin regions in macrophages,75c lending support to the idea that (1) histones, as well as nonhistone, chromatin-associated proteins, are PARylated at specific loci in vivo and (2) PARP-1’s catalytic activity can be stimulated by cellular signaling pathways. Conversely, the amino-terminal tails of core histones have been shown to regulate PARP-1 enzymatic activity, which could serve a regulatory role for PARylation by chromatin-bound PARP-1.11a,66a,76
Studies with the Drosophila H2A variant H2Av (a homologue of mammalian H2A variants H2Az and H2Ax) have shown that replacement of canonical H2A with H2Av alters the conformation of nucleosomes and promotes the binding of dPARP to H3 and H4.77 Phosphorylation of H2Av in response to cellular signaling pathways exposes the H4 N-terminal tail even further to activate dPARP catalytic activity, which in turn directs heat-shock-induced transcriptional activation and genotoxic stress-induced DNA repair.76,77 Studies with macroH2A, another H2A variant that has a macrodomain in its extended C-terminal domain, have shown that the 1.1 isoform, but not the 1.2 splice variant, binds both ADPR and PAR.78 The macrodomain may allow chromatin-bound macroH2A1.1 to bind to PAR produced locally by PARP-1, resulting in macroH2A1.1-dependent chromatin compaction.47a
Recently, Muthurajan and colleagues reported another interesting aspect of PARP-1 function in relation to the regulation of chromatin, where automodified PARP-1 functions as a histone chaperone.79 In this case, automodified PARP-1, which has reduced affinity for nucleosomes, binds to free histones and facilitates nucleosome assembly. Interestingly, the PAR-binding protein APLF also exhibits histone chaperone activities via its C-terminal acidic motif, which is homologous to a motif conserved in histone chaperones of the NAP1L family.80 These data suggest that PARP-1 may function as a histone chaperone by directly binding or recruiting other factors to facilitate nucleosome assembly.
In addition to its directs effects on components of chromatin, PARP-1 may also modulate the localization and activity of a broad array of histone-modifying and nucleosome remodeling enzymes. For example, PARylation of KDM5B, a histone lysine demethylase that acts on histone H3 lysine 4 trimethyl (H3K4me3), inhibits KDM5B binding and demethylase activity at specific sites in the genome.35c This leads to an increase in the levels of H3K4me3 at the promoters of PARP-1-regulated genes, supporting continued gene expression. Likewise, physical and functional interactions with PARP-1 can alter the activity of ATP-dependent nucleosome remodeling enzymes. For example, PARylation of Drosophila ISWI by dPARP inhibits ISWI nucleosome binding, ATPase, and chromatin condensation activities at heat-shock loci.35b In contrast, PAR-dependent interactions between PARP-1 and ALC1, a macrodomain-containing nucleosome remodeling enzyme, promote nucleosome remodeling by ALC1, as well as recruitment of ALC1 to specific loci in the genome.47b,81 Thus, as illustrated here, PARP-1 (and potentially other nuclear PARPs, such as PARP-2 and PARP-3) can modulate gene expression by altering chromatin structure to affect transcriptional outcomes.
3.1.2. PARP-1 as a Transcriptional Coregulator
In addition to its role as a modulator of chromatin structure to control gene expression, PARP-1 has also been shown to act as classical transcription factor-dependent coregulatory protein. As a coregulator, PARP-1 functions with the basal transcription machinery, other coregulators with enzymatic activities (e.g., histone-modifying enzymes and nucleosome remodelers), and sequence-specific DNA-binding transcription factors, such as NF-κB, HES1, Elk1, Sox2, and nuclear hormone receptors.11a,66 In this regard, two types of coregulatory functions have been ascribed to PARP-1: (1) scaffold and (2) exchange factor.
With respect to the former (i.e., scaffold), PARP-1 may act as scaffold by interacting with and promoting the recruitment of other coregulators independent of its DNA binding and catalytic activities. In this regard, PARP-1 has been shown to interact with the protein arginine methyltransferase PRMT1 and the protein acetyltransferase p300/CBP to support NF-κB-dependent gene expression.64a,82 With respect to the latter (i.e., exchange factor), PARP-1 has been shown to promote the release of inhibitory factors and the recruitment of stimulatory factors to DNA-bound transcription factors. PARP-1-dependent exchange of opposing pairs of factors has been observed for: (1) inactive Cdk8-positive Mediator, which is exchanged for an active Cdk8-negative Mediator during retinoic acid-regulated activation,83 (2) a TLE1-containing corepressor complex, which is exchanged for a HAT-containing coactivator complex during signal-dependent gene regulation in neuronal cells,84 and (3) a corepressor complex containing NCoR and HDAC3, which is exchanged for an activation complex containing topoisomerase IIβ (TopoIIβ) at steroid hormone-regulated promoters.85 The detailed mechanisms of how PARP-1 is recruited to specific transcription factors or specific genes to act as a transcriptional coregulator, however, are not well understood, but are likely to involve recruitment to specific sites in the genome by sequence-specific DNA binding transcription factors.7b,66b
These examples illustrate a common theme of coregulation by PARP-1, the modulation of protein complex formation, which may occur as a result of PARP-1 scaffolding functions or PARP-1 enzymatic activity.
3.1.3. PARP-1 as a Modulator of DNA Methylation
An emerging literature over the past decade or so has shown that PARP-1 can alter the methylation of genomic DNA.86 PARP-1 may mediate these effects by regulating the expression or activity of the DNA methyltransferase Dnmt1.86b,87 In this regard, PARP-1 may directly interact with Dnmt1 through newly synthesized PAR polymers to inhibit Dnmt1 DNA methyltransferase activity.88 PARP-1 has been shown to interact functionally with the methylcytosine dioxygenase Tet2, an enzyme that catalyzes the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (hmC).89 These interactions may play a key role during somatic cell reprogramming, acting to promote an epigenetic program that directs transcriptional induction at pluripotency loci; PARP-1 regulates the 5mC modification, while Tet2 promotes the early generation of 5hmC by the oxidation of 5mC.89 However, it is currently unknown if PARP-1 directly regulates Tet2 enzymatic activity. As these examples show, modulation of DNA methylation by PARP-1 can impact the genome and affect gene expression outcomes.
3.1.4. PARP-1 as a Regulator of RNA
A growing body of evidence indicates that PARP-1 plays an important role in various aspects of RNA biology. First, PARP-1 binds and acts in concert with noncoding pRNAs (promoter-associated RNAs; see below) to retain the silent rDNA chromatin in the nucleolus.90 Second, PARP-1 plays an essential role in ribosomal biogenesis in the nucleolus by PARylation of several nucleolar proteins in Drosophila.91 Third, PARP-1 was identified as a novel mRNA binding protein,92 as well as a factor in the human pre-mRNA 3′-end-processing complex.93 In addition, PARP-1 PARylates heterogeneous nuclear ribonucleoproteins (hnRNPs), which are involved in alternative splicing of pre-mRNAs and translation.94 Fourth, six PARPs, two PARG isoforms, and PAR are required for the cytoplasmic posttranscriptional regulation of mRNA in stress granules (SGs).67b Last, other PARP family members, such as PARP-13/ZAP and PARP-2, have been implicated in viral gene expression95 and in nucleolar processes,96 respectively.
3.2. Integration of Mechanisms in PARP-1-Dependent Gene Expression
Gene regulation by PARP-1 may (1) be stimulatory or inhibitory, (2) require or function independently of PARP-1 catalytic activity, (3) exhibit gene-specific, cell type-specific, cell state-specific effects, and (3) be altered by activation of cellular signaling pathways. As might be expected for a key regulatory enzymes that functions at the hub of many nuclear processes, PARP-1 is subject to the regulatory actions of upstream signaling pathways that can control PARP-1 activity and function.7 Although activation of PARP-1 by damaged DNA has been the most widely studied mechanism,11a other mechanisms for the regulation of PARP-1 enzymatic activity have also been identified: (1) interactions with DNA and protein binding partners, (2) posttranslational modifications, and (3) nuclear NAD+ metabolic enzymes, many of which can be influenced as the end point of cellular signaling pathways.7,13a The details of these modes of PARP-1 activation have been covered elsewhere within this Review, as well as in other published reviews.7,13a
4. The New Biology of PARPs in Gene Expression: Regulation of RNA
In this section, we describe in detail the growing awareness of the collective role of PARP-1, PAR, and other PARPs in the transcriptional and post-transcriptional regulation of gene expression through the modulation of RNA, which was introduced briefly above.
4.1. PARP-1 and Noncoding RNA in rRNA Synthesis
4.1.1. Noncoding RNA in Gene Regulation
Recently, genome-wide transcriptome analyses have revealed that many RNA species other than protein-coding mRNAs are produced and may have profound effects on cellular physiology and pathology.97 One such class of RNA known as noncoding RNA (ncRNA) is a functional RNA molecule that is not translated into protein.98 ncRNAs are involved in various biological processes, such as (1) transcription, splicing, translation, stability, and translation of mRNA; (2) chromosome maintenance and segregation and epigenetic memory as in X-chromosome inactivation; (3) various human diseases, including cancers and neurodegenerative, inflammatory, and cardiovascular diseases; (4) stress response; (5) scaffolding for the assembly of macromolecular complexes such as rRNA and chromatin complexes; and (6) embryonic pluripotency, differentiation, and development and reprogramming of somatic cells.97−99 ncRNAs include (1) transfer RNA (tRNA) and ribosomal RNA (rRNA), which are involved in messenger RNA (mRNA) translation; (2) antisense RNA (asRNA), long noncoding RNA (lncRNA), microRNA (miRNA), and small interfering RNA (siRNA), which are all involved in gene regulation; (3) small nuclear RNA (snRNA), which is involved in RNA splicing; (4) Y RNAs, which are components of the Ro60 ribonucleoprotein particle and are essential for the initiation of active chromosomal DNA replication forks through interactions with replication proteins; (5) small nucleolar RNA (snoRNA), which is involved in guiding site-specific methylation and pseudouridylation of rRNA nucleotides and promoting rRNA folding and stability in the nucleolus; and (6) piRNA, which binds to the PIWI subfamily of the Argonaute family of proteins and is involved in transposon repression via target degradation mechanisms.98b,99,100
Emerging evidence indicates that direct interaction of various ncRNAs with RNA-binding proteins (RBPs) contributes to the pathogenesis of several types of disease through the pathological dysregulation of gene expression. For instance, RBPs such as heterogeneous nuclear ribonucleoprotein K (hnRNP-K) regulate chromatin structure and gene expression through physical interaction with lncRNA-p21 in various disease models.101 In addition, the lncRNA HOTAIR (HOX antisense intergenic RNA) acts as a scaffold that binds polycomb-repressive complex 2 (PRC2) to sites of target genes, leading to altered histone H3 lysine 27 methylation and gene expression.102 Recently, it has been shown that SPT5L protein binds chromatin and works together with argonaute 4 (AGO4) in the recruitment of a complex composed of lncRNAs and the switch/sucrose nonfermentable (SWI/SNF) complex to mediate transcriptional silencing at silenced loci.103 These studies raise the interesting possibility of identifying new ncRNA–protein interactions and determining the role that these interactions contribute to the understanding of gene expression.
4.1.2. The Role of PARP-1 and Noncoding RNA in Silent rDNA Chromatin
Ribosomal RNA genes (rDNA) exist in two distinct chromatin states: (1) a permissive state for transcription and (2) a transcriptionally repressive state.104 A growing body of evidence indicates that rRNA genes exist in a heterochromatic, transcriptionally silent chromatin structure regulated by NoRC, the nucleolar remodeling complex, through epigenetic reprogramming such as DNA methylation, histone modification, and chromatin remodeling activities.104b,105 NoRC, consisting of the ATPase SNF2h and the nucleolar protein TTF-1-interacting protein-5 (TIP5), is recruited to rDNA promoters by the transcription termination factor TTF-I bound to a promoter-proximal terminator element.106 To modify histones, NoRC recruits and interacts with the histone deacetylase HDAC1, which mediates deacetylation of histone H4 tails as well as remodels rDNA chromatin. NoRC also recruits DNA methyltransferase (DNMT), which methylates CpG residues within the rDNA promoter.107 This epigenetic reprogramming leads to impaired binding of upstream binding factor (UBF), selectivity factor (SL1), and transcription initiation factor IA (TIF-IA) to the nucleosomal rDNA promoter and subsequent transcriptional silencing and heterochromatin formation.105,108
Recent studies have shown that PARP-1 interacts with NoRC-associated RNA (pRNA), which represses repeated rRNA gene transcription and establishes transcriptionally silent rDNA chromatin with TIP5 (Figure 10).90,109 pRNA is a 150–250-nucleotide ncRNA transcript that originates from an intergenic spacer promoter positioned upstream from the pre-rRNA transcription start site and is complementary in sequence to the rDNA promoter.109c PARP-1 also interacts with TIP5, the large subunit of the chromatin remodeling complex, NoRC, via the noncoding pRNA and binds to the promoter of silent rRNA genes and promotes the formation of silent rDNA chromatin. More specifically, PARP-1 PARylates TIP5 as well as newly synthesized rDNA chromatin, suggesting that PARylation is required for the establishment of silent rDNA chromatin.
Figure 10.
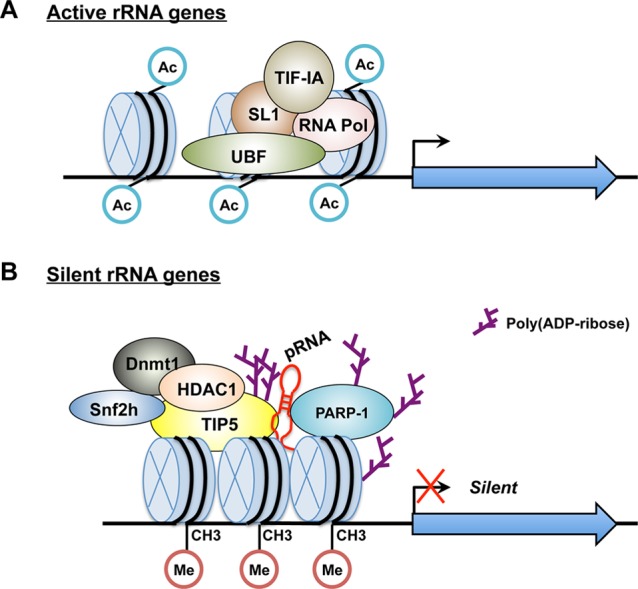
Functional interplay between PARP-1, the nucleolar remodeling complex, and ncRNA in epigenetically silenced rRNA genes. (A) Active rRNA genes exhibit an open chromatin structure and are characterized by DNA hypomethylation as well as acetylation of histone H4. This open chromatin structure leads to binding of the transcription factors upstream binding factor (UBF), selectivity factor (SL1), and transcription initiation factor IA (TIF-IA) to the nucleosomal rDNA promoter. (B) To establish the silent state, NoRC is recruited to the rRNA gene promoter by interaction with the transcription termination factor (TTF-I). In subsequent steps, NoRC recruits the histone deacetylase HDAC1, DNA methyltransferase (DNMT), and noncoding pRNA. PARP-1 also interacts with NoRC via the noncoding pRNA and binds to the promoters of silent rRNA genes, promoting the formation of silent rDNA chromatin through PARylation of TIP5, histone, and PARP-1 itself.90,220
pRNA mediates interactions between TIP5 and PARP-1, as well as their nucleolar localization, suggesting that PARP-1–RNA interactions are crucial for the regulation of rDNA transcription. For example, treatment of cells with RNase A results in the loss of PARP-1 from nucleoli, and TIP5–PARP-1 interactions are only detected in the presence of pRNA in vitro.90 In addition, the amino-terminal zinc-binding domain (FI, II, III) of PARP-1 mediates the interactions with pRNA.90 Both the nucleolar localization of PARP-1 and the association with TIP5 for the transcriptional regulation of rDNA are dependent on RNA interactions. TIP5–pRNA–PARP-1 complexes are formed at the rRNA genes, and then PARP-1 enzymatic activity enables the formation of silent rDNA chromatin and transcriptional silencing.90
The catalytically inactive PARP-1 E988K mutant leads to impaired repression of rRNA transcription and methylation of rDNA, suggesting that PARP-1-mediated PARylation plays an essential role in the regulation of silent rDNA chromatin formation.90 In addition, depletion of PARP-1 leads to (1) increased rRNA synthesis, (2) decreased methylated rDNA levels, and (3) decreased levels of H3K9me2 bound to rDNA, a histone mark associated with silent rDNA chromatin.90 Taken together, the studies described here indicate that PARP-1 and PARylation inhibit rRNA synthesis and maintain silent rDNA chromatin by direct interaction with pRNA and the chromatin-remodeling complex NoRC, causing epigenomic reprogramming during cell division. Because direct interaction of various ncRNAs with proteins contributes to regulation of gene expression in the various biological processes mentioned above, it will be interesting and informative to determine the underlying mechanism of the functional interactions between PARP-1 and ncRNAs in gene regulation.
4.2. PARP-1 and Ribosome Biogenesis in the Nucleolus
4.2.1. Ribosome Biogenesis in the Nucleolus
Ribosome biogenesis is required for assembly of four different rRNA molecules (25S/28S, 18S, 5.8S, and 5S) with more than 70 ribosomal proteins synthesized by RNA polymerase I, II, or III in eukaryotic cells.110 Ribosome biogenesis is a fundamental process required for cellular adaptation to changing environments, responses to changes in cellular growth rate and metabolic activity, and proliferation. Ribosomal biogenesis is a very complex process that involves the following series of events: (1) synthesis of ribosomal components by all three types of RNA polymerase, (2) pre-rRNA processing and modification, (3) assembly with ribosomal proteins and preribosomal particles, then (4) exit from the nucleus to become mature ribosomes in the cytoplasm.110,111
Ribosome biogenesis begins in the nucleolus, the site of rRNA transcription, maturation, and assembly with ribosomal proteins to form preribosomal particles. The nucleolus contains a cluster of tandemly repeated rRNAs as well as various classes of proteins.112 It is also associated with many RNA processing factors, such as RNA-modifying enzymes and various proteins involved in the production and assembly of ribosome subunits. Additionally, cell-cycle control, DNA replication and repair, transcription factors, splicing factors, chromatin-related factors, and RBPs such as hnRNPs and RNA helicase were identified as nucleolar proteins by bioinformatic analyses of proteomic data.112b
4.2.2. The Nucleolus and Various Human Diseases
The nucleolus has been implicated in a diverse range of genetic disorders, including Werner syndrome, Bloom syndrome, Treacher Collins syndrome, dyskeratosis congenital syndrome, and Rothmund–Thomson syndrome (RTS).112b,113 These diseases are associated with mutations of a gene encoding nuclear proteins that are known to be present in the nucleolus under specific cell-cycle stages or in response to specific stimuli. For example, the RECQ classes of DNA helicase (WRN and BLM, mutated in Bloom syndrome), which are expressed and localized in the nucleus during interphase, relocate and accumulate in the nucleolus during S phase. The RTS-associated protein RECQL4 accumulates in the nucleolus after oxidative stress, suggesting that nucleolus localization of certain mutated proteins is crucial in human genetic disorders.
Another protein associated with nucleolus localization has been implicated in various diseases such as cancer and neurodegenerative disorders. Parathyroid hormone-related protein (PTHrP) is a nuclear/nucleolar protein that (1) is frequently associated with the head, neck, breast, lung, and kidney, (2) involves the nuclear import receptor importin 1, and (3) accumulates in the nucleolus during G1 in response to mitogenic factors.113 It has also been shown that there is a relationship between the nucleolus and various neurodegenerative diseases, including Alzheimer’s disease, Huntington’s disease, and spinocerebellar ataxias. Hence, it will be interesting and informative to determine the underlying mechanism of the functional regulation of the set of nucleolar proteins in human disease.112b
4.2.3. Ribosomal Biogenesis and PARP-1 in the Nucleolus
A growing body of evidence indicates that PARP-1 plays an important role in ribosomal biogenesis in Drosophila nucleoli (Figure 11).91 PARP-1 is preferentially localized to the nucleolus; more specifically, PARP-1 is accumulated in the dense fibrillar component of nucleolar foci, where primary rRNA gene transcription and processing of pre-rRNA is initiated.114 Proteomics analyses using a combination of mass spectrometry and sequence database searches also showed the presence of PARP-1 in the nucleolus of HeLa cells.115 Interestingly, PARP-1 accumulation in nucleoli is altered upon treatment with an RNA synthesis inhibitor, indicating that PARP-1 localization is related to RNA synthesis in nucleoli.116
Figure 11.

Model of ribosomal biogenesis regulated by PARP-1. rDNA gene is transcribed into the 45S rRNA precursor (pre-rRNA), which is subsequently modified and processed into 5.8S, 18S, and 28S rRNAs by various rRNA-processing and rRNA-modifying factors. These rRNAs assemble with ribosomal proteins and preribosomal particles and then exit from the nucleus to become functional ribosomes in the cytoplasm. PARylated PARP-1 within rDNA contributes to formation of the dynamic PAR network in the nucleolus. This dynamic PAR network plays an essential role in rRNA processing, modification, and the loading of subsets of ribosomal proteins by regulating the localization of nucleolar-specific proteins in proximity to precursor rRNA in the nucleoli of Drosophila. PARP-1 mutants express a deletion in the PARP-1 protein or disruption of PARP-1 enzymatic activity, which leads to (1) altered subcellular localization of nucleolar-specific proteins, (2) nucleolar fragmentation, (3) delayed RNA processing, (4) a significant increase in rRNA intermediates, and (5) a decrease in ribosome levels.91,221
Nucleoli consist of three different components, the fibrillar center, the dense fibrillar component, and the granular component, and these regions are manifestations of major events such as rDNA transcription, processing, and ribosome assembly.112a In addition to ribosome biogenesis, the nucleolus has been implicated in cell-cycle regulation, storage of nuclear factors, and the processing of spliceosomal small nuclear U6 RNA, telomerase RNA, and signal recognition particle RNA.117
Interestingly, ADP-ribosylation reactions in nucleoli of exponentially growing HeLa cells indicate that PARP-1 PARylates a set of nucleolar proteins, including the nucleolar phosphoproteins numatrin/B23 and nucleolin/C23.118 These proteins have been implicated in rRNA transcription, rRNA maturation, ribosome assembly, and nucleo-cytoplasmic transport, suggesting that they are required in ribosome biogenesis.119 For example, nucleophosmin/B23 is localized in granular regions of the nucleolus and is associated with preribosomal particles and assembly of ribosomes, suggesting that it is a key regulator in ribosome biogenesis.119a Nucleolin/C23 is involved in the first rRNA processing step by interacting with the pre-rRNA substrate and U3 snoRNP to promote cleavage within the 5′ external transcribed spacer.119b In addition, PARP-1 contributes to the nucleolar localization of fibrillarin, a nucleolar protein known to be involved in pre-rRNA processing during ribosomal biogenesis.91 dPARP protein, dPARP activity, and PAR levels are significantly enriched in the nucleoli of Drosophila, suggesting that dPARP plays a role in nucleolar function.91 dPARP contributes to nucleolar integrity and localization of nucleolar-specific proteins in proximity to precursor rRNA in the nucleoli of Drosophila. Depletion of dPARP protein in the ParpCH1 mutant leads to altered subcellular localization of nucleolar-specific proteins such as fibrillarin, AJ1, and CCo1311.91 In addition, inhibition of dPARP activity by 3-aminobenzamide (3AB) caused nucleolar fragmentation of fibrillarin, suggesting that dPARP and its enzyme activity are required for the maintenance of Drosophila nucleolar structural integrity.91
The nucleoli of PARP-1 and PARG mutants reveal profoundly condensed areas positioned close to nuclear lamina, which may affect nucleolar functions such as rRNA processing and ribosome biogenesis. For example, the ParpC03256 mutant (which expresses a short isoform of the dPARP protein lacking the first zinc finger),120 and the Parg27.1 mutant (which lacks two-thirds of the PARG ORF, including the conserved catalytic domain),121 cause a delay in RNA processing and a significant increase in the levels of rRNA intermediates. The accumulation of rRNA intermediates, such as 47S and 36S rRNA transcripts, in PARP-1 mutants is caused by defects in RNA processing factors but not by altered transcriptional activity, because PARP-1 mutants reveal delocalization of nucleolar proteins required for rRNA processing. This accumulation results in a lack of polysomes, abnormal amounts of mature ribosomal subunits, and defects in mRNA translation. Interestingly, nucleolar rRNA processing and ribosome maturation-associated proteins, such as fibrillarin, nucleolin, AJ1, and nucleophosmin, interact with PAR, suggesting that PARP-1 enzyme activity plays an important role in ribosome biogenesis through PAR, which may act as a matrix for binding these nucleolar proteins and keeping them together in proximity to pre-rRNA.91 It has been shown that, upon DNA damage such as UV light exposure, γ radiation, and cross-linking by cisplatin, rRNA synthesis is blocked in cells.122 Inhibition of the DNA repair proteins such as DNA-dependent protein kinase (DNA-PK) or PARP-1 prevented DNA damage-induced block of rRNA synthesis, suggesting that PARP-1 activation by DNA damage also plays a key role in the regulation of rRNA synthesis.
4.2.4. PAR in Ribosome Biogenesis
CCCTC-binding factor (CTCF), which is bound to transposable element sequences within the rDNA, plays an important role in regulation of rDNA and nucleolar stability in Drosophila.123 Nucleolar accumulation of CTCF depends on rDNA transcription and protein synthesis, as well as its PARylation. The 180 kDal PARylated isoform of CTCF is predominantly localized in the nucleolus, where it inhibits RNA polymerase I transcription.117 Inhibition of PARylation is associated with reestablishment of active nucleolar transcription through reduced CTCF nucleolar translocation.117 CTCF inhibits nucleolar transcription, and its activity is regulated by PARylation, indicating that this may be essential for CTCF nucleolar localization and function.117 Knockdown of CTCF gene activity results in nucleolar fragmentation and increased rDNA expression as well as expression of rDNA-associated transposable elements, similar to that seen in disruption of PARylation. This finding suggests that PAR modification affects regulation of rDNA and nucleolar stability by modulating nucleolar localization of CTCF.
4.2.5. Other Factors in Ribosome Biogenesis
NMNAT-1 binds to PARP-1 at target gene promoters to support PARP-1-dependent PARylation through NAD+ production and thereby enhances the enzymatic activity of PARP-1 as mentioned above. During glucose deprivation, NMNAT-1 is recruited to the energy-dependent nucleolar silencing complex (eNoSC), which contains the nucleolar proteins nucleomethykin (NML), SIRT1, and SUV39H1. The interaction of NMNAT-1 with the eNoSC is responsible for the establishment of heterochromatin by modulating H3K9 dimethylation and subsequent repression of rDNA transcription.124 Depletion of NMNAT-1 leads to increased rRNA synthesis, implicating NMNAT-1 in the regulation of rRNA transcription and ribosome biogenesis during nutrient deprivation.63 NMNAT-1 recruitment into the NML–SIRT1 complex causes local NAD+ production that leads to SIRT1-mediated deacetylation reactions and subsequent repression of rRNA transcription. PARP-1 plays an important role in rRNA transcription and ribosome biogenesis, as mentioned above, suggesting that NMNAT-1 contributes to the regulation of rRNA biosynthesis by modulating PARP-1 catalytic activity.
4.3. PARP-1 and mRNA Regulation
4.3.1. The RNA-Binding Protein PARP-1
Eukaryotic cells encode >500 RNA binding proteins (RBPs), each containing unique RNA-binding activity and RNA-binding domains, and these RBPs play an important role in various aspects of RNA biology.125 RBPs physically interact with pre-mRNA and mRNA to form ribonucleoprotein (RNP) complexes.126 RBPs and RNPs are implicated in posttranscriptional control of gene expression, cellular homeostasis, and RNA biogenesis in eukaryotes. More specifically, they have been implicated in synthesis, stability, alternative splicing, polyadenylation, cellular localization, folding, translation, and translocation of mRNA.125,126 Interestingly, aberrant expression of RBPs affecting the temporal, spatial, and functional dynamics of RNAs has been shown to be involved in various human diseases, including neurodegenerative disorders and cancer.125 Although the range of mRNA types bound by PARP-1 has not been explored in detail, PARP-1 was identified as a novel RBP by a method that used covalent UV cross-linking of RBPs to RNA followed by proteomic analysis in human HeLa cells.92 This study raised the interesting possibility that PARP-1 plays crucial roles in many aspects of RNA processing to alter gene expression via regulation of cellular mRNAs. Taken together, the identification and characterization of PARP-1–mRNA interactions may provide important insights into the role of PARP-1 in mRNA regulation and subsequent human disease.
4.3.2. PARP-1 and Poly(A) Polymerase (PAP)
Polyadenylation is a process comprised of endonucleolytic cleavage of pre-mRNAs catalyzed by CPSF73 followed by the synthesis of a poly(A) tail onto the 5′-cleaved product by PAP.127 Additionally, the human pre-mRNA 3′-processing complex contains approximately 85 proteins, including core 3′-processing factors and over 50 proteins that may mediate crosstalk between pre-mRNA 3′-processing and other nuclear events.127 Key aspects of polyadenylation are associated with mRNA metabolism, including transcription termination, mRNA stability, mRNA export to the cytoplasm, and the efficiency of translation.127,128 Proteomic and structural analyses have identified PARP-1 as a factor in the human pre-mRNA 3′-end-processing complex, and studies imply a role for PARP-1 in its activation, assembly, and function.93 PARP-1 activation by NAD+ inhibits lengthening of the poly(A) tail in vitro by PARylation of PAP, which may inhibit its enzymatic activity. PARP-1 inhibition restores polyadenylation activity in vitro, suggesting that PARP-1 enzymatic activity is crucial for the regulation of PAP-induced polyadenylation.
Interestingly, PARP-1 is activated in vivo by heat shock, which is associated with significant inhibition of polyadenylation activity (Figure 12). In response to heat shock, PARP-1 PARylates PAP, which decreases its binding to the transcribed gene and results in the inhibition of polyadenylation and subsequent alteration of gene expression. By inhibiting polyadenylation, PARP-1 provides an additional layer of protection against cellular stress by preventing mRNAs from being translated into proteins that may misfold during heat shock.93 Emerging evidence indicates that PAP is modified by various posttranslational modifications, including phosphorylation, acetylation, and sumoylation,129 which result in important changes in its enzyme activity, nuclear localization, stability, and association with the 3′-processing complex.129,130 This study highlights the expanding role for PARP-1 as a regulator of polyadenylation by decreasing PAP enzyme activity. As a result, the enzyme loses its ability to bind to the 3′ end of mRNA precursors in PARylation-dependent mechanisms. Taken together, PARP-1 plays a crucial role in mRNA metabolism by directly interacting with and PARylating PAP.
Figure 12.

The role of PARP-1 as a regulator of polyadenylation during heat shock. (A) Lengthening of the polyadenylated 3′ end of an mRNA is associated with endonucleolytic cleavage of pre-mRNAs catalyzed by CPSF73, which recognizes the highly conserved AAUAAA hexamer, followed by the synthesis of a poly(A) tail onto the 5′ cleaved product by PAP under normal conditions.93,222 (B) PARP-1 activation by heat shock inhibits generation of the poly(A) tail by PARylation of the PAP, which is associated with significant inhibition of polyadenylation activity. PARylated PAP loses its ability to bind to the 3′ end of mRNA precursors, which subsequently arrests gene expression. The arrow represents the cleavage site.93,222
4.3.3. PARP-1 and Splicing
Cajal bodies (CBs), which are nuclear organelles, contain a variety of components, including small nuclear ribonucleoproteins (snRNPs), small nucleolar RNPs (snoRNPs), RNA polymerase II transcription factors, and nuclear proteins.131 CBs have been implicated in the formation of the spliceosome and the maturation of RNPs, both of which are essential for pre-mRNA splicing. They have also been shown to play a role in pre-rRNA processing.132 When PARP-1 is automodified by PARylation, it dissociates from chromatin and colocalizes with PAR to CBs to regulate their formation and disassembly.133 By promoting the noncovalent interaction of nuclear proteins with PAR, PARylated PARP-1 may act as a shuttle to deliver nuclear protein–PAR complexes into the CBs. For example, PARP-1 interacts with key protein components of CBs, such as coilin and fibrillarin, in a PAR-dependent manner and is required for their localization to the CB. PARP-1 loss-of-function mutations resulted in the dissociation of coilin and fibrillarin, fragmentation of coilin-containing bodies, and relocalization of fibrillarin from the nucleolus to the cytoplasm. These findings suggest that PARP-1 and PAR may play an important role in regulating the integrity of CBs, thereby affecting various processes, including transcription and splicing.
HnRNPs are well-known to bind directly to nascent transcripts and are involved in transcriptional regulation, alternative splicing of pre-mRNAs, pre-rRNA processing, nucleo-cytoplasmic transport localization, translation, and stability of mRNAs.134 Interestingly, 11 human hnRNP proteins (A1, A2/B1, C1/C2, G, H, K, E1, A3, L, M, U) contain a conserved PAR-binding motif (Table 2).135 Various proteomics approaches as well as pulse-chase experiments utilizing [32P]-labeled NAD+ as substrate have shown these proteins to be PARylated, suggesting a functional interplay between PAR and hnRNPs in RNA biology.135 Interestingly, PARylation of hnRNPs attenuates the RNA-binding ability of hnRNPs and results in the dissociation of hnRNPs from target RNA, suggesting that PARylation plays an important role in hnRNP regulation.94,135
Table 2. hnRNPs Contain Putative Poly(ADP-ribose)-Binding Motifsa.
| hnRNPs | functions | putative PAR-binding site |
|---|---|---|
| hnRNP A1 | splicing, mRNA transport, telomere biogenesis | 99-GAHLTVKKIF-108 |
| hnRNP A2/B1 | splicing, mRNA trafficking | 106-GAHVTVKKLF-115 |
| hnRNP C1/C2 | transcript packaging, splicing, mRNA stability | 92-RGKAGVKRSA-101 |
| hnRNP G | splicing | 118-GTRGPPSRGG-127 |
| hnRNP H | splicing | 210-YDRPGAGRGY-219 |
| hnRNP K | transcription, stability, translation | 261-FDRMPPGRGG-270 |
| hnRNP E1 | stability, translation | 25-VGSIIGKKGE-34 |
| hnRNP A3 | mRNA trafficking, telomere biogenesis | not known |
| hnRNP L | mRNA export, stability, riboswitch | not known |
| hnRNP M | splicing | 212-DYKVGWKKLK-221 |
| hnRNP U | nuclear retention | not known |
PARP-1 overexpression, PARG loss-of-function, and heat-shock treatment all result in increasing PARylation of hnRNPs, which alters their association with nascent RNAs and thus modulates alternative splicing pathways. For example, the Drosophila hnRNPs Squid/hrp40 and Hrb98DE/hrp38 are regulated in vivo by PARP-1 and PARG in a PARylation-dependent manner.94 It has been shown that these hnRNPs are similar to the human hnRNP A/B type and are involved in pre-mRNA splicing. Squid/hrp40 is also implicated in proper RNA localization. PARylation of Squid/hrp40 and Hrb98DE/hrp38 leads to diminished RNA-binding activity of the hnRNPs, which results in their dissociation from target RNA and alters their alternative splicing activities in vivo. More specifically, PARylation of Squid/hrp40 and Hrb98DE/hrp38 increases splicing of the intron in the Ddc pre-mRNA; however, splicing of the intron in the Hsrω-RC transcript is suppressed.94 These findings highlight the role of PAR and PARP-1 in regulating alternative splicing by modulating the binding of hnRNPs to their target RNAs.
4.4. Other PARPs and RNA Regulation
4.4.1. PARPs, Stress Granules, and Other RNA-Containing Granules
A growing body of evidence indicates that SGs play an important role in post-transcriptional regulation of gene expression under variable cellular stress conditions.136 SGs, which are transient cytoplasmic RNA–protein complexes, have been implicated in the translation and stability of mRNAs during the stress response in eukaryotic cells.137 These granules are formed under a variety of stress responses that are implicated in suppression of translation initiation, including heat–cold shock, oxidative stress,138 viral infection,139 energy deprivation,140 and glucose starvation.141 In addition, SGs contain nontranslating mRNAs and a variety of proteins, including those involved in translation initiation (eIF4E, eIF4G, eIF4A, eIF3, and eIF2), mRNA function (small ribosomal subunits and poly(A)-binding protein),136,142 and RBPs such as TIA-R, TIA-1, and G3BP.143
Using a library of GFP fusions to human PARPs and antibodies against each PARP, Leung et al. identified PAR, six PARPs (PARP-5a, PARP-12, PARP-13.1, PARP-13.2, PARP-14, and PARP-15), and two PARG isoforms (PARG99 and PARG102) as SG components associated with the assembly and disassembly of cytoplasmic SGs67b (Figure 13). The proteins identified also contribute to SG integrity upon occurrence of various stressors, including heat shock, glucose deprivation, and treatment with the proteasome inhibitor MG132 or with translation-initiation inhibitors.67b
Figure 13.

The proposed functions of PARPs and PAR in regulating SG assembly. SGs are formed under a variety of stress responses and contain nontranslating poly(A)+ mRNAs, RBPs, and stalled translation-initiation factors. Upon stress, (1) PARG activity decreases, and (2) multiple proteins, including Ago2, G3BP1, and TIA-1, are PARylated by SG-localized PARPs and exhibit significantly increased PARylation and subsequent enrichment in SGs. SG-localized PARPs (SG-PARPs; SG-PARPs) and PAR may play a crucial role in the assembly and maintenance of SG structure by functioning as a framework to join diverse mRNA–protein complexes together. RBPs are modified by PAR and become cross-linked to PAR-binding proteins, resulting in the assembly of SGs. In addition, PARylation of SG-PARPs and Ago2 by other SG-localized PARPs as well as a high local concentration of PAR near the Ago2–miRNA complex, affects the formation of Ago2–miRNA complexes or the accessibility of these complexes to their target mRNAs, resulting in alterations to miRNA-mediated translational repression and miRNA-directed mRNA cleavage.67b,223
A recent study has shown that the formation of RNA granules lacking a boundary membrane can be mimicked in vitro by incubating cell or tissue lysates with a biotinylated isoxazole reagent (b-isox).144 This chemical selectively precipitates a set of RBPs with significant overlap to the constituents of RNA granules.144b The mRNAs associated with these RBPs contain extended 3′ UTR sequences and are enriched in binding sites for known granule-associated proteins.144a Interestingly, the granule-associated proteins overlap significantly with proteomic data sets of PARylated proteins, suggesting that PAR may play a role for RNA granule assembly. On the basis of these findings, Leung et al. proposed that PAR plays crucial roles in the assembly of RNA granules to form cellular structures, such as SGs.145 RNA granule proteins, which are enriched for low complexity regions that aid self-assembly, are preferentially PARylated, promoting the formation of “droplets” by increasing the local concentration of proteins through noncovalent interactions.
Interestingly, PAR colocalizes with the miRNA-binding protein argonaute 2 (Ago 2), RNA decay factor G3BP1, translational suppressor TIA-1, and poly(A)-binding protein PABP, which are responsible for regulation of mRNA translation and decay in SGs upon arsenite-mediated oxidative stress.67b Moreover, Ago family members Ago1–4, G3BP1, and TIA-1, but not PABP, are modified by PAR in unstressed cells.67b Under stress conditions, these proteins exhibit significantly increased PARylation and are subsequently enriched in SGs, which suggests that PAR is required for the cytoplasmic posttranscriptional regulation of mRNA, potentially through modulation of cytoplasmic RBPs in SGs. These SG-localized PARPs and PARGs may play a crucial role in the assembly and maintenance of SG structure through modulation of local PAR concentration.
In particular, Argonaute family members play a crucial role in miRNA processing and posttranscriptional regulation of miRNA expression, and their association is required for SG localization.146 Upon stress in SGs, increased PARylation of PARP-13.2 and Argonaute by PARP-12 and PARP-15 alleviates miRNA silencing activity, suggesting that PAR modification affects the formation of Argonaute–miRNA complexes or accessibility of these complexes to their target mRNA, resulting in alterations of miRNA-mediated translational repression and miRNA-directed mRNA cleavage. The knockdown of two cytoplasmic isoforms of PARG induced PAR modification of Ago2, resulting in a decrease in miRNA-mediated repression. Taken together, PAR is implicated in miRNA-mediated translation repression and miRNA-directed mRNA cleavage by modification of miRNA-binding Ago family members, resulting in altered posttranscriptional gene expression in the SGs. These observations highlight the novel role of PARPs and PARG in posttranscriptional gene regulation via modulation of microRBPs in PAR-dependent mechanisms under the stress response.
4.4.2. Regulation of Retroviral RNA by PARP-13/ZAP
PARP-13/ZAP (CCCH-type zinc finger antiviral protein) was identified as an active antiviral cDNA that inhibits infection by a genetically marked retrovirus in virus-resistant cells.95 PARP-13/ZAP regulates viral gene expression through degradation of viral mRNAs and subsequent inhibition of virus replication by directly interacting with the viral RNA via four unusual CCCH-type zinc fingers in the cytoplasm. In addition, the shorter PARP-13 isoform (ZAPS) directly interacts with the RNA helicase RIG-I upon treatment with 5′-triphosphate-modified RNA.147 This interaction leads to increased RIG-I-mediated antiviral signaling and subsequent inhibition of viral replication after infection with RNA viruses through activation of IRF3 and NF-κB transcription factors. Consistent with these results, PARP-13/ZAP is implicated in the regulation of viral gene expression either by directly interacting with viral mRNAs or by activating the RIG-I pathway, which is responsible for degradation of viral RNA. Interestingly, PARP-13/ZAP is catalytically inactive,3b,67b,95,148 suggesting the possibility that PARP-13 function may require prior ADP-ribosylation by another PARP to “jumpstart” its activity. PARP-13 has two isoforms; the PARP-13.2 isoform lacks a PARP domain, while the PARP-13.1 isoform contains a PARP domain lacking the HYE motif (Figure 1).3b,67b,95,148
Most recently, evolutionary analyses of all primate PARPs have shown that additional PARPs, including PARP-4, PARP-9, PARP-14, and PARP-15, may be involved in host–virus interactions. However, it is still unclear whether these PARPs are implicated in the regulation of viral gene expression by directly associating with viral RNAs to carry out their functions.149
4.4.3. PARP-2 and RNA Biology
PARP-2 exhibits 69% homology with the catalytic domain in PARP-1, catalyzes PARylation of proteins in the cellular response to DNA damage, and is implicated in various physiological roles, including maintenance of genome integrity, heterochromatin integrity, cell death, differentiation, spermatogenesis, adipocyte differentiation, T cell development, and inflammation.150 The N-terminal SAF-A/B, Acinus, and PIAS DNA-binding (SAP) domain (a putative DNA–RNA binding motif) of PARP-2 (ARTD2) directly interacts with short rRNA and other single-stranded RNAs in nucleoli.96 PARP-2-dependent ADP-ribosylation is significantly activated by RNA binding, suggesting that RNA is a new activator of PARP-2 enzymatic activity. Hydrogen peroxide (H2O2) or N-methyl-N′-methyl-nitro-N-nitrosoguanidine (MNNG) in combination with actonimycin D treatment leads to the accumulation of short rRNA transcripts, resulting in activated PARP-2 activity and a subsequent increase in PAR formation in the nucleolus. This increased cellular PAR formation may play an important role in various nucleolar processes, including ribosome biogenesis, suggesting that PARP-2 activity regulated by RNA is an essential factor in nucleolar functions such as cellular adaptation to changing environments, response to changes in cellular growth rate and metabolic activity, and proliferation. The N-terminal domain of PARP-2 contains a nucleolar localization signal and localizes within the whole nucleolus independently of PARP-1.
Like PARP-1, PARP-2 partially colocalizes with nucleolar proteins, including nucleophosmin/B23, which is implicated in rRNA transcription and maturation, ribosome assembly, and rRNA processing.151 The nucleolar accumulations of PARP-1 and PARP-2, together with B23, are delocalized from the nucleolus with RNA polymerase I inhibitor treatment but not with RNA polymerase II inhibitors and are moderately affected upon oxidative or alkylated DNA damage, suggesting that their nucleolar localization depends on changes in nucleolar transcription processing. This study shows that murine fibroblasts deficient in PARP-1 or PARP-2 are not affected in ribosomal transcription, suggesting that nucleolar accumulations of PARP-1 and PARP-2 are not essential factors for rRNA transcription.96 However, a growing body of evidence indicates that PARP-1 plays an important role in formation of heterochromatin through regulation of rRNA transcription in nucleoli as well as modulation of ribosomal biogenesis in Drosophila nucleoli.90,91 Further mechanistic studies of PARPs and PAR will provide insight into the function of these factors in the regulation of ribosomal biogenesis that takes place in the nucleolus.
4.4.4. PARPs and RNA Splicing
The interactomes of PARP-1, PARP-2, and PARG, discovered by affinity-purification mass spectrometry (AP-MS) combined with gene ontology analysis, have revealed that PARP-1, PARP-2, and PARG have profound effects on RNA splicing.32b For instance, PARP-1 interacts with various hnRNPs, splicing factors, snRNPs, and THO/TREX, which are associated with the splicing machinery. In addition, PARP-2 and PARG interact with ATP-dependent RNA helicase, hnRNPs, polyadenylate-binding protein 1, and nuclease-sensitive element-binding protein 1 (YBX-1), which are involved in various types of RNA processing, including splicing. Moreover, PAR forms a complex with alternative splicing factor/splicing factor 2 (ASF/SF2, a prototypical SR-protein) and inhibits topoisomerase I-dependent phosphorylation of ASF/SF2, resulting in an altered incidence of alternative splicing and subsequent gene expression.152 These findings strongly indicate that PARP-1, PARP-2, and PARG play an important role in the regulation of RNA splicing in a PAR-dependent manner.
5. Physiology and Pathophysiology of PARP-1 and Other PARPs
Because of the important role of PARP-1 in the DNA damage response and transcriptional regulation, a substantial number of studies have been focused on determining the pathological and physiological outcomes of PARP-1 using various biological systems. Initially, the pathophysiology of PARP-1 was mainly focused on DNA damage and genome instability caused by genotoxic, oxidative, and oncogenic stresses. The known physiological role of PARP-1 has expanded more recently, and it has become implicated in various biological processes such as development, metabolism, the immune response, circadian rhythms, differentiation, and reprogramming. Development of the Parp1 knockout mouse has not only significantly contributed to determining the physiological functions of PARP-1, but has also generated contradictory results. In the section below, we will discuss several pathophysiological roles of PARP-1 in the context of animal studies and additionally highlight some of the most recent advances as well as remaining questions.
5.1. PARP-1 in Genome Maintenance and Carcinogenesis
During the 1990s, three independent groups generated PARP-1 null mice targeting different exons. Wang et al. reported the first Parp1 knockout mice by targeting exon 2 in a 129/Sv, C57BL/6 mixed background.153 The mice were viable and exhibited normal postnatal development with only a mild phenotype.153 This was surprising, because PARP-1 is one of the most immediate protein responders upon DNA damage and was expected to be lethal when genetically deleted. Subsequent generation of PARP-1 null mice by different groups targeting other exons (de Murcia et al.154 exon 4 in a 129/Sv, C57BL/6 mixed background; Masutani et al.155 exon 1 in an ICR, 129/Sv mixed background) also confirmed the absence of an obviously abnormal phenotype of Parp1 knockout mice. However, these mice are more sensitive to chemical- or γ irradiation-induced genotoxic stress153,154 and show genomic instability154,156 independent of the exon targeted or genetic background, suggesting that PARP-1 may play an important role in genome maintenance under stress conditions. Indeed, Parp1–/– mice show increased susceptibility to various carcinogens and frequent tumor formation as compared to Parp1+/+ mice,157 supporting the role of PARP-1 in genome stability.
Genome instability caused by loss of PARP-1 is enhanced when PARP-1 is depleted together with other tumor-suppressor genes. For example, PARP-1 deficiency (deleting exon 2) in p53 null mice (Parp1–/–p53–/–) enhances tumorigenesis as compared to Parp1+/+p53–/– mice, resulting in a high frequency of developing mammary gland, lung, prostate, skin, and brain tumors.158 Also, Parp1 knockout accelerated medulloblastoma and basal cell carcinoma development upon irradiation in the background of Patched gene (Ptc1) heterozygotes159 and increased frequency of spontaneous liver cancer formation when bred with Ku80 heterozygotes,160 supporting the role of PARP-1 in genome maintenance.
While increasing evidence indicates that the genome instability caused by PARP-1 deficiency is deleterious, whether it can lead to spontaneous cancer remains unclear. Several studies have reported that PARP-1 deficiency itself can lead to increased tumor formation,158b,160,161 while others have reported contradictory results.162 Moreover, Conde et al. also reported conflicting results using Parp1–/– (deleting exon 4) p53–/– mice, where the mice show delayed tumor formation, possibly due to abolition of nitric oxide synthase (iNOS) expression (Table 3).163 This disparity can possibly be explained by (1) different exon deletions in each study, which might generate truncated transcripts of PARP-1, or (2) using a mixed genetic background (129/Sv × C57BL/6), which might cause genetic variation. However, more careful studies are required to explain this discrepancy between different PARP-1-deficient phenotypes.
Table 3. Phenotypic Outcomes of Parp1 Knockout Mice upon Genotoxic Stressa.
| genotype | exon | genotoxic stress | phenotype (compare to wild type) |
|---|---|---|---|
| Parp1–/– | 2 | spontaneous | increased uterine, lung, breast, and hepatocellular carcinoma160,161 |
| γ-irradiation | hypersensitive to γ-irradiation156 | ||
| MNNG | decreased DNA damage repair153 | ||
| diethylnitrosamine | increased hepatocellular carcinoma160 | ||
| 4 | MNU | more susceptible to MNU injection154 | |
| γ-irradiation | hypersensitive to γ-irradiation154 | ||
| 1 | spontaneous | no difference162 | |
| MMS | hypersensitive to MMS treatment212 | ||
| γ-irradiation | hypersensitive to γ-irradiation212 | ||
| azoxymethane | increased colon, liver tumor213 | ||
| 4-nitroquinoline 1-oxide | no difference214 | ||
| IQ | no difference162b | ||
| Parp1–/– p53–/– | 2 | spontaneous | increased colon, breast, brain tumor158 |
| 4 | spontaneous | decreased thymic lymphoma163 | |
| Parp1–/– Ku80+/– | 2 | spontaneous | increased liver tumor160 |
| Parp1–/– Ptc+/– | 2 | spontaneous | increased medulloblastoma and basal cell carcinoma159 |
This table was adapted from Masutani and Fujimori, 2013.157
5.2. PARP-1 in Inflammation and Stress Responses
Subsequent studies over the past two decades have provided a more comprehensive understanding of PARP-1’s role in inflammatory responses. The initial observation of PARP-1-dependent inflammation regulation came from inhibitor studies where PARP-1 inhibition suppressed induction of inflammatory-response genes such as IL-6, TNF-α, and iNOS upon lipopolysaccharide (LPS) or TNF-α treatment in macrophages.164 These initial observations were further confirmed in vivo using Parp1 knockout mice, which show extreme resistance to LPS-induced endotoxic shock and attenuation of neutrophil recruitment followed by reduced organ injury.165 The clear anti-inflammatory phenotype of Parp1 knockout is obtained through regulation of the transcription factor NF-κB, a master regulator of inflammatory responses, in which the absence of PARP-1 reduces induction of genes downstream of NF-κB during immune responses.165b
Although it now seems clear that PARP-1 regulates NF-κB-dependent transcription, the clear mechanism of how PARP-1 regulates this transcription factor remains debatable. Hassa et al. reported that PARP-1 directly interacts with both p50 and p65 in vitro, but neither DNA- binding nor enzymatic activity were required for the activation of NF-κB, while acetylation of PARP-1 by p300/CBP was required for the regulation of NF-κB-dependent transcription.64a,82a,166 By contrast, different groups have shown that the enzymatic activity of PARP-1 is important for NF-κB activation, where PARylation of the p50/p65 dimer or automodification of PARP-1 are critical for their binding to DNA and subsequent transcription activation.167
PARP-1 also modulates the inflammatory response through regulating other important immune-related transcription factors. For example, PARP-1 positively regulates AP-1, a transcription factor responsible for cytokine production and T helper cell differentiation.168 In the absence of PARP-1, the DNA binding of AP-1 is inhibited through altered activation of JNK and MEK-4. PARP-1 has also been shown to modulate NFAT transcription factor activity in T cells by PARylating NFAT.169 Moreover, PARP-1 has recently been shown to be involved in the differentiation of regulatory T cells170 as well as TGF-β signaling in CD4+ T cells,171 suggesting an extensive role for PARP-1 in immune responses.
Regulation of both the DNA damage response and the inflammatory signaling by PARP-1 suggests its importance in multiple tissue damage caused by various stresses. Ischemia and reperfusion injury is a type of tissue damage triggered by transient disruption of the normal blood supply followed by reperfusion. During this process, tissue injury is mediated by reactive oxygen species (ROS)-induced DNA damage and also the rapid transcriptional activation of pro-inflammatory genes.172 A wide array of studies has revealed the role of PARP-1 in ischemia reperfusion injuries in various tissues. For instance, mice in which PARP-1 is inhibited or knocked out are resistant to N-methyl-d-aspartate (NMDA)-induced toxicity or ischemia reperfusion damage in the brain.173 PARP-1 also plays a major role during the process of tissue injury in heart, kidney, liver, intestine, and lung, where inhibition or genetic ablation of Parp1 dramatically reduces tissue damage caused by ischemia and reperfusion.174 The protective effect of PARP-1 inhibition is due to reducing the severe drop in NAD+ and ATP levels caused by ROS-induced DNA damage, as well as inhibiting pro-inflammatory gene expression.175
5.3. PARP-1 in Metabolism and Energy Expenditure
The initial observation of the role of PARP-1 in relation to metabolism came from knockout mouse studies where Parp1–/– mice were resistant to streptozotocin (STZ)-induced diabetes. This phenotype was consistent in two different types of Parp1 knockout mice (disruption of exon 1 or exon 2) in different genetic backgrounds (129/Sv × ICR and 129/Sv × C57BL/6, respectively).176 Only more recently, however, has the novel aspect of PARP-1 as a metabolic regulator been suggested. In an obesity-resistant (129/Sv) background, Parp1 knockout leads to age-related and diet-induced obesity. Hyperleptinemia, glucose intolerance, and insulin resistance were observed in Parp1–/– mice, suggesting a protective role for PARP-1 against metabolic stress.177
Bai et al., however, reported contradictory observations using Parp1 knockout mice in an obesity-prone (C57BL/6J) background, where Parp1 ablation led to protection against diet-induced obesity, decreased fat deposition, and improved glucose metabolism.178 In this study, Parp1 deficiency led to SIRT1 activation in muscle and brown adipose tissue. SIRT1 deacetylates PGC1-α and FOXO1, transcription factors responsible for oxidative metabolism, leading to mitochondrial biogenesis and enhanced energy expenditure.179 In line with this observation, chronic inhibition of PARP-1 also resulted in improved mitochondrial function and enhanced energy expenditure via SIRT1 activation, supporting the negative role of PARP-1 in metabolism.180 It has been suggested that activation of SIRT1 in Parp1–/– mice is due to increased NAD+ availability. Interestingly, mice fed with the NAD+ precursors NMN or NR resemble the metabolic phenotypes of Parp1–/– mice in the C57BL/6 background, resulting in enhanced oxidative metabolism and protection against diet-induced obesity and related metabolic disorders.181 SIRT1 is activated in both NMN- and NR-fed mice, supporting the possibility of SIRT1 activation via increased NAD+ accessibility.
The reason why Parp1–/– mice exhibit contrasting metabolic phenotypes is still elusive. It is possible that the genetic differences between the 129/Sv and the C57BL/6 strains affect the metabolic outcome of PARP-1 loss.7a Indeed, C57BL/6J mice harbor a loss-of-function deletion of the nicotinamide nucleotide transhydrogenase gene (NNT), which encodes a mitochondrial protein that catalyzes production of NAD+ through the reversible reduction of NADP+ by NADH, supporting the possibility of strain-specific effects.182 However, Erener et al. also challenged Parp1–/– mice in a C57BL/6 background with high-fat diet feeding and observed conflicting results. In this study, loss of PARP-1 led to lower body weight and reduced fat mass but impaired glucose metabolism, and hepatic lipid accumulation and dyslipidemia were also observed (summarized in Table 4).183 Thus, strain specificity is insufficient to explain the different metabolic phenotypes of Parp1–/– mice. Considering the ubiquitous expression of PARP-1 in various tissues, it is possible that systemic deletion of PARP-1, rather than strain specificity, affects the metabolic outcome of PARP-1. Further studies using tissue-specific knockout of Parp1 will be required to elucidate the role of PARP-1 in metabolism.
Table 4. Phenotypic Outcomes of Parp1–/– Mice upon Metabolic Stress.
| stress | exon | background | body weight | fat deposition | glucose metabolism | other phenotypes |
|---|---|---|---|---|---|---|
| STZ | 2 | 129/Sv × C57BL/6 | N/A | N/A | maintained | protection against STZ-induced diabetes176b |
| 1 | 129/Sv × ICR | N/A | N/A | maintained | protection against STZ-induced diabetes176a | |
| HFD | 2 | 129/SvlmJ | increased | increased | impaired | hyperleptinemia177 |
| 4 | C57BL/6 | decreased | decreased | improved | enhanced energy expenditure178 | |
| 2 | C57BL/6 | decreased | decreased | impaired | higher hepatic lipid accumulation and dyslipidemia183 |
5.4. PARP-1 in Circadian Rhythms
Circadian clocks in peripheral organs are controlled by the interplay between transcriptional activators and repressors, which generates a negative feedback loop of core clock gene expression. In peripheral tissues, metabolic cycles, such as feeding and fasting, regulate the oscillation of their internal clocks.184 The regulation of the circadian clock by NAD+-dependent transcriptional regulation was first suggested by an in vitro study where DNA binding activities of CLOCK–BMAL1, master transcription factors responsible for the clock oscillation, are strongly affected by NAD(P)+/NAD(P)H levels.185 Subsequent studies further revealed that NAMPT is expressed in a circadian manner and generates daily oscillations of NAD+ levels, affecting circadian gene expression through the covalent modification of clock transcription factors and chromatin-associated proteins by SIRT1.186
Recently, Asher et al. reported a profound role for PARP-1 in the regulation of circadian clocks in peripheral tissues. In the liver, PARP-1 PARylates CLOCK transcription factor, disrupting its DNA binding and promoting a phase-shift in the interaction of CLOCK–BMAL1 with the PER and CRY repressor proteins.40 Enzymatic activity of PARP-1 oscillates in response to feeding stimuli, and Parp1 knockout mice exhibit altered circadian rhythms and CLOCK–BMAL1-dependent gene expression.40 Although how PARP-1 is regulated by food intake still remains elusive, these results indicate that PARP-1 is a critical player in modulating the circadian clock in the liver connecting nutrient intake with gene expression.
Interestingly, a role for PAR in the regulation of circadian period length in the plant Arabidopsis has recently been described.187 Mutation of TEJ, a gene encoding a ., causes a long free-running period, affects clock-controlled transcription, and alters the timing of the photoperiod-dependent transition from vegetative growth to flowering. Treatment with a PARP inhibitor rescues the period phenotype of the tej mutant and shortens period length in wild-type plants. These results suggest that PARylation of an oscillator component may contribute to setting the period length of the central oscillator in Arabidopsis.
5.5. PARP-1 in Differentiation, Development, and Reprogramming
5.5.1. PARP-1 in Embryonic Development
Despite the absence of developmental defects in Parp1 knockout mice, a role for PARP-1 in development has been suggested by a number of studies. The developmental roles of PARP-1 are well illustrated in Drosophila larval development, where maximal activity of dPARP was observed at the prepupal stage.11b,133 The enzymatic activity of dPARP is required for modulating heterochromatin structure as well as chromatin loosening at the sites of ecdysone response genes during development, and dPARP loss-of-function causes larval lethality,188 indicating its pivotal role in developmental processes. PARP-1 has also been shown to regulate mammalian germ line development, especially oocyte maturation. Although Parp1 null female mice are fertile, isolated oocytes from Parp1 knockout mice display meiotic defects such as incomplete homologous chromosome synapsis and persistent histone H2AX phosphorylation.189 The lack of any embryonic developmental defect due to Parp1 knockout can be explained by functional redundancy with other PARPs, such as PARP-2, and the embryonic-lethal phenotype of Parp1/Parp2 double-null mice supports this possibility190 (see below).
5.5.2. PARP-1 in Cellular Differentiation
PARP-1 has been implicated in various cellular differentiation processes. Differentiation of cells requires a complex cascade of transcription factor actions, and PARP-1 coordinates these processes mainly through regulating the transcription factors involved. In immune cell differentiation, Parp1 genetic ablation reduces TH2 cell differentiation and enhances regulatory (CD4+/CD25+/Foxp3+) T cells.170,191 PARP-1 also regulates neuronal differentiation by exchanging corepressor complexes with coactivators at the target gene promoter. In neuronal stem cells, MASH1 gene expression is repressed by the transcription factor HES1, which subsequently recruits the TLE corepressor complex containing PARP-1 to the promoter of the MASH1 gene.84 During differentiation, PARP-1 mediates PDGF-dependent release of the TLE complex and further recruits HAT-containing coactivator complex and derepresses expression of the MASH1 gene.84 In addition, PARP-1 regulates adipocyte differentiation by regulating PPARγ-dependent gene expression,192 supporting the role of PARP-1 as a regulator of cellular differentiation processes.
PARP-1 also plays roles in embryonic stem (ES) cell differentiation. When injected into nude mice, Parp1–/– ES cells develop into teratocarcinoma-like tumors expressing sets of genes belonging to the trophoblast lineage.193 In line with tumor development, PARP-1-deficient ES cells also exhibit increased expression of trophoblast marker genes in culture, indicating that PARP-1 inhibits ES cell differentiation into trophectoderm lineages.193 Genome-wide alteration of gene expression upon PARP-1 depletion in ES cells is significantly greater than that in liver (about 10% in ES cells and 3% in liver),194 suggesting a significant role for PARP-1 in regulating the gene expression of ES cells. In ES cells, major transcription factors, such as Oct4, Sox2, and Nanog, coordinate maintenance of pluripotency and self-renewal as well as differentiation.195 PARP-1 PARylates Sox2 during ES cell differentiation inhibits Sox2 binding to the enhancer region of fibroblast growth factor 4 (FGF4), leading to enhanced FGF4 expression, and promoting differentiation.38a Collectively, the available data suggest that PARP-1 regulates various cellular differentiation processes through modulating transcription factors and subsequent gene expression.
5.5.3. PARP-1 in Somatic Cell Reprogramming
The breakthrough discovery by Takahashi and Yamanaka to reprogram somatic cells into pluripotent stem cells has promising therapeutic potential in regenerative medicine.196 Introducing four major transcription factors, Oct4, Sox2, Klf4, and c-Myc (together referred to as OSKM), into somatic cells enables these somatic cells to undergo epigenetic reprograming and generate inducible pluripotent stem cells (iPSCs).197 The first evidence of PARP-1’s role in this reprogramming process came from a functional screen of epigenetic modification factors that identified PARP-1 as a potent inducer of OSKM-mediated iPSC generation (Figure 14).89 During somatic cell reprogramming, PARP-1 is recruited to the Nanog and Esrrb loci, thereby establishing the active chromatin state, as well as promoting accessibility to the Oct4 transcription factor.89 Interestingly, Klf-4 or c-Myc can be replaced by PARP-1 in OSKM-induced iPSC generation,198 suggesting PARP-1 as a reprogramming factor. Mouse embryonic fibroblasts (MEFs) derived from Parp1 knockout mice significantly reduced reprogramming efficiency, and both DNA binding and enzymatic activity of PARP-1 are essential for iPSC generation,89,198 highlighting the importance of PARP-1 and PARylation during the reprogramming process. PARP-1 has also been shown to regulate Sox2 and subsequent FGF4 expression during iPSC generation.38b As in ES cell differentiation, PARP-1 acts by PARylating Sox2 transcription factor.38 Collectively, these data suggest that PARP-1 plays a profound role in cellular reprogramming and further suggest potential therapeutic applications for PARP inhibitors.
Figure 14.
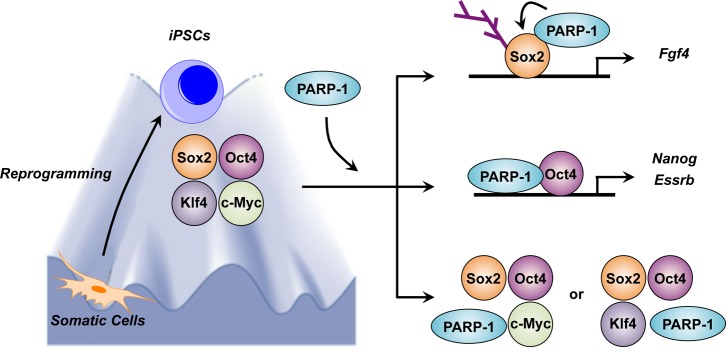
Regulation of somatic cell reprogramming by PARP-1. Somatic cells undergo epigenetic reprogramming to generate iPSCs when OSKM transcription factors are introduced. PARP-1 regulates iPSC generation by regulating Sox2 and subsequent FGF4 expression,38b and regulates Nanog and Essrb gene expression during reprogramming by promoting accessibility to the Oct4 transcription factor at the Nanog and Essrb loci.89 In addition, PARP-1 can also act as a direct reprogramming factor, replacing Klf-4 or c-Myc during OSKM-induced iPSC generation.198
5.6. Physiology and Pathophysiology of Other PARP Members
The PARP superfamily has 17 members, each sharing homology to the catalytic domain of PARP-1 containing a conserved β–α–loop−β–α NAD+-binding fold.3b Studies over the past decade have begun to reveal the physiological and pathological roles of the other PARP family members. Below, we highlight some of the key results in the context of the pathophysiological role of other PARPs.
5.6.1. PARP-2
PARP-2 (see Figure 1) was first discovered as a result of the residual PARP activity upon DNA damage in Parp1-deficient MEFs.150a Similar to PARP-1, PARP-2 is also activated by DNA-strand breaks and accumulates at sites of DNA damage.199Parp2–/– mice exhibit no obvious phenotype in the absence of genotoxic insults and are also viable and fertile. However, Parp2–/– mice are more susceptible to ionizing radiation190 and, like Parp1–/– mice, develop spontaneous tumors in a p53-deficient background.200 The similar phenotype observed in both Parp1 and Parp2 knockout mice, as well as the absence of any severe abnormalities, suggest functional redundancy between the two PARPs. In fact, mice deficient in both Parp1 and Parp2 are embryonic lethal prior to E8.0, while Parp1+/–Parp2–/– mice show female-specific lethality at E9.5, which is associated with X-chromosome instability.190 These results support the functional overlap of PARP-1 and PARP-2 in genome maintenance and embryonic development.
Although studies of PARP-2 are limited compared to PARP-1, PARP-2 has also been shown to regulate inflammatory responses. Parp2–/– mice show attenuated neuronal inflammation and lymphocyte infiltration in a mouse model of multiple sclerosis201 and are protected from focal cerebral ischemia.202 In addition, antisense oligonucleotide-mediated PARP-2 depletion reduces inflammation and leads to significant improvement of colitis in IL-10-deficient mice,203 illustrating the importance of PARP-2 in immune responses.
Recent reports have described PARP-2 as a novel regulator of various cellular differentiation programs. Parp2-deficient mice exhibit severely impaired spermatogenesis, thymopoiesis, and adipogenesis, supporting the role of PARP-2 in the regulation of differentiation. More specifically, Parp2–/– mice show defective meiotic sex chromosome inactivation, impaired chromosome segregation, and a defect in prophase of meiosis I due to massive apoptosis at pachytene and metaphase I stages, leading to subsequent spermatogenesis failure.204 Genetic ablation of Parp2 also affects thymopoiesis, leading to decreased numbers of CD4+ and CD8+ thymocytes as well as the total number of cells and thymus weight.205 In adipocyte differentiation, PARP-2 acts as a cofactor of the PPARγ transcription factor, controlling the expression of PPARγ target genes, which are involved in adipocyte function. PARP-2-deficient mice display reduced weight of white adipose tissue and reduced differentiation of preadipocytes to adipocytes.206 The functions of PARP-2 as a transcriptional coregulator of PPARγ raise the possibility of targeting PARP-2 in obesity and related metabolic disorders. Indeed, PARP-2 regulates energy expenditure in muscle and liver by modulating SIRT1 expression, supporting a potential role of PARP-2 as a metabolic regulator.207 Collectively, these data support the importance of PARP-2 in various pathophysiological processes.
5.6.2. PARP-3
Understanding the physiology and pathophysiology of PARP-3 (see Figure 1) has largely been focused on its role in DNA repair. Similar to PARP-1 and PARP-2, PARP-3-depleted cells exhibit a significant delay in the repair of DNA breaks upon chemical- or radiation-induced DNA damage.208 Interestingly, Parp3–/– mice are viable, fertile, and develop normally without any obvious phenotypes, similar to Parp1 or Parp2 null mice, suggesting the possibility that other PARPs may efficiently compensate for the loss of PARP-3.209 In line with this possibility, Parp1–/–Parp3–/– mice show increased sensitivity toward radiation-induced DNA damage as compared to either single disruption, supporting a functional synergistic crosstalk between both enzymes for DNA damage responses and genome maintenance.209
Upon DNA damage, PARP-3 uses a mechanism similar to that of PARP-1 to modulate DNA responses. Like PARP-1 (see above), PARP-3 is also recruited to laser-induced DNA damage sites and interacts with components of the classical nonhomologous end-joining (NHEJ) pathway including DNA-PKs, Ku70, Ku80, and DNA ligase IV.208 The enzymatic activity of PARP-3 is stimulated by DNA double-strand breaks, promoting the recruitment of APLF to DSB sites and accelerating DNA ligation during NHEJ.210 These studies support a role for PARP-3 as a DNA-damage response protein, which shows some similarities in function with PARP-1. Whether PARP-3 also regulates chromatin structure and gene expression like PARP-1, however, has yet to be determined.
6. Conclusions and Perspectives
The diverse subcellular locations and functions of PAR and the members of the PARP family provide many opportunities to impact molecular processes and biological outcomes. The functions of the enzymatically active PARP family members are intimately tied to the NAD+ biosynthetic pathways, which provide a ready supply of ADPR units for catalysis and targeting, and underlie some of the functional interplay observed with other NAD+-utilizing enzymes (e.g., SIRT1). Some of the best characterized functions of PARP-1 and other PARP family members are in DNA repair and the regulation of gene expression, the latter including modulation of chromatin structure, coregulatory functions, and alteration in DNA methylation. However, exciting recent studies have highlighted a role for PARPs in RNA biology, including rRNA synthesis, ribosome biogenesis, and mRNA regulation. Our understanding of the role of PARPs and PAR in the transcriptional and post-transcriptional regulation of gene expression through modulation of RNA is still in the early stages. Continued identification and characterization of functional interplay between PARPs and RNA may provide important insights into the role of PARPs in RNA regulation.
Although our understanding of PAR and PARPs has come a long way over the past 50 years, many unanswered questions remain. A greater understanding of the subcellular localization, enzymatic activities, and functions of the less well-characterized PARP family members will help to expand our understanding of the biology of this group of proteins. Proteomics and mass spectrometry are leading the way in identifying the sites of ADP-ribosylation on new PARP targets proteins. More needs to be done to understand why these proteins are targeted and how it affects their functions. Structural biology is helping us to understand the structure–function relationships of PARP family members and related enzymes, although we need structures of full-length proteins, as well as protein complexes, to understand the biophysical underpinnings of PARP (and PARG and NAD+ biosynthetic enzymes) activity and regulation (auto and allosteric). Such studies may help us to design more specific and more effective PARP inhibitors, perhaps those that can target sites on the protein outside of the catalytic domain. Furthermore, systematic approaches, combining metabolomics and genomics, will also help us to achieve a better understanding of the role of PARPs and NAD+ metabolism in transcription regulation, including the interplay between nuclear PARPs and SIRT1. Finally, a greater understanding of the physiology and pathophysiology of PARPs will help us to target them more effectively, using PARP inhibitors as therapeutics.
Acknowledgments
We thank members of the Kraus lab for critical feedback and thoughtful suggestions on this Review. We also thank Paul Todd from Editwallah for his help in editing the initial drafts of the manuscript. The PARP-related research in the Kraus lab is supported by an R01 grant from the NIH/NIDDK to W.L.K. and a postdoctoral fellowship from the DOD Breast Cancer Research Program to D.-S.K.
Biographies

Keun Woo Ryu is senior doctoral student at the University of Texas Southwestern Medical Center in the laboratory of Dr. W. Lee Kraus. He earned a B.S. degree from Seoul National University, Korea in 2009. Mr. Ryu’s research interests are in the role of nuclear NAD+ signaling in the control of transcription.

Dr. Dae-Seok Kim is a postdoctoral fellow at the University of Texas Southwestern Medical Center in the laboratory of Dr. W. Lee Kraus. He earned a B.S. degree (2003) and a M.S. degree (2005) in biosciences and biotechnology from the Hankuk University of Foreign Study, South Korea. He then earned a Ph.D. degree in molecular and cellular biology at Sungkyunkwan University, South Korea in 2011. Dr. Kim’s research interests are in the functional interactions between PARP-1 and RNAs, and how they determine cellular signaling outcomes in disease states. Dr. Kim’s research is supported by a postdoctoral fellowship from the United States Department of Defense Breast Cancer Research Program.

Dr. W. Lee Kraus is the Director of the Cecil H. and Ida Green Center for Reproductive Biology Sciences at the University of Texas Southwestern Medical Center. He is also Professor and Vice Chair for Basic Sciences in the Department of Obstetrics and Gynecology, and Professor of Pharmacology. Dr. Kraus earned a B.S. degree in Animal Physiology in 1989 from Cornell University in Ithaca, NY, followed by M.S. (1991) and Ph.D. (1994) degrees in Physiology and Biophysics from the University of Illinois, Urbana–Champaign, with Dr. Benita S. Katzenellenbogen, where he studied gene regulation by steroid hormone signaling pathways. He performed postdoctoral studies in biochemistry and molecular biology at the University of California, San Diego in 1998 with Dr. Jim Kadonaga, where he studied the mechanisms of estrogen-regulated transcription from chromatin. Dr. Kraus was on the faculty at Cornell University in Ithaca, NY from 1999 to 2010, rising through the ranks from assistant professor to full professor. Since July 2010, he has been at UT Southwestern Medical Center. Dr. Kraus’s research interests are related to the biochemistry, molecular biology, and physiology of signal-regulated gene expression. His research has led to new information about the connections between hormonal signaling and the gene-regulating effects of chromatin, which has implications for understanding and treating breast cancers. His work has also helped to characterize the estrogen-regulated transcriptome and identify thousands of novel noncoding RNAs. His recent work has led to some surprising new conclusions about the activity of poly(ADP-ribose) polymerase-1 (PARP-1), an NAD+-regulated nuclear factor that connects cellular NAD+ levels to nuclear signaling, chromatin structure, and gene expression. Dr. Kraus has been recognized for his research accomplishments by the Endocrine Society with the Richard E. Weitzman Memorial Award for research excellence in 2007 and the Ernst Oppenheimer Award for research excellence in 2014. Dr. Kraus holds the Cecil H. and Ida Green Distinguished Chair in Reproductive Biology Sciences at UT Southwestern Medical Center.
Author Contributions
∥ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Chambon P.; Weill J. D.; Mandel P. Biochem. Biophys. Res. Commun. 1963, 11, 39. [DOI] [PubMed] [Google Scholar]
- a Futai M.; Mizuno D.; Sugimura T. Biochem. Biophys. Res. Commun. 1967, 28, 395. [DOI] [PubMed] [Google Scholar]; b Nishizuka Y.; Ueda K.; Nakazawa K.; Hayaishi O. J. Biol. Chem. 1967, 242, 3164. [PubMed] [Google Scholar]; c Reeder R. H.; Ueda K.; Honjo T.; Nishizuka Y.; Hayaishi O. J. Biol. Chem. 1967, 242, 3172. [PubMed] [Google Scholar]; d Shimizu Y.; Hasegawa S.; Fujimura S.; Sugimura T. Biochem. Biophys. Res. Commun. 1967, 29, 80. [DOI] [PubMed] [Google Scholar]; e Sugimura T.; Fujimura S.; Hasegawa S.; Kawamura Y. Biochim. Biophys. Acta 1967, 138, 438. [DOI] [PubMed] [Google Scholar]
- a Ame J. C.; Spenlehauer C.; de Murcia G. BioEssays 2004, 26, 882. [DOI] [PubMed] [Google Scholar]; b Schreiber V.; Dantzer F.; Ame J. C.; de Murcia G. Nat. Rev. Mol. Cell Biol. 2006, 7, 517. [DOI] [PubMed] [Google Scholar]
- Vyas S.; Matic I.; Uchima L.; Rood J.; Zaja R.; Hay R. T.; Ahel I.; Chang P. Nat. Commun. 2014, 5, 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O.; Hassa P. O.; Luscher B.; Schuler H.; Koch-Nolte F. Trends Biochem. Sci. 2010, 35, 208. [DOI] [PubMed] [Google Scholar]
- a Chapman J. D.; Gagne J. P.; Poirier G. G.; Goodlett D. R. J. Proteome Res. 2013, 12, 1868. [DOI] [PubMed] [Google Scholar]; b Sharifi R.; Morra R.; Appel C. D.; Tallis M.; Chioza B.; Jankevicius G.; Simpson M. A.; Matic I.; Ozkan E.; Golia B.; Schellenberg M. J.; Weston R.; Williams J. G.; Rossi M. N.; Galehdari H.; Krahn J.; Wan A.; Trembath R. C.; Crosby A. H.; Ahel D.; Hay R.; Ladurner A. G.; Timinszky G.; Williams R. S.; Ahel I. EMBO J. 2013, 32, 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang Y.; Wang J.; Ding M.; Yu Y. Nat. Methods 2013, 10, 981. [DOI] [PubMed] [Google Scholar]
- a Luo X.; Kraus W. L. Genes Dev. 2012, 26, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Krishnakumar R.; Kraus W. L. Mol. Cell 2010, 39, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier M. F.; Riccio A. A.; Pascal J. M. Nucleic Acids Res. 2014, 42, 7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa P. O.; Hottiger M. O. Front. Biosci. 2008, 13, 3046. [DOI] [PubMed] [Google Scholar]
- Gibson B. A.; Kraus W. L. Nat. Rev. Mol. Cell Biol. 2012, 13, 411. [DOI] [PubMed] [Google Scholar]
- a D’Amours D.; Desnoyers S.; D’Silva I.; Poirier G. G. Biochem. J. 1999, 342, 249. [PMC free article] [PubMed] [Google Scholar]; b Ji Y.; Tulin A. V. Curr. Opin. Genet. Dev. 2010, 20, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hassa P. O.; Haenni S. S.; Elser M.; Hottiger M. O. Microbiol. Mol. Biol. Rev. 2006, 70, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kraus W. L.; Hottiger M. O. Mol. Aspects Med. 2013, 34, 1109. [DOI] [PubMed] [Google Scholar]
- a Hottiger M. O.; Boothby M.; Koch-Nolte F.; Luscher B.; Martin N. M.; Plummer R.; Wang Z. Q.; Ziegler M. Sci. Signaling 2011, 4, mr5. [DOI] [PubMed] [Google Scholar]; b Rosado M. M.; Bennici E.; Novelli F.; Pioli C. Immunology 2013, 139, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kim M. Y.; Zhang T.; Kraus W. L. Genes Dev. 2005, 19, 1951. [DOI] [PubMed] [Google Scholar]; b Zhang T.; Berrocal J. G.; Yao J.; DuMond M. E.; Krishnakumar R.; Ruhl D. D.; Ryu K. W.; Gamble M. J.; Kraus W. L. J. Biol. Chem. 2012, 287, 12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F.; Ramirez-Hernandez M. H.; Ziegler M. Trends Biochem. Sci. 2004, 29, 111. [DOI] [PubMed] [Google Scholar]
- Chiarugi A.; Dolle C.; Felici R.; Ziegler M. Nat. Rev. Cancer 2012, 12, 741. [DOI] [PubMed] [Google Scholar]
- Houtkooper R. H.; Canto C.; Wanders R. J.; Auwerx J. Endocr. Rev. 2010, 31, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli R.; Felczak K.; Cappellacci L. Curr. Med. Chem. 2011, 18, 1973. [DOI] [PubMed] [Google Scholar]
- Revollo J. R.; Grimm A. A.; Imai S. J. Biol. Chem. 2004, 279, 50754. [DOI] [PubMed] [Google Scholar]
- Kornberg A. J. Biol. Chem. 1948, 176, 1475. [PubMed] [Google Scholar]
- Kornberg A.; Pricer W. E. Jr. J. Biol. Chem. 1951, 191, 535. [PubMed] [Google Scholar]
- Berger F.; Lau C.; Dahlmann M.; Ziegler M. J. Biol. Chem. 2005, 280, 36334. [DOI] [PubMed] [Google Scholar]
- a Emanuelli M.; Carnevali F.; Saccucci F.; Pierella F.; Amici A.; Raffaelli N.; Magni G. J. Biol. Chem. 2001, 276, 406. [DOI] [PubMed] [Google Scholar]; b Raffaelli N.; Sorci L.; Amici A.; Emanuelli M.; Mazzola F.; Magni G. Biochem. Biophys. Res. Commun. 2002, 297, 835. [DOI] [PubMed] [Google Scholar]; c Zhang X.; Kurnasov O. V.; Karthikeyan S.; Grishin N. V.; Osterman A. L.; Zhang H. J. Biol. Chem. 2003, 278, 13503. [DOI] [PubMed] [Google Scholar]
- Schweiger M.; Hennig K.; Lerner F.; Niere M.; Hirsch-Kauffmann M.; Specht T.; Weise C.; Oei S. L.; Ziegler M. FEBS Lett. 2001, 492, 95. [DOI] [PubMed] [Google Scholar]
- Gomes A. P.; Price N. L.; Ling A. J.; Moslehi J. J.; Montgomery M. K.; Rajman L.; White J. P.; Teodoro J. S.; Wrann C. D.; Hubbard B. P.; Mercken E. M.; Palmeira C. M.; de Cabo R.; Rolo A. P.; Turner N.; Bell E. L.; Sinclair D. A. Cell 2013, 155, 1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L.; Janeckova L.; Wagner D.; Mazzola F.; Cialabrini L.; Di Stefano M.; Orsomando G.; Magni G.; Bendotti C.; Smyth N.; Coleman M. FEBS J. 2011, 278, 2666. [DOI] [PubMed] [Google Scholar]
- a Zhou T.; Kurnasov O.; Tomchick D. R.; Binns D. D.; Grishin N. V.; Marquez V. E.; Osterman A. L.; Zhang H. J. Biol. Chem. 2002, 277, 13148. [DOI] [PubMed] [Google Scholar]; b Garavaglia S.; D’Angelo I.; Emanuelli M.; Carnevali F.; Pierella F.; Magni G.; Rizzi M. J. Biol. Chem. 2002, 277, 8524. [DOI] [PubMed] [Google Scholar]; c Werner E.; Ziegler M.; Lerner F.; Schweiger M.; Heinemann U. FEBS Lett. 2002, 516, 239. [DOI] [PubMed] [Google Scholar]
- Steffen J. D.; Brody J. R.; Armen R. S.; Pascal J. M. Front. Oncol. 2013, 3, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf A.; de Murcia G.; Schulz G. E. Biochemistry 1998, 37, 3893. [DOI] [PubMed] [Google Scholar]
- Otto H.; Reche P. A.; Bazan F.; Dittmar K.; Haag F.; Koch-Nolte F. BMC Genomics 2005, 6, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf A.; Rolli V.; de Murcia G.; Schulz G. E. J. Mol. Biol. 1998, 278, 57. [DOI] [PubMed] [Google Scholar]
- Kiehlbauch C. C.; Aboul-Ela N.; Jacobson E. L.; Ringer D. P.; Jacobson M. K. Anal. Biochem. 1993, 208, 26. [DOI] [PubMed] [Google Scholar]
- a Gagne J. P.; Pic E.; Isabelle M.; Krietsch J.; Ethier C.; Paquet E.; Kelly I.; Boutin M.; Moon K. M.; Foster L. J.; Poirier G. G. Nucleic Acids Res. 2012, 40, 7788. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Isabelle M.; Moreel X.; Gagne J. P.; Rouleau M.; Ethier C.; Gagne P.; Hendzel M. J.; Poirier G. G. Proteome Sci. 2010, 8, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Daniels C. M.; Ong S. E.; Leung A. K. J. Proteome Res. 2014, 13, 3510. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vivelo C. A.; Leung A. K.; . Proteomics 2014, DOI: 10.1002/pmic.201400217. [DOI] [PMC free article] [PubMed]
- Carter-O’Connell I.; Jin H.; Morgan R. K.; David L. L.; Cohen M. S. J. Am. Chem. Soc. 2014, 136, 5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Huang J. Y.; Chen W. H.; Chang Y. L.; Wang H. T.; Chuang W. T.; Lee S. C. Nucleic Acids Res. 2006, 34, 2398. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Heo K.; Kim H.; Choi S. H.; Choi J.; Kim K.; Gu J.; Lieber M. R.; Yang A. S.; An W. Mol. Cell 2008, 30, 86. [DOI] [PubMed] [Google Scholar]
- a Khoury-Haddad H.; Guttmann-Raviv N.; Ipenberg I.; Huggins D.; Jeyasekharan A. D.; Ayoub N. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E728. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sala A.; La Rocca G.; Burgio G.; Kotova E.; Di Gesu D.; Collesano M.; Ingrassia A. M.; Tulin A. V.; Corona D. F. PLoS Biol. 2008, 6, e252. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Krishnakumar R.; Kraus W. L. Mol. Cell 2010, 39, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaniolo K.; Desnoyers S.; Leclerc S.; Guerin S. L. BMC Mol. Biol. 2007, 8, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabisi O. A.; Soto-Nieves N.; Nieves E.; Yang T. T.; Yang X.; Yu R. Y.; Suk H. Y.; Macian F.; Chow C. W. Mol. Cell. Biol. 2008, 28, 2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gao F.; Kwon S. W.; Zhao Y.; Jin Y. J. Biol. Chem. 2009, 284, 22263. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weber F. A.; Bartolomei G.; Hottiger M. O.; Cinelli P. Stem Cells 2013, 31, 2364. [DOI] [PubMed] [Google Scholar]
- Lonn P.; van der Heide L. P.; Dahl M.; Hellman U.; Heldin C. H.; Moustakas A. Mol. Cell 2010, 40, 521. [DOI] [PubMed] [Google Scholar]
- Asher G.; Reinke H.; Altmeyer M.; Gutierrez-Arcelus M.; Hottiger M. O.; Schibler U. Cell 2010, 142, 943. [DOI] [PubMed] [Google Scholar]
- Wang C.; Zhang F.; Wang L.; Zhang Y.; Li X.; Huang K.; Du M.; Liu F.; Huang S.; Guan Y.; Huang D.; Huang K. Mol. Cell. Biol. 2013, 33, 4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Wang Y.; Wang L.; Luo X.; Huang K.; Wang C.; Du M.; Liu F.; Luo T.; Huang D.; Huang K. J. Biol. Chem. 2013, 288, 11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietsch J.; Rouleau M.; Pic E.; Ethier C.; Dawson T. M.; Dawson V. L.; Masson J. Y.; Poirier G. G.; Gagne J. P. Mol. Aspects Med. 2013, 34, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Masson M.; Niedergang C.; Schreiber V.; Muller S.; Menissier-de Murcia J.; de Murcia G. Mol. Cell. Biol. 1998, 18, 3563. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Okano S.; Lan L.; Caldecott K. W.; Mori T.; Yasui A. Mol. Cell. Biol. 2003, 23, 3974. [DOI] [PMC free article] [PubMed] [Google Scholar]; c El-Khamisy S. F.; Masutani M.; Suzuki H.; Caldecott K. W. Nucleic Acids Res. 2003, 31, 5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou D. M.; Adamson B.; Dephoure N. E.; Tan X.; Nottke A. C.; Hurov K. E.; Gygi S. P.; Colaiacovo M. P.; Elledge S. J. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ahel I.; Ahel D.; Matsusaka T.; Clark A. J.; Pines J.; Boulton S. J.; West S. C. Nature 2008, 451, 81. [DOI] [PubMed] [Google Scholar]; b Li G. Y.; McCulloch R. D.; Fenton A. L.; Cheung M.; Meng L.; Ikura M.; Koch C. A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Timinszky G.; Till S.; Hassa P. O.; Hothorn M.; Kustatscher G.; Nijmeijer B.; Colombelli J.; Altmeyer M.; Stelzer E. H.; Scheffzek K.; Hottiger M. O.; Ladurner A. G. Nat. Struct. Mol. Biol. 2009, 16, 923. [DOI] [PubMed] [Google Scholar]; b Ahel D.; Horejsi Z.; Wiechens N.; Polo S. E.; Garcia-Wilson E.; Ahel I.; Flynn H.; Skehel M.; West S. C.; Jackson S. P.; Owen-Hughes T.; Boulton S. J. Science 2009, 325, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus W. L. Nat. Struct. Mol. Biol. 2009, 16, 904. [DOI] [PubMed] [Google Scholar]
- a Li M.; Lu L. Y.; Yang C. Y.; Wang S.; Yu X. Genes Dev. 2013, 27, 1752. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li M.; Yu X. Cancer Cell 2013, 23, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Chen Y.; Li M.; Yu X. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kang H. C.; Lee Y. I.; Shin J. H.; Andrabi S. A.; Chi Z.; Gagne J. P.; Lee Y.; Ko H. S.; Lee B. D.; Poirier G. G.; Dawson V. L.; Dawson T. M. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b DaRosa P. A.; Wang Z.; Jiang X.; Pruneda J. N.; Cong F.; Klevit R. E.; Xu W.. Nature, 2014, DOI: 10.1038/nature13826. [DOI] [PMC free article] [PubMed]
- Wang Z.; Michaud G. A.; Cheng Z.; Zhang Y.; Hinds T. R.; Fan E.; Cong F.; Xu W. Genes Dev. 2012, 26, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi S. A.; Kang H. C.; Haince J. F.; Lee Y. I.; Zhang J.; Chi Z.; West A. B.; Koehler R. C.; Poirier G. G.; Dawson T. M.; Dawson V. L. Nat. Med. 2011, 17, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang Y.; Liu S.; Mickanin C.; Feng Y.; Charlat O.; Michaud G. A.; Schirle M.; Shi X.; Hild M.; Bauer A.; Myer V. E.; Finan P. M.; Porter J. A.; Huang S. M.; Cong F. Nat. Cell Biol. 2011, 13, 623. [DOI] [PubMed] [Google Scholar]; b Huang S. M.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G. A.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. Nature 2009, 461, 614. [DOI] [PubMed] [Google Scholar]
- a Jackson T. M.; Rawling J. M.; Roebuck B. D.; Kirkland J. B. J. Nutr. 1995, 125, 1455. [DOI] [PubMed] [Google Scholar]; b Sauve A. A.; Schramm V. L. Biochemistry 2003, 42, 9249. [DOI] [PubMed] [Google Scholar]; c Preiss J.; Schlaeger R.; Hilz H. FEBS Lett. 1971, 19, 244. [DOI] [PubMed] [Google Scholar]
- Coleman M. Nat. Rev. Neurosci. 2005, 6, 889. [DOI] [PubMed] [Google Scholar]
- Mack T. G.; Reiner M.; Beirowski B.; Mi W.; Emanuelli M.; Wagner D.; Thomson D.; Gillingwater T.; Court F.; Conforti L.; Fernando F. S.; Tarlton A.; Andressen C.; Addicks K.; Magni G.; Ribchester R. R.; Perry V. H.; Coleman M. P. Nat. Neurosci. 2001, 4, 1199. [DOI] [PubMed] [Google Scholar]
- a Araki T.; Sasaki Y.; Milbrandt J. Science 2004, 305, 1010. [DOI] [PubMed] [Google Scholar]; b Press C.; Milbrandt J. J. Neurosci. 2008, 28, 4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sasaki Y.; Araki T.; Milbrandt J. J. Neurosci. 2006, 26, 8484. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fang E. F.; Scheibye-Knudsen M.; Brace L. E.; Kassahun H.; Sengupta T.; Nilsen H.; Mitchell J. R.; Croteau D. L.; Bohr V. A. Cell 2014, 157, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F.; Lau C.; Ziegler M. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Piston D. W.; Goodman R. H. Science 2002, 295, 1895. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Berrocal J. G.; Frizzell K. M.; Gamble M. J.; DuMond M. E.; Krishnakumar R.; Yang T.; Sauve A. A.; Kraus W. L. J. Biol. Chem. 2009, 284, 20408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T.; Yang L.; Kabra N.; Chen L.; Koomen J.; Haura E. B.; Chen J. J. Biol. Chem. 2013, 288, 20908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hassa P. O.; Haenni S. S.; Buerki C.; Meier N. I.; Lane W. S.; Owen H.; Gersbach M.; Imhof R.; Hottiger M. O. J. Biol. Chem. 2005, 280, 40450. [DOI] [PubMed] [Google Scholar]; b Rajamohan S. B.; Pillai V. B.; Gupta M.; Sundaresan N. R.; Birukov K. G.; Samant S.; Hottiger M. O.; Gupta M. P. Mol. Cell. Biol. 2009, 29, 4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ko H. L.; Ren E. C. Biomolecules 2012, 2, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Herceg Z.; Wang Z. Q. Mutat. Res. 2001, 477, 97. [DOI] [PubMed] [Google Scholar]; c Javle M.; Curtin N. J. Br. J. Cancer 2011, 105, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jeggo P. A. Curr. Biol. 1998, 8, R49. [DOI] [PubMed] [Google Scholar]; e Smulson M. E.; Simbulan-Rosenthal C. M.; Boulares A. H.; Yakovlev A.; Stoica B.; Iyer S.; Luo R.; Haddad B.; Wang Z. Q.; Pang T.; Jung M.; Dritschilo A.; Rosenthal D. S. Adv. Enzyme Regul. 2000, 40, 183. [DOI] [PubMed] [Google Scholar]
- a Kraus W. L.; Lis J. T. Cell 2003, 113, 677. [DOI] [PubMed] [Google Scholar]; b Kraus W. L. Curr. Opin. Cell Biol. 2008, 20, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Leung A.; Todorova T.; Ando Y.; Chang P. RNA Biol. 2012, 9, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Leung A. K.; Vyas S.; Rood J. E.; Bhutkar A.; Sharp P. A.; Chang P. Mol. Cell 2011, 42, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Widom J. Curr. Biol. 1998, 8, R788. [DOI] [PubMed] [Google Scholar]; b Wolffe A. P.; Guschin D. J. Struct. Biol. 2000, 129, 102. [DOI] [PubMed] [Google Scholar]
- a Petesch S. J.; Lis J. T.. Mol. Cell 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tulin A.; Spradling A. Science 2003, 299, 560. [DOI] [PubMed] [Google Scholar]
- Poirier G. G.; de Murcia G.; Jongstra-Bilen J.; Niedergang C.; Mandel P. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kim M. Y.; Mauro S.; Gevry N.; Lis J. T.; Kraus W. L. Cell 2004, 119, 803. [DOI] [PubMed] [Google Scholar]; b Krishnakumar R.; Gamble M.; Frizzell K.; Berrocal J.; Kininis M.; Kraus W. Science 2008, 319, 819. [DOI] [PubMed] [Google Scholar]
- a Bonicalzi M. E.; Haince J. F.; Droit A.; Poirier G. G. Cell. Mol. Life Sci. 2005, 62, 739. [DOI] [PubMed] [Google Scholar]; b Min W.; Wang Z. Q. Front. Biosci., Landmark Ed. 2009, 14, 1619. [DOI] [PubMed] [Google Scholar]
- a Burkle A.; Virag L. Mol. Aspects Med. 2013, 34, 1046. [DOI] [PubMed] [Google Scholar]; b Mashimo M.; Kato J.; Moss J. DNA Repair 2014, 23November 201488–94. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Virag L.; Robaszkiewicz A.; Rodriguez-Vargas J. M.; Oliver F. J. Mol. Aspects Med. 2013, 34, 1153. [DOI] [PubMed] [Google Scholar]
- Feijs K. L.; Verheugd P.; Luscher B. FEBS J. 2013, 280, 3519. [DOI] [PubMed] [Google Scholar]
- a Cohen-Armon M.; Visochek L.; Rozensal D.; Kalal A.; Geistrikh I.; Klein R.; Bendetz-Nezer S.; Yao Z.; Seger R. Mol. Cell 2007, 25, 297. [DOI] [PubMed] [Google Scholar]; b Geistrikh I.; Visochek L.; Klein R.; Miller L.; Mittelman L.; Shainberg A.; Cohen-Armon M. Biochem. J. 2011, 438, 337. [DOI] [PubMed] [Google Scholar]; c Martinez-Zamudio R.; Ha H. C. Mol. Cell. Biol. 2012, 32, 2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnola A.; Naumova N.; Shah M.; Tulin A. V. J. Biol. Chem. 2007, 282, 32511. [DOI] [PubMed] [Google Scholar]
- Kotova E.; Lodhi N.; Jarnik M.; Pinnola A. D.; Ji Y.; Tulin A. V. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Karras G. I.; Kustatscher G.; Buhecha H. R.; Allen M. D.; Pugieux C.; Sait F.; Bycroft M.; Ladurner A. G. EMBO J. 2005, 24, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kustatscher G.; Hothorn M.; Pugieux C.; Scheffzek K.; Ladurner A. G. Nat. Struct. Mol. Biol. 2005, 12, 624. [DOI] [PubMed] [Google Scholar]
- Muthurajan U. M.; Hepler M. R.; Hieb A. R.; Clark N. J.; Kramer M.; Yao T.; Luger K. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra P. V.; Ahel D.; Ryan D. P.; Weston R.; Wiechens N.; Kraehenbuehl R.; Owen-Hughes T.; Ahel I. Mol. Cell 2011, 41, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk A. J.; Timinszky G.; Kong S. E.; Jin J.; Cai Y.; Swanson S. K.; Washburn M. P.; Florens L.; Ladurner A. G.; Conaway J. W.; Conaway R. C. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hassa P. O.; Buerki C.; Lombardi C.; Imhof R.; Hottiger M. O. J. Biol. Chem. 2003, 278, 45145. [DOI] [PubMed] [Google Scholar]; b Hassa P. O.; Covic M.; Bedford M. T.; Hottiger M. O. J. Mol. Biol. 2008, 377, 668. [DOI] [PubMed] [Google Scholar]
- Pavri R.; Lewis B.; Kim T. K.; Dilworth F. J.; Erdjument-Bromage H.; Tempst P.; de Murcia G.; Evans R.; Chambon P.; Reinberg D. Mol. Cell 2005, 18, 83. [DOI] [PubMed] [Google Scholar]
- Ju B. G.; Solum D.; Song E. J.; Lee K. J.; Rose D. W.; Glass C. K.; Rosenfeld M. G. Cell 2004, 119, 815. [DOI] [PubMed] [Google Scholar]
- Ju B. G.; Lunyak V. V.; Perissi V.; Garcia-Bassets I.; Rose D. W.; Glass C. K.; Rosenfeld M. G. Science 2006, 312, 1798. [DOI] [PubMed] [Google Scholar]
- a Attwood J. T.; Yung R. L.; Richardson B. C. Cell. Mol. Life Sci. 2002, 59, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Caiafa P.; Zampieri M. J. Cell. Biochem. 2005, 94, 257. [DOI] [PubMed] [Google Scholar]
- Caiafa P.; Guastafierro T.; Zampieri M. FASEB J. 2009, 23, 672. [DOI] [PubMed] [Google Scholar]
- Reale A.; Matteis G. D.; Galleazzi G.; Zampieri M.; Caiafa P. Oncogene 2005, 24, 13. [DOI] [PubMed] [Google Scholar]
- Doege C. A.; Inoue K.; Yamashita T.; Rhee D. B.; Travis S.; Fujita R.; Guarnieri P.; Bhagat G.; Vanti W. B.; Shih A.; Levine R. L.; Nik S.; Chen E. I.; Abeliovich A. Nature 2012, 488, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guetg C.; Scheifele F.; Rosenthal F.; Hottiger M. O.; Santoro R. Mol. Cell 2012, 45, 790. [DOI] [PubMed] [Google Scholar]
- Boamah E. K.; Kotova E.; Garabedian M.; Jarnik M.; Tulin A. V. PLoS Genet. 2012, 8, e1002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A.; Fischer B.; Eichelbaum K.; Horos R.; Beckmann B. M.; Strein C.; Davey N. E.; Humphreys D. T.; Preiss T.; Steinmetz L. M.; Krijgsveld J.; Hentze M. W. Cell 2012, 149, 1393. [DOI] [PubMed] [Google Scholar]
- Di Giammartino D. C.; Shi Y.; Manley J. L. Mol. Cell 2013, 49, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y.; Tulin A. V. Nucleic Acids Res. 2009, 37, 3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G.; Guo X.; Goff S. P. Science 2002, 297, 1703. [DOI] [PubMed] [Google Scholar]
- Leger K.; Bar D.; Savic N.; Santoro R.; Hottiger M. O. Nucleic Acids Res. 2014, 42, 5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick J. S.; Makunin I. V. Hum. Mol. Genet. 2006, 15 Spec No 1, R17. [DOI] [PubMed] [Google Scholar]
- a Goodrich J. A.; Kugel J. F. Nat. Rev. Mol. Cell Biol. 2006, 7, 612. [DOI] [PubMed] [Google Scholar]; b Mercer T. R.; Mattick J. S. Nat. Struct. Mol. Biol. 2013, 20, 300. [DOI] [PubMed] [Google Scholar]
- Esteller M. Nat. Rev. Genet. 2011, 12, 861. [DOI] [PubMed] [Google Scholar]
- a Szymanski M.; Barciszewska M. Z.; Erdmann V. A.; Barciszewski J. Biochim. Biophys. Acta 2005, 1756, 65. [DOI] [PubMed] [Google Scholar]; b Kaikkonen M. U.; Lam M. T.; Glass C. K. Cardiovasc. Res. 2011, 90, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee J. T. Science 2012, 338, 1435. [DOI] [PubMed] [Google Scholar]; d Brantl S. Curr. Opin. Microbiol. 2007, 10, 102. [DOI] [PubMed] [Google Scholar]; e McKeown M. Curr. Opin. Cell Biol. 1993, 5, 448. [DOI] [PubMed] [Google Scholar]; f Perreault J.; Perreault J. P.; Boire G. Mol. Biol. Evol. 2007, 24, 1678. [DOI] [PubMed] [Google Scholar]
- Huarte M.; Guttman M.; Feldser D.; Garber M.; Koziol M. J.; Kenzelmann-Broz D.; Khalil A. M.; Zuk O.; Amit I.; Rabani M.; Attardi L. D.; Regev A.; Lander E. S.; Jacks T.; Rinn J. L. Cell 2010, 142, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M. C.; Manor O.; Wan Y.; Mosammaparast N.; Wang J. K.; Lan F.; Shi Y.; Segal E.; Chang H. Y. Science 2010, 329, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Rowley M. J.; Bohmdorfer G.; Wierzbicki A. T. Mol. Cell 2013, 49, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Murano K.; Okuwaki M.; Hisaoka M.; Nagata K. Mol. Cell. Biol. 2008, 28, 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Grummt I.; Pikaard C. S. Nat. Rev. Mol. Cell Biol. 2003, 4, 641. [DOI] [PubMed] [Google Scholar]
- Santoro R. Cell. Mol. Life Sci. 2005, 62, 2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro R.; Li J.; Grummt I. Nat. Genet. 2002, 32, 393. [DOI] [PubMed] [Google Scholar]
- Santoro R.; Grummt I. Mol. Cell 2001, 8, 719. [DOI] [PubMed] [Google Scholar]
- Grummt I. Hum. Mol. Genet. 2007, 16 Spec No 1, R21. [DOI] [PubMed] [Google Scholar]
- a Mayer C.; Neubert M.; Grummt I. EMBO Rep. 2008, 9, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Santoro R.; Schmitz K. M.; Sandoval J.; Grummt I. EMBO Rep. 2010, 11, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mayer C.; Schmitz K. M.; Li J.; Grummt I.; Santoro R. Mol. Cell 2006, 22, 351. [DOI] [PubMed] [Google Scholar]
- Tschochner H.; Hurt E. Trends Cell Biol. 2003, 13, 255. [DOI] [PubMed] [Google Scholar]
- a Leary D. J.; Huang S. FEBS Lett. 2001, 509, 145. [DOI] [PubMed] [Google Scholar]; b Scheer U.; Hock R. Curr. Opin. Cell Biol. 1999, 11, 385. [DOI] [PubMed] [Google Scholar]
- a Nazar R. N. IUBMB Life 2004, 56, 457. [DOI] [PubMed] [Google Scholar]; b Boisvert F. M.; van Koningsbruggen S.; Navascues J.; Lamond A. I. Nat. Rev. Mol. Cell Biol. 2007, 8, 574. [DOI] [PubMed] [Google Scholar]
- Lam M. H.; Thomas R. J.; Martin T. J.; Gillespie M. T.; Jans D. A. Immunol. Cell Biol. 2000, 78, 395. [DOI] [PubMed] [Google Scholar]
- Mosgoeller W.; Steiner M.; Hozak P.; Penner E.; Wesierska-Gadek J. J. Cell Sci. 1996, 109, 409. [DOI] [PubMed] [Google Scholar]
- a Andersen J. S.; Lyon C. E.; Fox A. H.; Leung A. K.; Lam Y. W.; Steen H.; Mann M.; Lamond A. I. Curr. Biol. 2002, 12, 1. [DOI] [PubMed] [Google Scholar]; b Scherl A.; Coute Y.; Deon C.; Calle A.; Kindbeiter K.; Sanchez J. C.; Greco A.; Hochstrasser D.; Diaz J. J. Mol. Biol. Cell 2002, 13, 4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnoyers S.; Kaufmann S. H.; Poirier G. G. Exp. Cell Res. 1996, 227, 146. [DOI] [PubMed] [Google Scholar]
- Torrano V.; Navascues J.; Docquier F.; Zhang R.; Burke L. J.; Chernukhin I.; Farrar D.; Leon J.; Berciano M. T.; Renkawitz R.; Klenova E.; Lafarga M.; Delgado M. D. J. Cell Sci. 2006, 119, 1746. [DOI] [PubMed] [Google Scholar]
- Leitinger N.; Wesierska-Gadek J. J. Cell. Biochem. 1993, 52, 153. [DOI] [PubMed] [Google Scholar]
- a Lindstrom M. S. Biochem. Res. Int. 2011, 2011, 195209. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ginisty H.; Amalric F.; Bouvet P. EMBO J. 1998, 17, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotova E.; Jarnik M.; Tulin A. V. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai S.; Kanai M.; Ohashi S.; Okamoto K.; Yamada M.; Takahashi H.; Miwa M. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins A. S.; Iglehart J. D.; Lazaro J. B. Nucleic Acids Res. 2013, 41, 7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero P. A.; Maggert K. A. PLoS One 2011, 6, e16401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama A.; Ohmori K.; Fujimura A.; Minami H.; Yasuzawa-Tanaka K.; Kuroda T.; Oie S.; Daitoku H.; Okuwaki M.; Nagata K.; Fukamizu A.; Kimura K.; Shimizu T.; Yanagisawa J. Cell 2008, 133, 627. [DOI] [PubMed] [Google Scholar]
- Lukong K. E.; Chang K. W.; Khandjian E. W.; Richard S. Trends Genet. 2008, 24, 416. [DOI] [PubMed] [Google Scholar]
- Glisovic T.; Bachorik J. L.; Yong J.; Dreyfuss G. FEBS Lett. 2008, 582, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Di Giammartino D. C.; Taylor D.; Sarkeshik A.; Rice W. J.; Yates J. R. 3rd; Frank J.; Manley J. L. Mol. Cell 2009, 33, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B.; Hu J.; Zhang H.; Lutz C. S. Nucleic Acids Res. 2005, 33, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vethantham V.; Rao N.; Manley J. L. Genes Dev. 2008, 22, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; Manley J. L. Mol. Cell. Biol. 1998, 18, 5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S. C.; Lamond A. I. J. Cell Biol. 2002, 159, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. E. Biochim. Biophys. Acta 2008, 1783, 2108. [DOI] [PubMed] [Google Scholar]
- Kotova E.; Jarnik M.; Tulin A. V. PLoS Genet. 2009, 5, e1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mayeda A.; Krainer A. R. Cell 1992, 68, 365. [DOI] [PubMed] [Google Scholar]; b Dreyfuss G.; Kim V. N.; Kataoka N. Nat. Rev. Mol. Cell Biol. 2002, 3, 195. [DOI] [PubMed] [Google Scholar]
- Ji Y.; Tulin A. V. Int. J. Mol. Sci. 2013, 14, 16168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P.; Kedersha N. Trends Biochem. Sci. 2008, 33, 141. [DOI] [PubMed] [Google Scholar]
- Buchan J. R.; Parker R. Mol. Cell 2009, 36, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W.; Denman R. B. Mol. Biol. Int. 2011, 2011, 137459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd R. E. PLoS Pathog. 2012, 8, e1002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N.; Anderson P. Biochem. Soc. Trans. 2002, 30, 963. [DOI] [PubMed] [Google Scholar]
- Hoyle N. P.; Castelli L. M.; Campbell S. G.; Holmes L. E.; Ashe M. P. J. Cell Biol. 2007, 179, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagopal V.; Parker R. Curr. Opin. Cell Biol. 2009, 21, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks N.; Kedersha N.; Ayodele M.; Shen L.; Stoecklin G.; Dember L. M.; Anderson P. Mol. Biol. Cell 2004, 15, 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Han T. W.; Kato M.; Xie S.; Wu L. C.; Mirzaei H.; Pei J.; Chen M.; Xie Y.; Allen J.; Xiao G.; McKnight S. L. Cell 2012, 149, 768. [DOI] [PubMed] [Google Scholar]; b Kato M.; Han T. W.; Xie S.; Shi K.; Du X.; Wu L. C.; Mirzaei H.; Goldsmith E. J.; Longgood J.; Pei J.; Grishin N. V.; Frantz D. E.; Schneider J. W.; Chen S.; Li L.; Sawaya M. R.; Eisenberg D.; Tycko R.; McKnight S. L. Cell 2012, 149, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung A. K. J. Cell Biol. 2014, 205, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung A. K.; Calabrese J. M.; Sharp P. A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa S.; Shiratori S.; Yamato H.; Kameyama T.; Kitatsuji C.; Kashigi F.; Goto S.; Kameoka S.; Fujikura D.; Yamada T.; Mizutani T.; Kazumata M.; Sato M.; Tanaka J.; Asaka M.; Ohba Y.; Miyazaki T.; Imamura M.; Takaoka A. Nat. Immunol. 2011, 12, 37. [DOI] [PubMed] [Google Scholar]
- Aguiar R. C.; Takeyama K.; He C.; Kreinbrink K.; Shipp M. A. J. Biol. Chem. 2005, 280, 33756. [DOI] [PubMed] [Google Scholar]
- Daugherty M. D.; Young J. M.; Kerns J. A.; Malik H. S. PLoS Genet. 2014, 10, e1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yelamos J.; Schreiber V.; Dantzer F. Trends Mol. Med. 2008, 14, 169. [DOI] [PubMed] [Google Scholar]; b Ame J. C.; Rolli V.; Schreiber V.; Niedergang C.; Apiou F.; Decker P.; Muller S.; Hoger T.; Menissier-de Murcia J.; de Murcia G. J. Biol. Chem. 1999, 274, 17860. [DOI] [PubMed] [Google Scholar]
- Meder V. S.; Boeglin M.; de Murcia G.; Schreiber V. J. Cell Sci. 2005, 118, 211. [DOI] [PubMed] [Google Scholar]
- Malanga M.; Czubaty A.; Girstun A.; Staron K.; Althaus F. R. J. Biol. Chem. 2008, 283, 19991. [DOI] [PubMed] [Google Scholar]
- Wang Z. Q.; Auer B.; Stingl L.; Berghammer H.; Haidacher D.; Schweiger M.; Wagner E. F. Genes Dev. 1995, 9, 509. [DOI] [PubMed] [Google Scholar]
- de Murcia J. M.; Niedergang C.; Trucco C.; Ricoul M.; Dutrillaux B.; Mark M.; Oliver F. J.; Masson M.; Dierich A.; LeMeur M.; Walztinger C.; Chambon P.; de Murcia G. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani M.; Nozaki T.; Nishiyama E.; Shimokawa T.; Tachi Y.; Suzuki H.; Nakagama H.; Wakabayashi K.; Sugimura T. Mol. Cell. Biochem. 1999, 193, 149. [PubMed] [Google Scholar]
- Wang Z. Q.; Stingl L.; Morrison C.; Jantsch M.; Los M.; Schulze-Osthoff K.; Wagner E. F. Genes Dev. 1997, 11, 2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani M.; Fujimori H. Mol. Aspects Med. 2013, 34, 1202. [DOI] [PubMed] [Google Scholar]
- a Tong W. M.; Hande M. P.; Lansdorp P. M.; Wang Z. Q. Mol. Cell. Biol. 2001, 21, 4046. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tong W. M.; Yang Y. G.; Cao W. H.; Galendo D.; Frappart L.; Shen Y.; Wang Z. Q. Oncogene 2007, 26, 3857. [DOI] [PubMed] [Google Scholar]
- Tanori M.; Mancuso M.; Pasquali E.; Leonardi S.; Rebessi S.; Di Majo V.; Guilly M. N.; Giangaspero F.; Covelli V.; Pazzaglia S.; Saran A. Carcinogenesis 2008, 29, 1911. [DOI] [PubMed] [Google Scholar]
- Tong W. M.; Cortes U.; Hande M. P.; Ohgaki H.; Cavalli L. R.; Lansdorp P. M.; Haddad B. R.; Wang Z. Q. Cancer Res. 2002, 62, 6990. [PubMed] [Google Scholar]
- Piskunova T. S.; Zabezhinskii M. A.; Popovich I. G.; Tyndyk M. L.; Iurova M. N.; Anisimov V. N. Vopr. Onkol. 2009, 55, 608. [PubMed] [Google Scholar]
- a Tsutsumi M.; Masutani M.; Nozaki T.; Kusuoka O.; Tsujiuchi T.; Nakagama H.; Suzuki H.; Konishi Y.; Sugimura T. Carcinogenesis 2001, 22, 1. [DOI] [PubMed] [Google Scholar]; b Ogawa K.; Masutani M.; Kato K.; Tang M.; Kamada N.; Suzuki H.; Nakagama H.; Sugimura T.; Shirai T. Cancer Lett. 2006, 236, 32. [DOI] [PubMed] [Google Scholar]
- Conde C.; Mark M.; Oliver F. J.; Huber A.; de Murcia G.; Menissier-de Murcia J. EMBO J. 2001, 20, 3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hauschildt S.; Scheipers P.; Bessler W. G. Biochem. Biophys. Res. Commun. 1991, 179, 865. [DOI] [PubMed] [Google Scholar]; b Pellat-Deceunynck C.; Wietzerbin J.; Drapier J. C. Biochem. J. 1994, 297, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Szabo C.; Lim L. H.; Cuzzocrea S.; Getting S. J.; Zingarelli B.; Flower R. J.; Salzman A. L.; Perretti M. J. Exp. Med. 1997, 186, 1041. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Oliver F. J.; Menissier-de Murcia J.; Nacci C.; Decker P.; Andriantsitohaina R.; Muller S.; de la Rubia G.; Stoclet J. C.; de Murcia G. EMBO J. 1999, 18, 4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa P. O.; Covic M.; Hasan S.; Imhof R.; Hottiger M. O. J. Biol. Chem. 2001, 276, 45588. [DOI] [PubMed] [Google Scholar]
- a Kameoka M.; Ota K.; Tetsuka T.; Tanaka Y.; Itaya A.; Okamoto T.; Yoshihara K. Biochem. J. 2000, 346, 641. [PMC free article] [PubMed] [Google Scholar]; b Nakajima H.; Nagaso H.; Kakui N.; Ishikawa M.; Hiranuma T.; Hoshiko S. J. Biol. Chem. 2004, 279, 42774. [DOI] [PubMed] [Google Scholar]
- Andreone T. L.; O’Connor M.; Denenberg A.; Hake P. W.; Zingarelli B. J. Immunol. 2003, 170, 2113. [DOI] [PubMed] [Google Scholar]
- Valdor R.; Schreiber V.; Saenz L.; Martinez T.; Munoz-Suano A.; Dominguez-Villar M.; Ramirez P.; Parrilla P.; Aguado E.; Garcia-Cozar F.; Yelamos J. Mol. Immunol. 2008, 45, 1863. [DOI] [PubMed] [Google Scholar]
- Nasta F.; Laudisi F.; Sambucci M.; Rosado M. M.; Pioli C. J. Immunol. 2010, 184, 3470. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Nakatsukasa H.; Tu E.; Kasagi S.; Cui K.; Ishikawa M.; Konkel J. E.; Maruyama T.; Wei G.; Abbatiello B.; Wang Z. Q.; Zhao K.; Chen W. Blood 2013, 122, 2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P.; Szabo C. Cardiovasc. Drug Rev. 2007, 25, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson M. J.; Sampei K.; Mandir A. S.; Hurn P. D.; Traystman R. J.; Bao J.; Pieper A.; Wang Z. Q.; Dawson T. M.; Snyder S. H.; Dawson V. L. Nat. Med. 1997, 3, 1089. [DOI] [PubMed] [Google Scholar]
- Beneke S. Exp. Gerontol. 2008, 43, 605. [DOI] [PubMed] [Google Scholar]
- Chiarugi A. Pharmacol. Res. 2005, 52, 15. [DOI] [PubMed] [Google Scholar]
- a Masutani M.; Suzuki H.; Kamada N.; Watanabe M.; Ueda O.; Nozaki T.; Jishage K.; Watanabe T.; Sugimoto T.; Nakagama H.; Ochiya T.; Sugimura T. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 2301. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pieper A. A.; Brat D. J.; Krug D. K.; Watkins C. C.; Gupta A.; Blackshaw S.; Verma A.; Wang Z. Q.; Snyder S. H. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3059. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Burkart V.; Wang Z. Q.; Radons J.; Heller B.; Herceg Z.; Stingl L.; Wagner E. F.; Kolb H. Nat. Med. 1999, 5, 314. [DOI] [PubMed] [Google Scholar]
- Devalaraja-Narashimha K.; Padanilam B. J. J. Endocrinol. 2010, 205, 243. [DOI] [PubMed] [Google Scholar]
- Bai P.; Canto C.; Oudart H.; Brunyanszki A.; Cen Y.; Thomas C.; Yamamoto H.; Huber A.; Kiss B.; Houtkooper R. H.; Schoonjans K.; Schreiber V.; Sauve A. A.; Menissier-de Murcia J.; Auwerx J. Cell Metab. 2011, 13, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. C.; Guarente L. Trends Endocrinol. Metab. 2014, 25, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirinen E.; Canto C.; Jo Y. S.; Morato L.; Zhang H.; Menzies K. J.; Williams E. G.; Mouchiroud L.; Moullan N.; Hagberg C.; Li W.; Timmers S.; Imhof R.; Verbeek J.; Pujol A.; van Loon B.; Viscomi C.; Zeviani M.; Schrauwen P.; Sauve A. A.; Schoonjans K.; Auwerx J. Cell Metab. 2014, 19, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yoshino J.; Mills K. F.; Yoon M. J.; Imai S. Cell Metab. 2011, 14, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Canto C.; Houtkooper R. H.; Pirinen E.; Youn D. Y.; Oosterveer M. H.; Cen Y.; Fernandez-Marcos P. J.; Yamamoto H.; Andreux P. A.; Cettour-Rose P.; Gademann K.; Rinsch C.; Schoonjans K.; Sauve A. A.; Auwerx J. Cell Metab. 2012, 15, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cerutti R.; Pirinen E.; Lamperti C.; Marchet S.; Sauve A. A.; Li W.; Leoni V.; Schon E. A.; Dantzer F.; Auwerx J.; Viscomi C.; Zeviani M. Cell Metab. 2014, 19, 1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman H. C.; Hugill A.; Dear N. T.; Ashcroft F. M.; Cox R. D. Diabetes 2006, 55, 2153. [DOI] [PubMed] [Google Scholar]
- Erener S.; Mirsaidi A.; Hesse M.; Tiaden A. N.; Ellingsgaard H.; Kostadinova R.; Donath M. Y.; Richards P. J.; Hottiger M. O. FASEB J. 2012, 26, 2631. [DOI] [PubMed] [Google Scholar]
- Maury E.; Ramsey K. M.; Bass J. Circ. Res. 2010, 106, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter J.; Reick M.; Wu L. C.; McKnight S. L. Science 2001, 293, 510. [DOI] [PubMed] [Google Scholar]
- a Asher G.; Gatfield D.; Stratmann M.; Reinke H.; Dibner C.; Kreppel F.; Mostoslavsky R.; Alt F. W.; Schibler U. Cell 2008, 134, 317. [DOI] [PubMed] [Google Scholar]; b Nakahata Y.; Sahar S.; Astarita G.; Kaluzova M.; Sassone-Corsi P. Science 2009, 324, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ramsey K. M.; Yoshino J.; Brace C. S.; Abrassart D.; Kobayashi Y.; Marcheva B.; Hong H. K.; Chong J. L.; Buhr E. D.; Lee C.; Takahashi J. S.; Imai S.; Bass J. Science 2009, 324, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S.; Poirier G. G.; Kay S. A. Dev. Cell 2002, 3, 51. [DOI] [PubMed] [Google Scholar]
- Tulin A.; Stewart D.; Spradling A. C. Genes Dev. 2002, 16, 2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Baumann C.; De La Fuente R. Dev. Biol. 2009, 331, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menissier de Murcia J.; Ricoul M.; Tartier L.; Niedergang C.; Huber A.; Dantzer F.; Schreiber V.; Ame J. C.; Dierich A.; LeMeur M.; Sabatier L.; Chambon P.; de Murcia G. EMBO J. 2003, 22, 2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambucci M.; Laudisi F.; Novelli F.; Bennici E.; Rosado M. M.; Pioli C. Sci. World J. 2013, 2013, 375024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erener S.; Hesse M.; Kostadinova R.; Hottiger M. O. Mol. Endocrinol. 2012, 26, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemberger M.; Nozaki T.; Winterhager E.; Yamamoto H.; Nakagama H.; Kamada N.; Suzuki H.; Ohta T.; Ohki M.; Masutani M.; Cross J. C. Dev. Biol. 2003, 257, 371. [DOI] [PubMed] [Google Scholar]
- Ogino H.; Nozaki T.; Gunji A.; Maeda M.; Suzuki H.; Ohta T.; Murakami Y.; Nakagama H.; Sugimura T.; Masutani M. BMC Genomics 2007, 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. A. Cell 2011, 144, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K.; Yamanaka S. Cell 2006, 126, 663. [DOI] [PubMed] [Google Scholar]
- Yamanaka S.; Blau H. M. Nature 2010, 465, 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou S. H.; Jiang B. H.; Yu Y. L.; Chou S. J.; Tsai P. H.; Chang W. C.; Chen L. K.; Chen L. H.; Chien Y.; Chiou G. Y. J. Exp. Med. 2013, 210, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szanto M.; Brunyanszki A.; Kiss B.; Nagy L.; Gergely P.; Virag L.; Bai P. Cell. Mol. Life Sci. 2012, 69, 4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas L.; Martinez C.; Baro C.; Rodriguez M.; Baroja-Mazo A.; Sole F.; Flores J. M.; Ampurdanes C.; Dantzer F.; Martin-Caballero J.; Aparicio P.; Yelamos J. Oncogene 2010, 29, 2877. [DOI] [PubMed] [Google Scholar]
- Kamboj A.; Lu P.; Cossoy M. B.; Stobart J. L.; Dolhun B. A.; Kauppinen T. M.; de Murcia G.; Anderson C. M. J. Neuroinflammation 2013, 10, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler J.; Otsuka T.; Zhang Z.; Noppens R.; Grafe M. R.; Koh D. W.; Dawson V. L.; de Murcia J. M.; Hurn P. D.; Traystman R. J. J. Cereb. Blood Flow Metab. 2006, 26, 135. [DOI] [PubMed] [Google Scholar]
- Popoff I.; Jijon H.; Monia B.; Tavernini M.; Ma M.; McKay R.; Madsen K. J. Pharmacol. Exp. Ther. 2002, 303, 1145. [DOI] [PubMed] [Google Scholar]
- Dantzer F.; Mark M.; Quenet D.; Scherthan H.; Huber A.; Liebe B.; Monaco L.; Chicheportiche A.; Sassone-Corsi P.; de Murcia G.; Menissier-de Murcia J. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelamos J.; Monreal Y.; Saenz L.; Aguado E.; Schreiber V.; Mota R.; Fuente T.; Minguela A.; Parrilla P.; de Murcia G.; Almarza E.; Aparicio P.; Menissier-de Murcia J. EMBO J. 2006, 25, 4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P.; Houten S. M.; Huber A.; Schreiber V.; Watanabe M.; Kiss B.; de Murcia G.; Auwerx J.; Menissier-de Murcia J. J. Biol. Chem. 2007, 282, 37738. [DOI] [PubMed] [Google Scholar]
- Bai P.; Canto C.; Brunyanszki A.; Huber A.; Szanto M.; Cen Y.; Yamamoto H.; Houten S. M.; Kiss B.; Oudart H.; Gergely P.; Menissier-de Murcia J.; Schreiber V.; Sauve A. A.; Auwerx J. Cell Metab. 2011, 13, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck C.; Robert I.; Reina-San-Martin B.; Schreiber V.; Dantzer F. Exp. Cell Res. 2014, 329118–25. [DOI] [PubMed] [Google Scholar]
- Boehler C.; Gauthier L. R.; Mortusewicz O.; Biard D. S.; Saliou J. M.; Bresson A.; Sanglier-Cianferani S.; Smith S.; Schreiber V.; Boussin F.; Dantzer F. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten S. L.; Fisher A. E.; Robert I.; Zuma M. C.; Rouleau M.; Ju L.; Poirier G.; Reina-San-Martin B.; Caldecott K. W. Mol. Cell 2011, 41, 33. [DOI] [PubMed] [Google Scholar]
- Gagne J. P.; Hunter J. M.; Labrecque B.; Chabot B.; Poirier G. G. Biochem. J. 2003, 371, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani M.; Nozaki T.; Nakamoto K.; Nakagama H.; Suzuki H.; Kusuoka O.; Tsutsumi M.; Sugimura T. Mutat. Res. 2000, 462, 159. [DOI] [PubMed] [Google Scholar]
- Nozaki T.; Fujihara H.; Watanabe M.; Tsutsumi M.; Nakamoto K.; Kusuoka O.; Kamada N.; Suzuki H.; Nakagama H.; Sugimura T.; Masutani M. Cancer Sci. 2003, 94, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunji A.; Uemura A.; Tsutsumi M.; Nozaki T.; Kusuoka O.; Omura K.; Suzuki H.; Nakagama H.; Sugimura T.; Masutani M. Cancer Lett. 2006, 241, 87. [DOI] [PubMed] [Google Scholar]
- Rouleau M.; McDonald D.; Gagne P.; Ouellet M. E.; Droit A.; Hunter J. M.; Dutertre S.; Prigent C.; Hendzel M. J.; Poirier G. G. J. Cell. Biochem. 2007, 100, 385. [DOI] [PubMed] [Google Scholar]
- Langelier M. F.; Planck J. L.; Roy S.; Pascal J. M. Science 2012, 336, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustermann S.; Brockmann C.; Mehrotra P. V.; Yang J. C.; Loakes D.; West S. C.; Ahel I.; Neuhaus D. Nat. Struct. Mol. Biol. 2010, 17, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi J.; Richards M. W.; Crumpler S.; Brown N.; Blagg J.; Bayliss R. J. Biol. Chem. 2010, 285, 39348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forst A. H.; Karlberg T.; Herzog N.; Thorsell A. G.; Gross A.; Feijs K. L.; Verheugd P.; Kursula P.; Nijmeijer B.; Kremmer E.; Kleine H.; Ladurner A. G.; Schuler H.; Luscher B. Structure 2013, 21, 462. [DOI] [PubMed] [Google Scholar]
- Grummt I. FEBS J. 2010, 277, 4626. [DOI] [PubMed] [Google Scholar]
- Freed E. F.; Bleichert F.; Dutca L. M.; Baserga S. J. Mol. BioSyst. 2010, 6, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K.; Calvo O.; Manley J. L. RNA 2004, 10, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. R.; King O. D.; Shorter J.; Gitler A. D. J. Cell Biol. 2013, 201, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]