

Abstract
Mucopolysaccharidosis (MPS) VII is a lysosomal storage disorder caused by the deficiency of the enzyme β-glucuronidase (Gusb-/-) and results in glycosaminoglycan (GAG) accumulation. Skeletal abnormalities include stunted long bones and bone degeneration. GAGs have been hypothesized to activate toll-like receptor 4 (Tlr4) signaling and the complement pathway, resulting in upregulation of inflammatory cytokines that suppress growth and cause degeneration of bone. Gusb-/- mice were bred with Tlr4- and complement component 3 (C3)-deficient mice, and the skeletal manifestations of the doubly- and triply-deficient mice were compared to those of purebred Gusb-/- mice. Radiographs showed that purebred Gusb-/- mice had shorter tibias and femurs, and wider femurs, compared to normal mice. No improvement was seen in Tlr4, C3, or Tlr4/C3-deficient Gusb-/- mice. The glenoid cavity and humerus were scored on a scale from 0 (normal) to +3 (severely abnormal) for dysplasia and bone irregularities, and the joint space was measured. No improvement was seen in Tlr4, C3, or Tlr4/C3-deficient Gusb-/- mice, and their joint space remained abnormally wide. Gusb-/- mice treated neonatally with an intravenous retroviral vector (RV) had thinner femurs, longer legs, and a narrowed joint space compared with untreated purebred Gusb-/- mice, but no improvement in glenohumeral degeneration. We conclude that Tlr4- and/or C3- deficiency fail to ameliorate skeletal abnormalities, and other pathways may be involved. RV treatment improves some but not all aspects of bone disease. Radiographs may be an efficient method for future evaluation, as they readily show glenohumeral joint abnormalities.
Keywords: Mucopolysaccharidosis, toll-like receptor, complement, degenerative joint disease, dysostosis multiplex, gene therapy
1. Introduction
The mucopolysaccharidoses (MPS)1 are a family of lysosomal storage disorders characterized by deficiencies in enzymes that contribute to the degradation of glycosaminoglycans (GAGs) [1]. The resulting accumulation of GAGs leads to bone and joint disease, respiratory and cardiovascular complications, mental retardation, and hearing and vision impairment. Skeletal abnormalities are referred to as dysostosis multiplex, which results in a limited range of motion, stunted long bones, and difficulty ambulating due to degenerative joint disease (DJD) or loss of joint stability.
MPS VII is a type of MPS due to the deficiency of the enzyme β-glucuronidase (Gusb), and will be referred to hereafter as Gusb-/-. Skeletal disease is severe in Gusb-/- patients [2-6], dogs [6-9], and mice [10-11]. The pathogenesis of skeletal disease is still being investigated, and one hypothesis is that GAGs bind to toll-like receptor 4 (Tlr4), resulting in upregulation of inflammatory cytokines such as tumor necrosis factor-α (Tnf), interleukin-1β (IL1b), and chemokine (C-C motif) ligand 3 [Ccl3 or macrophage inflammatory protein 1α (MIP1α)] [12-14]. Indeed, GAGs are structurally similar to the canonical ligand for Tlr4, lipopolysaccharide (LPS), and the upregulation of Tnf, IL1b, and Ccl3 by GAGs was reduced in microglial cells of Tlr4-deficient mice [12]. These inflammatory cytokines are upregulated in blood, synovial fluid and/or cultured fibroblast- like synoviocytes of MPS animals [13-18], and induce expression of destructive proteases [19] such as matrix metalloproteinases (MMPs) and cathepsins that are associated with DJD [20-23]. MPS has also been associated with proliferation of fibroblast-like synoviocytes, upregulation of proteases, and chondrocyte apoptosis in MPS VI rats [13, 16], which likely contribute to the synovial hyperplasia and joint and cartilage degeneration seen in MPS [8-9, 24-25]. The reduced proliferation in the growth plate of Gusb-/- mice [26] is likely responsible for the stunting of long bones. It was previously reported that Gusb-/- mice that were also deficient in Tlr4 had improvements in bone lengths and a reduction in synovial Tnf RNA levels [14]. However, the effect on degenerative changes in the bones was not assessed.
Another hypothesis for the pathogenesis of disease in MPS is that GAGs activate the complement pathway. We previously demonstrated that complement was activated in the aorta of Gusb-/- mice and proposed that the pathogenesis of aortic dilatation might involve complement activation [27], as a variety of carbohydrates can activate complement [28]. Complement is important for immune-complex-induced arthritis [29] and plays a role in Tlr4 signaling, as deficiency of an inhibitor of complement, Cd55, markedly potentiates the effect of LPS on Tlr4-dependent cytokine signaling [30-31]. Complement component 3 (C3) is central to the classical, alternative, and lectin pathways, and inhibition of the complement pathway can reduce Tnf levels in inflammation [32].
Current treatments for some types of MPS patients include hematopoietic stem cell transplantation (HSCT) and/or enzyme replacement therapy (ERT) [33-35]. Neither has prevented the skeletal abnormalities associated with MPS, although ERT has improved ambulation in some MPS I [36], MPS II [37], and MPS VI [38-40] patients. Gene therapy is being tested in animal models [41]. One method involves a neonatal intravenous (IV) injection of a gamma retroviral vector (RV), which transduces liver cells that express the desired enzyme. The enzyme is modified with mannose 6-phosphate (M6P) and secreted into the blood, after which it diffuses to tissues and is taken up by cells via the M6P receptor. Previously, we have observed that a neonatal injection of an RV increased bone lengths and reduced degeneration in Gusb-/- dogs [8-9] and increased bone lengths in Gusb-/- mice [10]. However, the effect of gene therapy on DJD was not evaluated in Gusb-/- mice. The goal of this study was to evaluate the effects of Tlr4- and C3-deficiency and RV-treatment on the development of skeletal disease in Gusb-/- mice.
2. Materials and methods
2.1 Animal care and genotyping
Guidelines set by the National Institutes of Health for the care and use of animals in research were followed. All mice were on a C57Bl/6 background. Genotyping for Gusb was done as previously described using a Taqman PCR assay [27] sensitive to the single bp insertion in exon 10 [42]. C3-/- mice had a neomycin-resistance gene inserted into the promoter region of C3 [43] and were generously provided by Drs. Xiabo Wu and John Atkinson of Washington University in St. Louis. Genotyping for C3 deficiency used a SYBR green mastermix from KAPA Biosystems (Wilmington, MA), and primers that recognized the wild-type C3 gene (Forward: 5′-TGTTGCCCCAGGTTTGTGA-3′ and Reverse: 5′-CCAGGGACTGCCCAAAATTT-3′) at 61°C, or the C3 gene with a neomycin insertion (Forward: 5′-CGACAAGACCGGCTTCCA-3′ and Reverse: 5′-AAGCGAAACATCGCATCGA-3′) at 61°C. Tlr4-/- mice had a 74 kb deletion that included the Tlr4 coding sequence [44], and were obtained from Jackson labs (Bar Harbor, ME; B6.B10ScN-Tlr4lps-del/JthJ; stock number #007227). Genotyping for Tlr4 used SYBR green primers that recognized the wild-type Tlr4 gene (Forward: 5′- AGAAATTCCTGCAGTGGGTCA-3′ Reverse: 5′-TCTCTACAGGTGTTGCACATGTCA-3′) at 61°C, or the Tlr4 mutation (Forward: 5′-GCAAGTTTCTATATGCATTCTC-3′ and Reverse: 5′-CCTCCATTTCCAATAGGTAG-3′) at 63°C. Some Gusb-/- mice were injected IV at 2-3 days after birth with 1×1010 transducing units/kg of the RV designated hAAT- cGusb-WPRE that expresses canine Gusb and contains the human α1-antitrypsin promoter [45], which allowed them to survive and breed. Some RV-treated Gusb-/- mice were bred with C3-/- or with Tlr4-/- mice to generate obligate heterozygotes, which were then crossed to generate Gusb-/- Tlr4-/-, Gusb-/- C3-/-, or Gusb-/- Tlr4-/- C3-/- mice.
2.2 Radiographs
At the time of sacrifice, mice were anaesthesized by injection of ketamine/zylazine as reported previously [27], perfused with 20 ml of PBS, and died of exsanguination. Bones were frozen at -20°C, and radiographs were obtained and scanned into the computer. Femur and tibia measurements were obtained as shown in Supplemental Fig. 1A-C. Arm radiographs were blinded as to the genotype, and the glenohumeral joint space was measured as shown in Supplemental Fig. 1D and scored for dysplasia and irregularities as shown in Supplemental Fig. 2. For dysplasia of the proximal humerus, 0 represents a perfectly spherical ball shape; 1 represents a slightly flattened sphere but the ball shape was still apparent; 2 represents an oval shape; and 3 represents a shallow oval with an almost completely flat surface. For glenoid cavity dysplasia, 0 represents a flattening of 0-4% of the surface of the glenoid cavity; 1 represents a 5-29% flattening; 2 represents a 30-59% flattening; and 3 represents a 60-100% flattening. Irregularities of both the proximal humerus and glenoid cavity were scored with the following criteria: 0 represents irregularity of 0-10% of the subchondral surface; 1 represents 11-29% irregularity of the surface; 2 represents 30-60% irregularity; and 3 represents more than 60% irregularity.
2.3 Histochemistry
Bones were fixed for 10-14 days in a solution of phosphate buffered saline with 10% formalin and decalcified for ∼24 hours in Formical-2000 (Decal-Bone, Talman, NY) until the bones were pliable. Tissues were embedded in paraffin and 6-µm sections were stained with Masson's Trichrome. Slides were photographed with an Olympus Nanozoomer 2.0-HT system and NDP imaging software.
2.4 Statistical Analysis
Significance for values that were continuous (leg measurements and joint space) was assessed using One-Way ANOVA with Holm-Sidak post-hoc analysis using SigmaPlot 12.0 (Sigma-Aldrich, St. Louis, MO). Significance for values that were non- continuous (glenohumeral DJD scores) was compared using ANOVA on ranks with Dunn's post-hoc analysis.
3. Results
Gusb-/- mice were bred with Tlr4-/- mice and C3-/- mice to generate doubly and triply-deficient mice. Gusb+/- Tlr4+/+ C3+/+ mice will be referred to hereafter as normal mice, as previous studies have shown that mice that are heterozygous for Gusb are phenotypically normal [46]. Gusb-/- Tlr4+/+ C3+/+ mice will be referred to hereafter as purebred Gusb-/- mice. Gusb-/- mice that were also deficient in Tlr4 but were normal for the C3 locus (Gusb-/- Tlr4-/- C3+/+) will be referred to hereafter as Gusb-Tlr4 DKO (double knock-out) mice, although neither of these mutant strains were actually generated with homologous recombination. Gusb-/- Tlr4+/+ C3-/- mice will be referred to as Gusb-C3 DKO mice. Gusb-/- Tlr4-/- C3-/- mice will be referred to as Gusb-Tlr4-C3 TKO (triple knock-out) mice.
3.1 Radiographs of the femur and tibia
Representative radiographs of the femurs at 3 months of age are shown in Fig. 1A, where mice of all Gusb-/- genotypes had significantly shorter femurs than normal mice. Fig. 1B shows that purebred Gusb-/- mice had femurs that were only 77±1% as long as purebred normal mice (p<0.001). Gusb-Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice had no improvement in lengths compared with purebred Gusb-/- mice, and bones remained statistically shorter than in purebred normal mice (p<0.001). Similarly, purebred Gusb-/- tibias were 84±5% the length of normal tibias (p<0.001), and there was no improvement in the lengths of Gusb-Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice compared to purebred Gusb-/- mice. Purebred Gusb-/- mice had femurs that were 124±5% as wide as normal mice (p<0.001) (Fig. 1D), and the femurs of Gusb-Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice remained wide (p<0.001 vs. normal). Gusb-/-mice that were heterozygous for Tlr4 and/or C3 had values similar to those of purebred Gusb-/- mice (data not shown). Similarly, normal Gusb+/- mice that were +/- or -/- for Tlr4 and/or C3 had similar values to purebred Gusb+/- mice (data not shown). Degeneration of the stifle joint was difficult to observe on radiographs in purebred Gusb-/- mice compared with normal mice (data not shown).
Fig. 1. Radiographical evaluation of the femur and tibia.
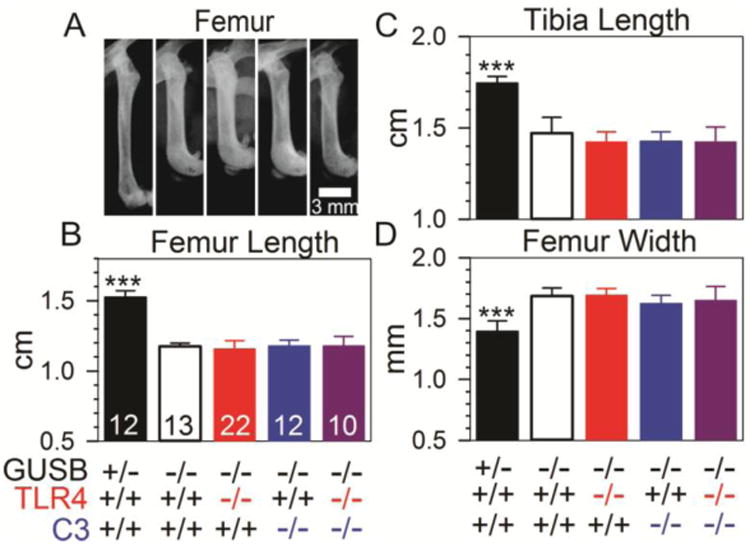
Radiographs of the legs were obtained. A. Representative radiographs of the femur of male mice of the indicated genotype at 3 months of age are shown. The proximal femur is at the top. B-D. Measurements of femur length, tibia length, and femur width at 3-6 months of age are shown for the indicated number of mice, which included both males and females. Statistical comparison between purebred Gusb-/- mice and the other groups used ANOVA with Holm-Sidak post-hoc analysis, and *** signifies a p-value <0.001.
3.2 Histochemistry of the stifle joint
Histochemistry was evaluated to determine bone and cartilage quality in the stifle joint. Normal mice (Fig. 2A-B) displayed a strong line of solid subchondral bone, with normal-sized chondrocytes in the articular cartilage and growth plate. Purebred Gusb-/- mice (Fig. 2C-D) displayed thinning of the subchondral bone in both the distal femur and proximal tibia, and the articular cartilage and growth plate were thickened due to GAG accumulation. Gusb-Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice (Fig. 2E-J) exhibited similar bone thinning and thickening of articular cartilage and the growth plate as did purebred Gusb-/- mice.
Fig 2. Histochemistry of the femur and tibia.
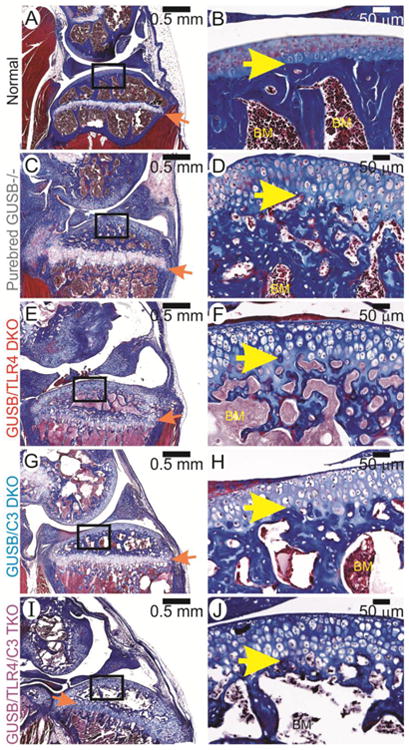
Sections of stifle joints from male mice at 3 months of age were stained with Masson's Trichrome as stated in the methods. Representative photographs were taken at low (left) and high (right) power. The groups are indicated on the far left. In the left column, the distal femur is at the top while the proximal tibia is at the bottom. The orange arrows indicate the growth plate. The boxes outline the area magnified in the right column, in which the bone marrow (BM) is indicated, and the yellow arrows point out the junction between articular cartilage and bone.
3.3 Radiographs of the glenohumeral joint
3.3.1 Joint space measurements
Radiographs of mouse arms displayed shortened humeri and ulna/radii lengths in all Gusb-/- genotypes compared to normal mice (data not shown). Representative examples of radiographs of the glenohumeral joint are shown in Fig. 3A to 3E, and joint space measurements were obtained to determine the distance between the bones of the proximal humeri and the bones of the glenoid cavity as shown in Supplemental Fig. 1D, and the average values for several animals are shown in Fig. 3F. Normal mice had an average joint space of 0.10±0.03 mm, which was significantly narrower than for purebred Gusb-/- mice with an average joint space of 0.46±0.06 mm (p<0.001). The joint space of Gusb-Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice was not significantly improved compared to that of purebred Gusb-/- mice.
Fig 3. Radiographical evaluation of the humerus and glenoid cavity.
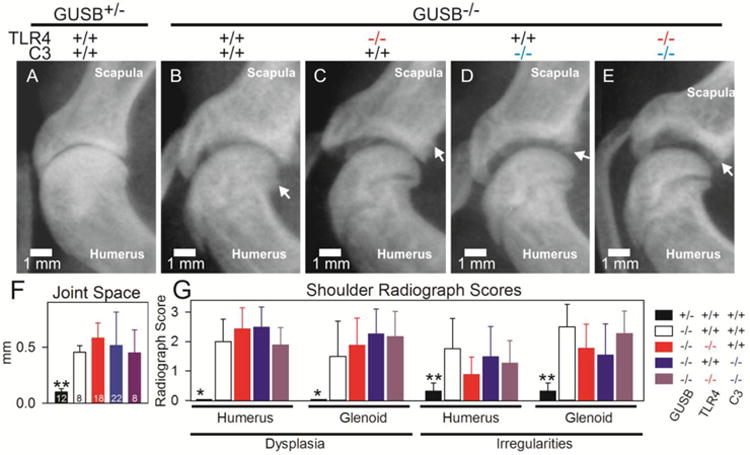
Radiographs of the arms were obtained at 3 months of age. A-E. Representative radiographs of the glenohumeral joint of male mice of the indicated genotype are shown. The joint space appears as the black region between the glenoid cavity of the scapula and the humerus. Arrows indicate irregular regions of the subchondral bone. F. Joint space measurements were performed for the indicated number of mice, which included both males and females. Statistical comparisons between purebred Gusb-/- mice and the other groups were done using One-way ANOVA with Holm Sidak's post-hoc analysis. * signifies a p-value between 0.01 and 0.05,** signifies a p-value of 0.001 to 0.01, and *** represents a p value<0.001. G. Dysplasia and irregularities of the subchondral bone were scored for the same mice as in panel F for the humerus and glenoid cavity as detailed in the Methods section and illustrated in Supplemental Fig. 2. A score of 0 is normal and +3 is severely abnormal. Statistical comparisons were done using Kruskal-Wallis ANOVA on ranks with Dunn's post-hoc analysis.
3.3.2 Dysplasia
Dysplasia of the proximal humerus and glenoid cavity was scored as flattening of the articular surfaces from 0 (normal) to +3 (severely abnormal) as stated in the methods and shown in Supplemental Fig. 2. For the proximal humerus, normal mice scored an average of 0.0±0.0. Purebred Gusb-/- mice scored significantly higher than normal mice with an average of 2.0±0.8 (p<0.05), and there was no significant improvement in Gusb/Tlr4 DKOs (2.4±0.7), Gusb/C3 DKOs (2.5±0.7), or Gusb/Tlr4/C3 TKOs (1.9±0.6) compared with purebred Gusb-/- mice (p>0.05). For dysplasia of the glenoid cavity, normal mice scored an average of 0.0±0.0, which was significantly lower than for purebred Gusb-/- mice (1.5±1.2; p<0.05). Again, no significant improvement was seen in Gusb/Tlr4 DKO (1.9±0.9), Gusb/C3 DKO (2.3±0.8), or Gusb/Tlr4/C3 TKO (2.2±0.9) mice compared to purebred Gusb-/- mice (p>0.05).
3.3.3 Surface irregularities
Irregularities of the subchondral bone are another manifestation of MPS that appear on radiographs as a lack of a solid, traceable edge or osteophyte formation, and can be due to inadequate bone formation, erosions, or new bone formation. Irregularities were scored as stated in the methods and as shown in Supplemental Fig. 2. For both the proximal humerus and the glenoid cavity, normal mice scored an average of 0.3±0.3. Purebred Gusb-/- mice scored significantly higher in both areas with an average of 1.8±1.0 for the humerus (p<0.05 vs. normal), and 2.5±1.8 for the glenoid cavity (p<0.01 vs. normal). No significant improvement was seen in either area between Gusb/Tlr4 DKO, Gusb/C3 DKO, or Gusb/Tlr4/C3 TKO mice and purebred Gusb-/- mice.
3.4 Histochemistry of the humerus and glenoid cavity
3.4.1 Articular cartilage and bone quality
As was observed in the stifle joint, normal mice (Fig. 4A-B) exhibit a strong rim of bone underlying the articular cartilage, with no excess GAGs in either the articular cartilage or the growth plate. In contrast, purebred Gusb-/- mice (Fig. 4C-D) lack a solid rim of subchondral bone, and display thickened articular cartilage and growth plate due to the accumulation of GAGs. No improvement was seen in Gusb-Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice (Fig. 4E-J), as they similarly lacked the strong line of bone in normal mice, and displayed thickened cartilage.
Fig 4. Histochemistry of the humerus and glenoid cavity.
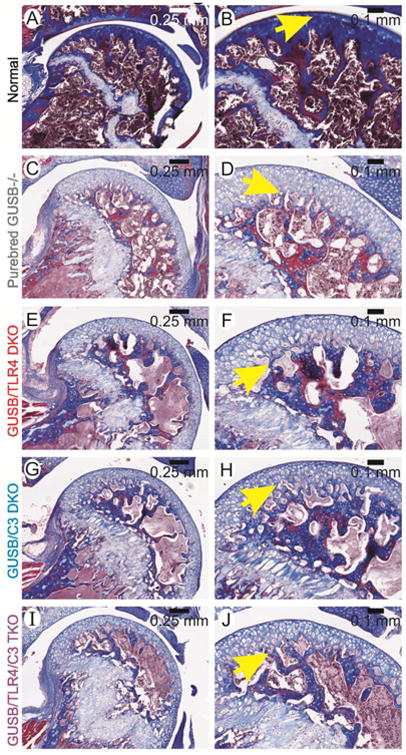
Glenohumeral joints from mice at 3 months of age were stained with Masson's Trichrome. Genotypes are indicated on the far left. Representative photographs were taken at low (left) and high (right) power. In the left column, the proximal humerus is shown, and is magnified in the right column panels, where the yellow arrows indicate the junction between articular cartilage and bone.
3.4.2 Synovial hyperplasia
In addition to differences in the bone and cartilage, differences in the synovium can also be observed between normal and purebred Gusb-/- mice, as shown in Supplemental Fig. 3. The synovium of normal mice is a few cell layers thick, while the hyperplastic synovium in purebred Gusb-/- mice is several cell layers thick, especially in the condylar neck region of the humerus. The synovium appears similarly hyperplastic in Gusb-Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice, although not all mice had a region on the slide that contained synovium, making this difficult to score.
3.5 Effect of RV-treatment on bones
During the breeding process, some Gusb-/- mice that were +/+ or +/- for Tlr4 and C3 were treated with a neonatal IV injection of RV. The average serum Gusb activity of these RV-treated mice was 1191±1201 Units/ml, which is 85-fold the value in heterozygous normal mice (Fig. 5A). Radiographs of these RV-treated mice at an average age of 8.4 months of age were compared with those of purebred Gusb-/- and Gusb+/- normal mice at 6 months of age for bone lengths and joint degeneration. Representative radiographs of the femur and evaluation of bone measurements are shown in Fig. 5B-E. Purebred Gusb-/- mice had femurs that were 124±6% the width of normal femurs (p<0.001). RV-treated mice had significantly narrower femurs than purebred Gusb-/- mice (p<0.001) and were 103±4% of normal (p=0.324). Purebred Gusb-/- mice had femurs and tibias that were 76±1% and 81±3%, respectively, the length of normal femurs and tibias (p<0.001 vs. normal for both). Although RV-treated mice had significantly longer femurs and tibias at 84±4% and 89±2% of normal, respectively, (p<0.001 for both vs. untreated purebred Gusb-/- mice), they were significantly shorter than those of normal mice (p<0.001 for both). Degeneration in the stifle joint could not be clearly seen on radiographs, so was not evaluated.
Fig. 5. Radiographical evaluation of RV-treated mice.
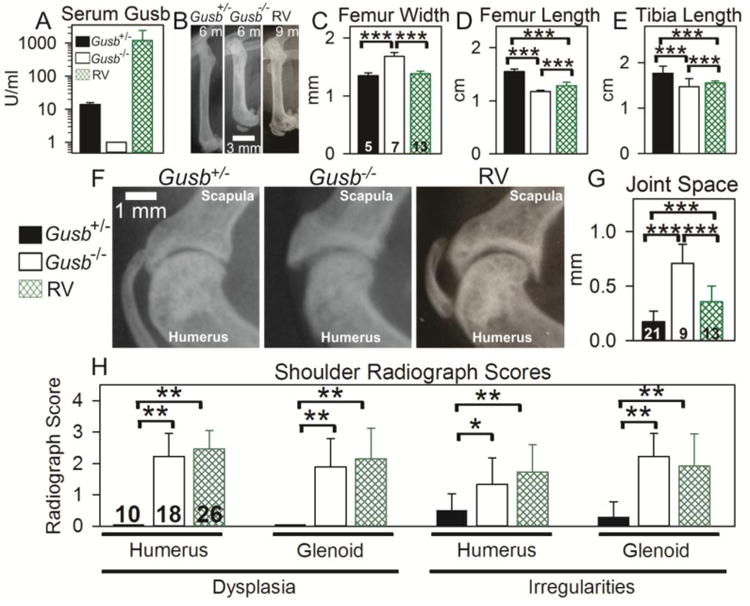
Some Gusb-/- mice were injected with RV at 2 to 3 days after birth (RV), and bones were evaluated at an average age of 8.4 months. These mice were either +/- or +/+ for Tlr4 and C3. Other Gusb-/- and Gusb+/- mice were untreated and bones were evaluated at the younger age of 6 months. A. Serum Gusb activity. Average Gusb serum activity was tested at 2 months of age. B. Representative radiographs of femurs with the groups indicated above the radiographs. The proximal femur is at the top and the age is shown in months (m). C-E. Femur width and length and tibia length were measured for the indicated number of mice. F. Representative radiographs of the glenohumeral joint are shown. G. Joint space was measured for the indicated number of mice. H. Dysplasia and surface irregularities were scored as stated in the methods and illustrated in Supplemental Fig.2, where 0 indicates normal and 3 indicates severely abnormal. The indicated number of mice were evaluated. Statistical comparison for panels C-E and panel G used one-way ANOVA with Holm-Sidak post-hoc analysis, and *** signifies a p-value <0.001. Statistical comparisons for panel H used Kruskal-Wallis ANOVA on ranks with Dunn's post-hoc analysis, and *indicates a p value of 0.01 to 0.05 and ** indicates p<0.01.
The arms of RV-treated mice were longer than those of purebred Gusb-/- mice, but were not as long as normal mice (data not shown), as has been previously reported [10]. The joint space was measured as shown in Supplemental Fig. 1D, and as shown in Fig. 5F and 5G. Purebred Gusb-/- mice had an average joint space of 0.71±0.18 mm, which was significantly wider than that of normal mice with 0.17±0.10 mm (p<0.001 vs. Gusb-/-) and that of RV-treated mice with 0.40±0.18 mm (p<0.001 vs. Gusb-/-; p<0.001 vs. normal). Radiographs of the glenohumeral joint were scored on a scale from 0 (normal) to +3 (severely abnormal) for dysplasia and surface irregularities, and values are shown in Fig. 5H. Purebred Gusb-/- mice scored significantly higher than normal mice in dysplasia and surface irregularities of the humerus and glenoid cavity (p<0.01). RV- treated mice scored significantly higher than normal mice in every aspect that was scored (p<0.01). The only significance found between RV-treated and purebred Gusb-/- mice was in irregularities of the humerus, where RV-treated mice scored more severely at 1.7±0.9 compared to purebred Gusb-/- mice at 1.3±0.8 (p<0.05). This may relate to the fact that RV-treated Gusb-/- mice were older than untreated mice, and thus had more time to develop degenerative changes.
4. Discussion
This study was conducted to evaluate the effects of Tlr4- and C3-deficiency, and RV-treatment on skeletal abnormalities in Gusb-/- mice. Skeletal disease is still prevalent in MPS patients after HSCT or ERT, which greatly reduces their quality of life. The failure to correct skeletal disease is likely due to the inability of blood-derived cells or enzyme to enter avascular regions such as cartilage. A hypothesis of this project is that GAGs bind to Tlr4 and/or activate the complement pathway, resulting in up-regulation of cytokines that induce the expression of proteases that contribute to DJD. Confirmation of the role of these pathways in MPS joint disease could lead to their inhibition through drugs as a possible treatment.
4.1. Deficiency of Tlr4 and/or C3 does not prevent bone and joint disease in MPS VII mice
To test the role of Tlr4 and C3 in the pathogenesis of joint disease, Gusb-/- mice were bred with mice that were deficient in Tlr4 and C3 to generate doubly and triply deficient mice, and the effects on bone disease were determined. In this study, Gusb- Tlr4 DKO, Gusb-C3 DKO, and Gusb-Tlr4-C3 TKO mice showed no significant improvement in lengths of the long bones of the arms and legs, in width of the femur, or in scores of dysplasia and bone irregularities of the glenohumeral joint when compared with purebred Gusb-/- mice.
The failure to prevent bone shortening in Gusb-Tlr4 DKO mice in this study is in contrast to the study of Simonaro et al. [14], which reported that the lengths of femurs and tibias in male Gusb-Tlr4 DKO mice were approximately 1.3-fold that in purebred Gusb-/- mice, and approached the values found in Gusb+/+ Tlr-/- mice. Both studies used Gusb-/- mice derived from Dr. Mark Sands at Washington University, and Tlr4-/- mice of the strain indicated in the Methods section from Jackson Laboratories (Calogera Simonaro, personal communication). It is unlikely that inaccurate genotyping on our part is responsible for this difference, as some Gusb-Tlr4 DKO mice that were bred and genotyped independently by Dr. William Sly using a different PCR technique but were radiographed by our laboratory had similar bone lengths and DJD scores as the animals bred, genotyped, and radiographed in our laboratory (data not shown). Although we pooled results for male and female mice in this study, our groups had a similar percentage of mice of each gender, and we failed to observe significant differences between male and female mice or between groups when animals of the same gender were analyzed separately (data not shown). Our study did evaluate mice that were older (3 to 6 months) than in the study by Simonaro et al. (1.5 months), so it is possible that the age difference was responsible for the discrepancy. It is also possible that genetic drift occurred in either the Gusb-/- or the Tlr4-/- colonies from the time when each was obtained from their source, which could contribute to the different results of the two studies. Indeed, the Gusb-/- mice in our study had femur lengths of 1.2 cm at 3 months, while the Gusb-/- mice in the previous study had femur lengths of ∼1 cm at 6 weeks [14], suggesting that differences could have been due to age or genetic background.
We conclude that Tlr4-deficiency does not protect Gusb-/- mice from stunted bones or DJD, and that inhibition of Tlr4 would not be effective against skeletal disease should such drugs be developed in the future. Indeed, the work of Ausseil et al. [12] demonstrated that although Ccl3 expression in response to GAGs in cultured microglial cells was only ∼20% as high in Tlr4-/- cells as in normal cells, Ccl3 expression in Tlr4-/-cells that were stimulated with GAGs remained ∼20-fold that of cells that were not stimulated with GAGs, suggesting that the Tlr4 pathway was not the only GAG-activated pathway. Furthermore, although Ccl3 mRNA in brain was reduced at 3 months or earlier in MPS IIIB mice that were deficient in Tlr4 compared to MPS IIIB mice normal for Tlr4, that difference was no longer present at 8 months of age, again suggesting that Tlr4 is not the only GAG-activated pathway that is inducing cytokines.
Thus, although GAGs can clearly induce cytokine expression via the Tlr4 pathway, they can also induce cytokines independently of Tlr4, which may explain why Tlr4- deficiency did not ameliorate bone disease in Gusb-/- mice in our study. Tnf is an important mediator of Tlr4 signaling, and inhibition of Tnf with an inhibitory antibody can reduce bone disease in MPS VI rats [14-15]. It would have been informative to test Gusb- Tlr4 DKO or Gusb-Tlr4-C3 TKO mice for Tnf RNA or protein levels in the synovium, bone, and cartilage, as Tnf can be induced by other signaling pathways. However, isolation of these tissues from mice is problematic due to their small size and was not attempted in this study.
Complement is another pathway that can be activated by GAGs [28] and is activated in the aorta of Gusb-/- mice [27], and C3 is critical component of the classical, alternative, and lectin pathways. However, deficiency of C3 alone or combined deficiency of Tlr4 and C3 did not ameliorate bone and joint disease in this study. We had previously postulated that GAGs might activate fibroblast growth factor receptor 3 (Fgfr3), as GAGs are important in the interaction of Fgfr3 with its ligand, and mutations that increase activation of Fgfr3 result in achondroplastic dwarfism. However, deficiency of Fgfr3 also did not prevent stunting of bones in Gusb-/- mice [26]. It is possible that GAGs are activating still another signaling pathway, which explains why deficiency of Tlr4, C3, or Fgfr3 did not prevent skeletal manifestations of disease.
4.2. Possible role of downstream mediators in bone and joint disease
An alternative explanation is that GAGs can cause bone and joint disease via a more direct mechanism such as activation of cathepsin K (CtsK), which is a protease that degrades bone and cartilage. Since CtsK is activated directly by chondroitin sulfate [47], a GAG that accumulates in MPS VII, it may be activated independently of signaling pathways. Indeed, CtsK mRNA and/or enzyme activity were elevated in the annulus fibrosus of the spine of MPS VII dogs [6] and in the aorta of MPS VII mice [27]. Our future studies will test if bone disease can be reduced in MPS VII mice by the CtsK- specific inhibitor odanacitib, which is currently in clinical trials for treating osteoporosis [48]. Interestingly, pentosan polysulfate, a drug that promotes chondrogenesis, was recently reported to reduce bone disease in MPS VI rats [17-18]. Although the authors demonstrated a reduction in serum cytokines and hypothesized that its mechanism reduced inflammation, it may be interesting to test if pentosan polysaccharide directly affects CtsK. Deficiency of other proteases such as cathepsin S or MMP12 did not ameliorate bone disease in Gusb-/- mice [27].
4.3. RV-treatment
Gusb-/- mice that were treated neonatally with an IV injection of an RV expressing canine Gusb were evaluated to determine the effects on bone disease. Bone lengths were partially improved relative to untreated Gusb-/- mice, which was consistent with our previous results in mice treated with the same vector that had similar levels of expression [10] and with our results in RV-treated Gusb-/- dogs [8-9]. In contrast, manifestations of DJD such as dysplasia and bone irregularities were not prevented in the RV-treated mice. Similarly, osteophyte formation was not prevented at any sites that was evaluated and dysplasia still occurred in the proximal femur, proximal tibia, and cervical spine of RV-treated Gusb-/- dogs, although dysplasia was reduced in the acetabulum and distal femur [8-9]. Subchondral bone irregularities were improved at 1 year but not at 8 years in RV-treated Gusb-/- dogs [8-9]. Thus, this study shows that evaluation of Gusb-/- mice for DJD using plain radiographs of the glenohumeral joint was quite informative, and demonstrated results in RV-treated Gusb-/- mice that were largely consistent with those in the RV-treated Gusb-/- dogs. Similar radiographic evaluation may therefore be useful for future studies that test the efficacy of a particular treatment on DJD in mice with MPS.
The failure to prevent most manifestations of DJD after neonatal gene therapy with an RV in Gusb-/- mice leads to our prediction that ERT will be similarly limited in its efficacy, even if started in the neonatal period. Although Gusb enzyme can diffuse to the synovium of Gusb-/- dogs [9] and to the edge of the cortex of bone of Gusb-/- mice [10] after neonatal gene therapy with an RV, articular cartilage of RV-treated Gusb-/- dogs did not have Gusb activity detected with a histochemical stain (E. Xing, K. Ponder, unpublished data). Low levels of enzyme in cartilage in RV-treated dogs may be due to the low levels of Gusb activity in synovial fluid at 7±5 U/ml (2±2% of the level in blood) [9] and the dense nature of cartilage that reduces diffusion, as well as the lack of a blood supply.
4.3. Future implications
This study suggests that neither Tlr4 nor C3 is essential for the pathogenesis of bone and joint disease in MPS VII mice, and that inhibition of these signaling pathways will not prevent stunted bones or DJD in patients. Neonatal IV injection of an RV can partially improve bone lengths, but has no effect on DJD. Our future studies will inject vectors directly into joints of MPS dogs and monitor the effects on skeletal disease, as intra-articular injection of enzyme has reduced lysosomal storage in MPS I dogs [49].
Supplementary Material
Research highlights.
Deficiency of Tlr4 or Complement C3 does not ameliorate bone disease in MPS VII mice.
Neonatal gene therapy with an IV injection of a retroviral vector improves bone lengths but does not prevent degenerative changes in MPS VII mice.
Acknowledgments
This work was supported by grants from the NIH (DK66448, DK054481, HD061879, and RR02512). We thank Dr. William Sly for sharing Gusb-/- Tlr4-/- mice, and Drs. Xiabo Wu and John Atkinson for sharing C3-/- mice with us.
Footnotes
Abbreviations: mucopolysaccharidosis (MPS); β-glucuronidase (Gusb); glycosaminoglycan (GAG); toll-like receptor 4 (Tlr4); complement component 3 (C3); retroviral vector (RV); degenerative joint disease (DJD); tumor necrosis factor-α (Tnf); interleukin-1β (IL1b); chemokine (C-C motif); ligand 3 (Ccl3); macrophage inflammatory protein 1α (MIP1α); lipopolysaccharide (LPS); matrix metalloproteinase (MMP); hematopoietic stem cell transplantation (HSCT); enzyme replacement therapy (ERT); intravenous (IV); mannose 6-phosphate (M6P); double knock-out (DKO); triple knock-out (TKO); fibroblast growth factor receptor 3 (Fgfr3); cathepsin K (CtsK).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- 1.Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford) 2011 Dec;50(Suppl 5):v4–12. doi: 10.1093/rheumatology/ker394. [DOI] [PubMed] [Google Scholar]
- 2.Sly WS, Quinton BA, McAlister WH, Rimoin DL. Beta glucuronidase deficiency: report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82:249–257. doi: 10.1016/s0022-3476(73)80162-3. [DOI] [PubMed] [Google Scholar]
- 3.Beaudet AL, DiFerrante NM, Ferry GD, Nichols BL, Jr, Mullins CE. Variation in the phenotypic expression of beta-glucuronidase deficiency. J Pediatr. 1975;86:388–394. doi: 10.1016/s0022-3476(75)80968-1. [DOI] [PubMed] [Google Scholar]
- 4.Vogler C, Levy B, Kyle JW, Sly WS, Williamson J, Whyte MP. Mucopolysaccharidosis VII: postmortem biochemical and pathological findings in a young adult with beta-glucuronidase deficiency. Mod Pathol. 1994;7:132–137. [PubMed] [Google Scholar]
- 5.de Kremer RD, Givogri I, Argarana CE, Hliba E, Conci R, Boldini CD, Capra AP. Mucopolysaccharidosis type VII (beta-glucuronidase deficiency): a chronic variant with an oligosymptomatic severe skeletal dysplasia. Am J Med Genet. 1992;44:145–152. doi: 10.1002/ajmg.1320440206. [DOI] [PubMed] [Google Scholar]
- 6.Smith LJ, Baldo G, Wu S, Liu Y, Whyte MP, Giugliani R, Elliott DM, Haskins ME, Ponder KP. Pathogenesis of lumbar spine disease in mucopolysaccharidosis VII. Mol Genet Metab. 2012;107:153–60. doi: 10.1016/j.ymgme.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ponder KP, Melniczek JR, Xu L, Weil MA, O'Malley TM, O'Donnell P, Knox VW, Aguirre GD, Mazrier H, Ellinwood NM, Sleeper M, Maguire AM, Volk SW, Mango RL, Zweigle J, Wolfe JH, Haskins ME. Therapeutic neonatal hepatic gene therapy in mucopolysaccharidosis VII dogs. Proc Natl Acad Sci U S A. 2002;99:13102–13107. doi: 10.1073/pnas.192353499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herati RS, Knox VW, O'Donnell P, D'Angelo M, Haskins ME, Ponder KP. Radiographic evaluation of bones and joints in mucopolysaccharidosis I and VII dogs after neonatal gene therapy. Mol Genet Metab. 2008;95:142–151. doi: 10.1016/j.ymgme.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing EM, Knox VW, O'Donnell PA, Sikura T, Liu Y, Wu S, Casal ML, Haskins ME, Ponder KP. The effect of neonatal gene therapy on skeletal manifestations in mucopolysaccharidosis VII dogs after a decade. Mol Genet Metab. 2013;109:183–93. doi: 10.1016/j.ymgme.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mango RL, Xu L, Sands MS, Vogler C, Seiler G, Schwarz T, Haskins ME, Ponder KP. Neonatal retroviral vector-mediated hepatic gene therapy reduces bone, joint, and cartilage disease in mucopolysaccharidosis VII mice and dogs. Mol Genet Metab. 2004;82:4–19. doi: 10.1016/j.ymgme.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Rowan DJ, Tomatsu S, Grubb JH, Montaño AM, Sly WS. Assessment of bone dysplasia by micro-CT and glycosaminoglycan levels in mouse models for mucopolysaccharidosis type I, IIIA, IVA, and VII. J Inherit Metab Dis. 2013;36:235–246. doi: 10.1007/s10545-012-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ausseil J, Desmaris N, Bigou S, Attali R, Corbineau S, Vitry S, Parent M, Cheillan D, Fuller M, Maire I, Vanier MT, Heard JM. Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice. PLoS One. 2008;3 doi: 10.1371/journal.pone.0002296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonaro CM, D'Angelo M, He X, Eliyahu E, Shtraizent N, Haskins ME, Schuchman EH. Mechanism of glycosaminoglycan-mediated bone and joint disease: implications for the mucopolysaccharidoses and other connective tissue diseases. Am J Pathol. 2008;172:112–22. doi: 10.2353/ajpath.2008.070564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simonaro CM, Ge Y, Eliyahu E, He X, Jepsen KJ, Schuchman EH. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc Natl Acad Sci U S A. 2010;107:222–7. doi: 10.1073/pnas.0912937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eliyahu E, Wolfson T, Ge Y, Jepsen KJ, Schuchman EH, Simonaro CM. Anti-TNF-Alpha Therapy Enhances the Effects of Enzyme Replacement Therapy in Rats with Mucopolysaccharidosis Type VI. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0022447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simonaro CM, Haskins ME, Schuchman EH. Articular chondrocytes from animals with a dermatan sulfate storage disease undergo a high rate of apoptosis and release nitric oxide and inflammatory cytokines: a possible mechanism underlying degenerative joint disease in the mucopolysaccharidoses. Lab Invest. 2001;81:1319–28. doi: 10.1038/labinvest.3780345. [DOI] [PubMed] [Google Scholar]
- 17.Frohbergh M, Ge Y, Meng F, Karabul N, Solyom A, Lai A, Iatridis J, Schuchman EH, Simonaro CM. Dose Responsive Effects of Subcutaneous Pentosan Polysulfate Injection in Mucopolysaccharidosis Type VI Rats and Comparison to Oral Treatment. PLoS One. 2014;9 doi: 10.1371/journal.pone.0100882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuchman EH, Ge Y, Lai A, Borisov Y, Faillace M, Eliyahu E, He X, Iatridis J, Vlassara H, Striker G, Simonaro CM. Pentosan polysulfate: a novel therapy for the mucopolysaccharidoses. PLoS One. 2013;8 doi: 10.1371/journal.pone.0054459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller RE, Lu Y, Tortorella MD, Malfait AM. Genetically Engineered Mouse Models Reveal the Importance of Proteases as Osteoarthritis Drug Targets. Curr Rheumatol Rep. 2013;15:350. doi: 10.1007/s11926-013-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P, Schwarz EM. The TNF-alpha transgenic mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25:19–33. doi: 10.1007/s00281-003-0125-3. [DOI] [PubMed] [Google Scholar]
- 21.Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, Sung WK, Lipovich L, Vega VB, Chen J, Shahab A, Zhao XD, Hibberd M, Wei CL, Lim B, Ng HH, Ruan Y, Chin KC. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–35. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 22.Zhang R, Sun P, Jiang Y, Chen Z, Huang C, Zhang X, Zhang R. Genome- wide haplotype association analysis and gene prioritization identify CCL3 as a risk locus for rheumatoid arthritis. Int J Immunogenet. 2010;37:273–8. doi: 10.1111/j.1744-313X.2010.00920.x. [DOI] [PubMed] [Google Scholar]
- 23.García-Hernández MH, González-Amaro R, Portales-Pérez DP. Specific therapy to regulate inflammation in rheumatoid arthritis: molecular aspects. Immunotherapy. 2014;6:623–36. doi: 10.2217/imt.14.26. [DOI] [PubMed] [Google Scholar]
- 24.Xu L L, Mango RL, Sands MS, Haskins ME, Ellinwood NM, Ponder KP. Evaluation of pathological manifestations of disease in mucopolysaccharidosis VII mice after neonatal hepatic gene therapy. Mol Ther. 2002;6:745–758. doi: 10.1006/mthe.2002.0809. [DOI] [PubMed] [Google Scholar]
- 25.O'Brien A, Bompadre V, Hale S, White KK. Musculoskeletal Function in Patients With Mucopolysaccharidosis Using the Pediatric Outcomes Data Collection Instrument. J Pediatr Orthop. 2014 doi: 10.1097/BPO.0000000000000168. [DOI] [PubMed] [Google Scholar]
- 26.Metcalf JA, Zhang Y, Hilton MJ, Long F, Ponder KP. Mechanism of shortened bones in mucopolysaccharidosis VII. Mol Genet Metab. 2009;97:202–11. doi: 10.1016/j.ymgme.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldo G, Wu S, Howe RA, Ramamoothy M, Knutsen RH, Fang J, Mecham RP, Liu Y, Wu X, Atkinson JP, Ponder KP. Pathogenesis of aortic dilatation in mucopolysaccharidosis VII mice may involve complement activation. Mol Genet Metab. 2011;104:608–19. doi: 10.1016/j.ymgme.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hashimoto M, Hirota K, Yoshitomi H, Maeda S, Teradaira S, Akizuki S. Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J Exp Med. 2010;207:1135–1143. doi: 10.1084/jem.20092301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, Wetsel RA, Miwa T, Song WC. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–36. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010;31:154–63. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nymo S, Niyonzima N, Espevik T, Mollnes TE. Cholesterol crystal-induced endothelial cell activation is complement-dependent and mediated by TNF. Immunobiology. 2014 doi: 10.1016/j.imbio.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Valayannopoulos V, Wijburg FA. Therapy for the mucopolysaccharidoses. Rheumatology (Oxford) 2011;50(Suppl 5):v49–v59. doi: 10.1093/rheumatology/ker396. [DOI] [PubMed] [Google Scholar]
- 34.Beck M. Therapy for lysosomal storage disorders. IUBMB Life. 2010;62:33–40. doi: 10.1002/iub.284. [DOI] [PubMed] [Google Scholar]
- 35.van der Linden MH, Kruyt MC, Sakkers RJ, de Koning TJ, Oner FC, Castelein RM. Orthopaedic management of Hurler's disease after hematopoietic stem cell transplantation: a systematic review. J Inherit Metab Dis. 2011;34:657–669. doi: 10.1007/s10545-011-9304-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M, Okazaki S, Huff K, Cox GF, Swiedler SJ, Kakkis ED. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab. 2007;90:171–180. doi: 10.1016/j.ymgme.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 37.White KK, Hale S, Goldberg MJ. Musculoskeletal health in Hunter disease (MPS II): ERT improves functional outcomes. J Pediatr Rehabil Med. 2010;3:101–107. doi: 10.3233/PRM-2010-0112. [DOI] [PubMed] [Google Scholar]
- 38.Harmatz P, Giugliani R, Schwartz IV, Guffon N, Teles EL, Miranda MC, Wraith JE, Beck M, Arash L, Scarpa M, Ketteridge D, Hopwood JJ, Plecko B, Steiner R, Whitley CB, Kaplan P, Yu ZF, Swiedler SJ, Decker C MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4- sulfatase. Mol Genet Metab. 2008;94:469–475. doi: 10.1016/j.ymgme.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Furujo M, Kubo T, Kosuga M, Okuyama T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab. 2011;104:597–602. doi: 10.1016/j.ymgme.2011.08.029. [DOI] [PubMed] [Google Scholar]
- 40.McGill JJ, Inwood AC, Coman DJ, Lipke ML, de Lore D, Swiedler SJ, Hopwood JJ. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age--a sibling control study. Clin Genet. 2010;77:492–498. doi: 10.1111/j.1399-0004.2009.01324.x. [DOI] [PubMed] [Google Scholar]
- 41.Ponder KP, Haskins ME. Gene therapy for mucopolysaccharidosis. Expert Opin Biol Ther. 2007;7:1333–1345. doi: 10.1517/14712598.7.9.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sands MS, Birkenmeier EH. A single-base-pair deletion in the beta-glucuronidase gene accounts for the phenotype of murine mucopolysaccharidosis type VII. Proc Natl Acad Sci U S A. 1993;90:6567–71. doi: 10.1073/pnas.90.14.6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, Briles DE, Volanakis JE, Wetsel RA, Colten HR. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology. 1999;42:135–49. doi: 10.1016/s0162-3109(99)00021-1. [DOI] [PubMed] [Google Scholar]
- 44.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (Tlr4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for Tlr4 as the Lps gene product. J Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 45.Xu L, Haskins ME, Melniczek JR, Gao C, Weil MA, O'Malley TM, O'Donnell PA, Mazrier H, Ellinwood NM, Zweigle J, Wolfe JH, Ponder KP. Transduction of hepatocytes after neonatal delivery of a Moloney murine leukemia virus based retroviral vector results in long-term expression of beta-glucuronidase in mucopolysaccharidosis VII dogs. Mol Ther. 2002;5:141–153. doi: 10.1006/mthe.2002.0527. [DOI] [PubMed] [Google Scholar]
- 46.Birkenmeier EH, Davisson MT, Beamer WG, Ganschow RE, Vogler CA, Gwynn B, Lyford KA, Maltais LM, Wawrzyniak CJ. Murine mucopolysaccharidosis type VII. Characterization of a mouse with beta-glucuronidase deficiency. J Clin Invest. 1989;83:1258–66. doi: 10.1172/JCI114010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemaire PA, Huang L, Zhuo Y, Lu J, Bahnck C, Stachel SJ, Carroll SS, Duong LT. Chondroitin Sulfate Promotes Activation of Cathepsin K. J Biol Chem. 2014 doi: 10.1074/jbc.M114.559898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuo Y, Gauthier JY, Black WC, Percival MD, Duong LT. Inhibition of bone resorption by the cathepsin k inhibitor odanacatib is fully reversible. Bone. 2014 doi: 10.1016/j.bone.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Wang RY, Aminian A, McEntee MF, Kan SH, Simonaro CM, Lamanna WC, Lawrence R, Ellinwood NM, Guerra C, Le SQ, Dickson PI, Esko JD. Intra-articular enzyme replacement therapy with rhIDUA is safe, well-tolerated, and reduces articular GAG storage in the canine model of mucopolysaccharidosis type I. Mol Genet Metab. 2014 Aug;112(4):286–93. doi: 10.1016/j.ymgme.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.