


Abstract
Here, we present an update on the Genome-Wide Repository of Associations between SNPs and Phenotypes (GRASP) database version 2.0 (http://apps.nhlbi.nih.gov/Grasp/Overview.aspx). GRASP is a centralized repository of publically available genome-wide association study (GWAS) results. GRASP v2.0 contains ∼8.87 million SNP associations reported in 2082 studies, an increase of ∼2.59 million SNP associations (41.4% increase) and 693 studies (48.9% increase) from our previous version. Our goal in developing and maintaining GRASP is to provide a user-friendly means for diverse sets of researchers to query reported SNP associations (P ≤ 0.05) with human traits, including methylation and expression quantitative trait loci (QTL) studies. Therefore, in addition to making the full database available for download, we developed a user-friendly web interface that allows for direct querying of GRASP. We provide details on the use of this web interface and what information may be gleaned from using this interactive option. Additionally, we describe potential uses of GRASP and how the scientific community may benefit from the convenient availability of all SNP association results from GWAS (P ≤ 0.05). We plan to continue updating GRASP with newly published GWAS and increased annotation depth.
INTRODUCTION
Over the past decade, genome-wide association studies (GWAS) have fundamentally changed how clinicians and researchers attempt to identify genes that contribute to human health and disease (1). With GWAS, investigators can scan the entire genome with hundreds of thousands (and now millions) of single nucleotide polymorphisms (SNPs) in the search of genes that contribute to complex traits. Thousands of GWAS have been completed on a wide variety of traits, ranging from human disease (e.g. obesity, hypertension and bipolar disorder) to non-disease-related human traits (e.g. height, smell sensitivity and political preference). Even as the use of next-generation sequencing studies has become more widespread, GWAS remains a common tool in the search of genes associated with human disease and traits.
These studies have generated millions of genetic association results with a litany of diverse phenotypes. Most GWAS publications focus on the strongest and/or most logical findings with the remaining results tucked away in the Supplementary tables and figures. Many of these associations do not meet more stringent criteria, including correction for multiple testing and replication in independent samples, imposed by the field to prevent the reporting of false positives (2). However, many traits and diseases are highly polygenic and suggestive association results are noteworthy and, when examined in follow-up studies, often do show evidence for replication or biological relevance. Initial GWAS publications are often limited in their efforts to pursue suggestive signals for reasons including lack of availability of additional samples, budget and publication strategies. Independent investigators may find these ‘non-significant’ results useful, particularly in the interpretation of other GWAS findings, translation of results to/from basic science experimentation and in the generation of new hypotheses to be tested. Unfortunately, these suggestive associations are often missed in literature searches and are generally under-annotated in existing genetic databases.
With this in mind, our group recently constructed a new database called the Genome-Wide Repository of Associations between SNPs and Phenotypes (GRASP) (3). GRASP v1.0 represented an update from our earlier open access GWAS results database, a centralized catalog of published SNP-based genetic associations (P ≤ 0.05) from GWAS (4). At the time of publication and launch, GRASP v1.0 consisted of 1389 studies published through December 31, 2011. Here, we present an update of GRASP (version 2.0), which includes an addition of 693 more studies completed through mid-2013, totaling ∼8.87 million association results from 2082 studies. GRASP v2.0 is freely available for download, but may also be accessed through our interface (http://apps.nhlbi.nih.gov/Grasp/Overview.aspx). Here, we provide details on our recent update to GRASP and how users can employ the GRASP interface to obtain GWAS information of interest.
DATA ACQUISITION AND EXTRACTION
In order to identify potential studies for inclusion in the new GRASP update, we first performed controlled literature searches on PubMed. Search terms are located in Supplementary File 1. Resulting studies were then further reviewed to determine whether they met inclusion/exclusion criteria. Studies were required to include association testing of ≥25 000 SNP markers for one or more human traits. Gene expression, methylation and metabolomic-based SNP association studies were considered to meet these criteria. Only single SNP main effects were included; all SNP–SNP, gene–gene and SNP–environment interactive effects, as well as conditional analyses, were excluded. Additionally, we did not include any gene-based or pathway-based results, unless these led to highlighting of single SNP statistics. All SNP-level association results with P ≤ 0.05 were extracted from the text, tables, figures and supplement from each article, with the location of each association result within the manuscript noted. Specific phenotypes for each association were recorded. Further details regarding data extraction and inclusion/exclusion criteria are available at the GRASP website (http://apps.nhlbi.nih.gov/Grasp/Overview.aspx) (3).
Following the extraction of all SNP associations, results were reviewed for quality control purposes and via computer programs to reduce duplicate entries for each SNP, ensure valid SNP identifiers (rsIDs) and limit to results with P ≤ 0.05. Study-level information was noted for each study included. Some study-level information, including sample size and descriptions of markers analyzed (e.g. genotyping platform, number of markers and use of imputation), was derived using the National Human Genome Research Institute (NHGRI) GWAS catalog when available and added manually when information was not available (5). The sample size of replication samples and the use of any gender-specific discovery samples were also noted. In addition to the specific phenotypes reported, we grouped phenotypes into narrow and broad categories at the study level. Studies were permitted to be categorized into multiple groups in order to most accurately describe the trait(s) analyzed in each publication. These categories allow for more convenient phenotype-based searching for users.
SNPs were mapped to genome position in human genome B37 (hg19) using dbSNP Build 141. For SNPs without dbSNP identifiers (e.g. array-specific IDs, chromosome and position coordinates), various bioinformatics search strategies were applied in attempts to obtain a unique identifier, chromosome and position. If valid identifiers and positions could not be identified, results were excluded. In several cases, errors in the original report SNP identifiers were detected and fixed. Various SNP-level annotations were applied to each SNP, including: dbSNP functional category, minor allele frequency in dbSNP, validation status in dbSNP and PolyPhen2/SIFT prediction results for non-synonymous SNPs (6,7). In addition, we noted whether an SNP overlapped in genomic position with respect to: large intergenic non-coding RNAs (lincRNAs), micro RNA (miRNA) regions from miRbase, miRNA-binding sites from PolymiRTS 2.0, regulatory annotations from ORegAnno, conserved predicted transcription factor sites from UCSC and validated human enhancer sequences from VistaEnhancer (8–12). Finally, protein function regions from UniProtKB were mapped to corresponding 3-based genomic positions and checked for overlap with SNPs (13).
DESCRIPTION OF ASSOCIATION RESULTS
In this update of GRASP, we have added ∼2.59 million SNP association results from 693 GWAS publications, totaling 8.87 million SNP association results and 2082 studies all together. This represents a 41.4% and 48.9% increase in association results and studies, respectively. Nine of the 693 new publications (1.29%) did not disclose any association results with P ≤ 0.05 but did meet our study inclusion criteria. Of the 2.59 million new association results, 277 372 were from 23 methylation, metabolomics, expression, glycomics or protein quantitative trait loci (QTL) studies. Eighteen (2.60%) of the newly added studies, three of which were QTL studies, made greater than 25 000 SNP association results with P ≤ 0.05 available, totaling 2.37 million results or 91.19% of the newly added results. These proportions are similar to those of the 2.88% of studies reporting greater than 25 000 association results, comprising 92.14% of the 6.28 million results, reported in GRASPv1.0 (3). In the list of studies included in GRASP, we have now added a column describing the number of markers tested and the number of associations with P ≤ 0.05 reported, so users can gauge the overall testing burden and results disclosure for specific studies. These low proportions indicate that authors typically do not make the vast majority of association results (P ≤ 0.05) publically available, although the number of markers tested must be taken into context (14). The NIH GWAS Sharing Policy and the increased use of the database of Genotypes and Phenotypes (dbGaP) provide protected means of sharing GWAS data and results, while largely maintaining the privacy and rights of subjects as well as the scientific integrity of the data (15). Increased sharing and availability of GWAS data and/or results appears to augment scientific discovery and productivity (15). GRASP is an open access database that provides a middle ground for results queries without formal data access committee applications, while being limited to the results authors’ choose to disclose and which are chronologically curated.
Traits and phenotype categories are diverse and numerous including 177 categories (14.9% growth over GRASP 1.0). Those categories represented across >5% of studies in the entire database are: quantitative traits (33.62% of studies), neurological-related (23.72% of studies), blood-related (20.46% of studies), cardiovascular disease risk factor (13.59% of studies), cancer (12.20% of studies), gender-specific (11.14% of studies), inflammation (10.81% of studies), behavioral (8.07%), female gender (7.97%), drug response (7.78% of studies), cardiovascular disease (6.39%), developmental (5.72%), pulmonary (5.67%) and aging-related (5.09%). The overlap of unique SNPs in GRASP 2.0 with selected annotation features is described at a liberal (P < 0.05), moderate (P < 1.0 × 10−5) and stringent (P < 5.0 × 10−8) statistical threshold in Table 1. Because of their atypical nature in GWAS (3), QTL studies and the major histocompatibility (MHC) region (6p21.3) are excluded from these statistics. As previously reported, a substantial proportion of SNPs showing association (P < 0.05) with a human-health-related phenotype were within genes (4). This proportion grows at more stringent P-value thresholds reaching 56.8% of associated SNPs mapping within a USCC RefSeq gene at the P < 5.0 × 10−8 threshold. Proportions of SNPs coinciding with several other functional categories examined increase (miRNA, miRNA predicted binding sites, missense, nonsense, intronic, 5′/3′ regions, synonymous variants) at more stringent statistical thresholds. A notable exception is that more highly associated SNPs appear depleted in overlaps with lincRNAs, similar to a trend previously noted among significant eQTL results (16).
Table 1. Annotation of SNPs included in GRASP v2.0 at three P-value threshold levels after excluding QTL studies and MHC region (6p21.3) associations.
| P ≤ 0.05 2 425 944 unique SNPs | P ≤ 1.0 × 10−5 91 615 unique SNPs | P ≤ 5.0 × 10−8 37 607 unique SNPs | ||||
|---|---|---|---|---|---|---|
| Number | Percentage | Number | Percentage | Number | Percentage | |
| Gene | 1 012 182 | 41.72 | 49 116 | 53.61 | 21 368 | 56.82 |
| LincRNA | 120 181 | 4.95 | 3251 | 3.55 | 1140 | 3.03 |
| miRNA | 61 | 0.0025 | 3 | 0.0033 | 3 | 0.0080 |
| miRNA predicted binding site | 6402 | 0.26 | 365 | 0.40 | 166 | 0.44 |
| 5′UTRa | 10 460 | 0.43 | 1103 | 1.20 | 585 | 1.56 |
| 3′UTRb | 23 935 | 0.99 | 1517 | 1.66 | 678 | 1.80 |
| Nonsense | 91 | 0.0038 | 16 | 0.017 | 10 | 0.027 |
| Intron | 984 902 | 40.60 | 46 079 | 50.30 | 19 826 | 52.72 |
| Missense | 12 679 | 0.52 | 1,333 | 1.46 | 706 | 1.88 |
| Synonymous | 11 398 | 0.47 | 849 | 0.93 | 436 | 1.16 |
| Multiple/otherc | 13 448 | 0.55 | 330 | 0.36 | 190 | 0.51 |
aIn the 5′untranslated region (5'UTR) or within 2000 bp of the 5′ end of gene.
bIn the 3′untranslated region (3'UTR) or within 500 bp of the 3′ end of gene.
cSNPs had multiple annotations and/or included other categories.
HOW TO USE GRASP
The entire GRASP database can be freely downloaded from the GRASP website. The full download allows for in-house querying, integration with other data sets and resources and analysis of the cataloged GWAS results. However, for those researchers who may only be interested in a subset of markers, genomic loci, genes or phenotypes, we have developed a user-friendly interface that allows for specific, easy querying for particular (i) SNPs (identified by refSNP identifiers (rsIDs)), (ii) gene names, (iii) chromosomal locations and (iv) phenotypes/traits. Additionally, a minimum P-value threshold filter more stringent than P ≤ 0.05 can be imposed, if desired. Results are organized into broad phenotype Categories and narrower specific Traits. Searches through the drop-down interface can be conducted against broad Categories, or against more specific Traits.
Figure 1a and b displays an example of a search for the gene APOE at a P-value threshold of P < 1.0 × 10−5. The search function will return all SNP associations with P < 1.0 × 10−5 in the GRASP database for SNPs within or in close proximity to the query gene as defined by current NCBI dbSNP locus definitions. The resulting output from the search in Figure 1a is shown in Figure 1b. Within the search tool, various pieces of information are given including: (i) NHLBI Key, a unique identifier for each SNP association result in the database; (ii) SNP ID, the dbSNP ID identifier in the current dbSNP Build; (ii) P-value, the reported P-value for this particular SNP association; (iv) PMID, the PubMed ID for the publication where this SNP association was reported with hyperlink to the abstract; (v) Location, where in the publication the results are reported; (vi) Phenotype, the trait the reported SNP is associated with; (vii) Phenotype Category, a general category or categories of where the trait falls into; (viii) Chr, chromosomal location of the SNP; (ix) Position, base pair position of the SNP (automatically updated by NCBI as genomic builds are updated); and (x) InGene, the gene the SNP is within if located in a gene region (based on current NCBI definitions). The results of these searches, with additional SNP and regional annotations, are available to copy and paste, or for export in .csv or Excel formats. An example using the APOE at a P-value threshold of P < 1.0 × 10−5 search can be found online in Supplementary File 2.
Figure 1.
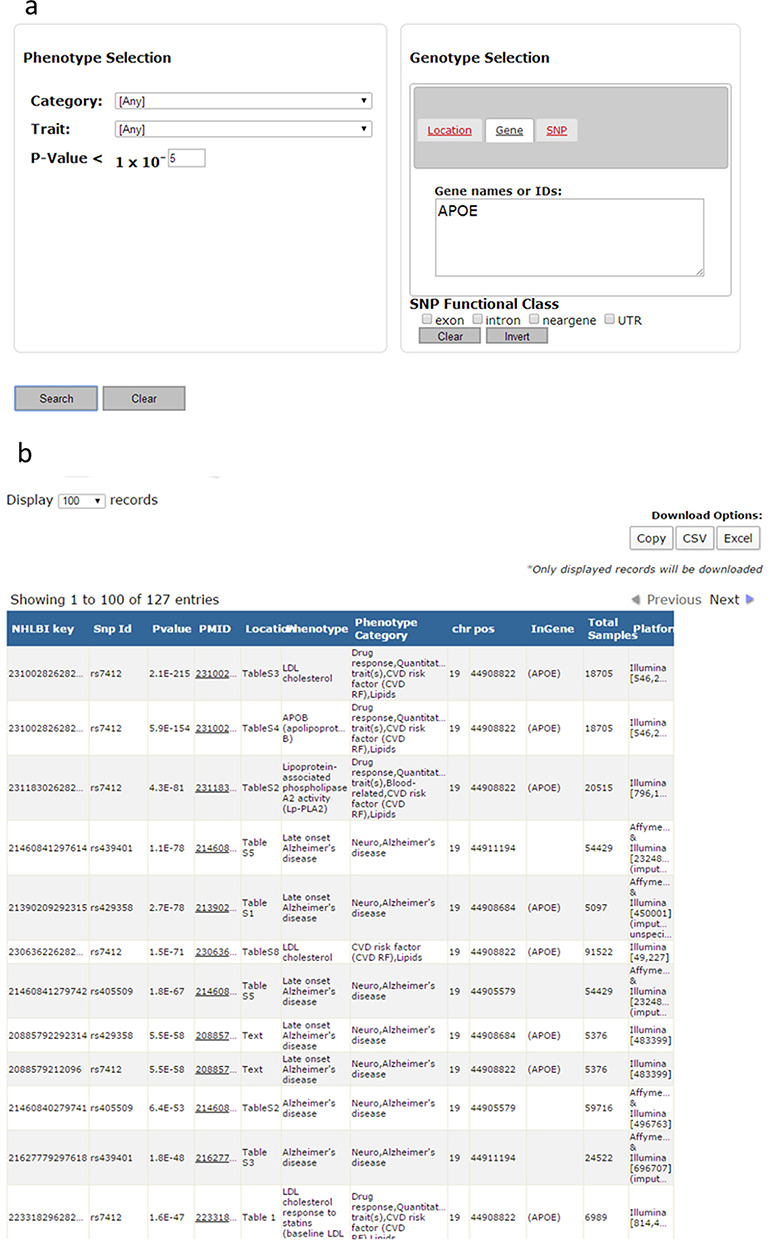
(a) Example of a GRASP search by gene (APOE) and imposing a P-value threshold (P < 1.0 × 10−5). (b) The results of the example GRASP search in (a). The full results of this example search can be found in the Supplementary materials.
The search functionality of GRASP is to serve as a quick, user-friendly tool for researchers to query publically available GWAS results for SNPs, genes, loci and phenotypes of interest. However, due to the sheer number of SNP-expression (expression QTL), SNP-methylation (methylation QTL), SNP-metabolomic (metabolome QTL), SNP-proteomic (protein QTL) and SNP-glycomic phenotype names, these associations cannot be accommodated in the drop-down menu phenotype search feature. These results are available in full download of GRASP.
POTENTIAL USES OF GRASP
The centralization of such a vast number of GWAS results enables the research community to complete various qualitative and quantitative analyses. First, publication trends of GWAS reports by year, journal, types of markers, use of imputation, among other things, can be ascertained (14). Second, researchers completing GWAS or other gene hunting studies can identify other associations of SNPs or genes with related traits. This can allow for better interpretation of findings as well as identify potential collaborators for replication, meta-analyses and/or follow-up functional analyses. Additionally, this knowledge can inform follow-up biologic experiments and analyses by identifying previous associations with sub-clinical disease factors or endophenotypes. Third, the results stored in GRASP (summary statistics from GWAS) could be leveraged for new analyses, whether in meta-analyses, pathway analyses or the use of new analytical methods. However, additional data (e.g. ascertainment of tested alleles) may need to be extracted from the original study in addition to the data extracted from GRASP. Finally, observations from GRASP may allow for the generation of new hypotheses, particularly regarding gene pleiotropy. In that vein, most repositories for SNP association results only include associations that meet stringent statistical thresholds. Although the imposition of stringent corrections has reduced the number of false positives in genetic association studies and could be applied in GRASP via P-value filtering, many suggestive associations that do not meet these stringent criteria may represent true associations. GRASP includes these suggestive associations (P ≤ 0.05) to allow the research community to reference these associations, provide context for future association studies and allow for the examination of genetic associations that may be overlooked. In comparison to other GWAS catalogs at both identical historic time-points and stringent threshold (e.g. P < 5.0 × 10−8), GRASP contains much greater proportions of studies and genetic results than other existing catalogs. The above represent a small number of potential utilities of GRASP; a wide range of applications for the repository exist depending on the initiative and creativity of the user community.
FUTURE WORK IN GRASP
In the future, our primary goal is to continue to update GRASP with SNP associations from newly published GWAS reports. Currently, we have incorporated results from GWAS published of manuscripts up to mid-2013. We plan on extracting and adding the remaining studies through the end of 2014 as well as regularly updating GRASP with new GWAS publications as they become available. We also plan to accept voluntary results submission from researchers in future GRASP builds for public posting of summary P-value results in a citable database. In addition, with the expanding knowledge of the human genome, we hope to increase the depth of annotations for associated SNPs. For example, the importance in human health and disease of non-coding RNAs, such as miRNAs, small nucleolar RNAs (snoRNAs) and lincRNAs, among others, has become evident (17). In addition to reporting whether an SNP occurs within a gene, we plan to expand the catalog of SNP associations that intersect known noncoding RNA elements. However, a significant minority of SNPs found in GWAS studies do not occur in loci that encode transcripts (5,18). The ENCODE project, among other efforts, has been working to improve annotation of the large proportion of the human genome that does not encode for proteins (19–21). These annotations include epigenetic modifications (e.g. acetylation or methylation sites) and genome-wide characterization of transcription factor binding sites, among others. The inclusion of these annotations will further inform researchers on possible functional implications of associated SNPs.
Finally, newer and cheaper sequencing technologies are making larger sequencing-based association studies more common. Currently, GRASP has not included the results of sequencing studies. We are currently working on a mechanism to efficiently and accurately report the findings of sequencing-based association studies within the GRASP framework. Burden-tests (e.g. SKAT) and other multi-SNP tests are frequently used in these association studies further complicating the incorporation of these studies into GRASP (22). Along with the enumerated updates above, we plan on making other systemic and contextual changes. The authors welcome suggestions for modification of the GRASP database from the research community.
CONCLUSION
Here, we present an update to the GRASP database with a 48.9% increase (n = 693) in the number of studies. The resulting database, GRASP v2.0, now contains ∼8.8 million SNP–trait association results from GWAS. These results are located in a central repository that is freely accessible and downloadable for the entire research community. Additionally, we have provided a convenient web-based search tool for quick queries of SNPs, genes, chromosomal loci and phenotypes of interest. GRASP will allow for efficient examination of past GWAS results for researchers, allowing for better data interpretation, communication between possible collaborators and new hypotheses to be generated. New annotations and new studies will be added to the GRASP framework with future updates. Updates to GRASP will be made available on our website (http://apps.nhlbi.nih.gov/Grasp/Overview.aspx) with details and corrections reported to those registered to the GRASP mailing list in future reports.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Mina Youssef, Ashish Mehta and the NHLBI Center for Biomedical Informatics, and Masato Kimura, Ming Xu and Michael Feolo from NCBI for help in creating and maintaining the web interface.
FUNDING
National Heart, Lung, and Blood Institute (NHLBI) intramural program. Funding for open access charge: National Institutes of Health/NHLBI.
Conflict of interest statement. None declared.
REFERENCES
- 1.Manolio T.A. Cohort studies and the genetics of complex disease. Nat. Genet. 2009;41:5–6. doi: 10.1038/ng0109-5. [DOI] [PubMed] [Google Scholar]
- 2.Pearson T.A., Manolio T.A. How to interpret a genome-wide association study. JAMA. 2008;299:1335–1344. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- 3.Leslie R., O'Donnell C.J., Johnson A.D. GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics. 2014;30:i185–i194. doi: 10.1093/bioinformatics/btu273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson A.D., O'Donnell C.J. An open access database of genome-wide association results. BMC Med. Genet. 2009;10:1–17. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. U.S.A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 7.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziebarth J.D., Bhattacharya A., Chen A., Cui Y. PolymiRTS Database 2.0: linking polymorphisms in microRNA target sites with human diseases and complex traits. Nucleic Acids Res. 2012;40:D216–D221. doi: 10.1093/nar/gkr1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffith O.L., Montgomery S.B., Bernier B., Chu B., Kasaian K., Aerts S., Mahony S., Sleumer M.C., Bilenky M., Haeussler M., et al. ORegAnno: an open-access community-driven resource for regulatory annotation. Nucleic Acids Res. 2008;36:D107–D113. doi: 10.1093/nar/gkm967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Doekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Visel A., Minovitsky S., Dubchak I., Pennacchio L.A. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson A.D., Leslie R., O'Donnell CJ. Temporal trends in results availability form genome-wide association studies. PLoS Genet. 2011;7:e1002269. doi: 10.1371/journal.pgen.1002269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paltoo D.N., Rodriguez L.L., Feolo M., Gillanders E., Ramos E.M., Rutter J.L., Sherry S., Wang V.O., Bailey A., Baker R., et al. Data use under the NIH GWAS Data Sharing Policy and future directions. Nat. Genet. 2014;46:934–938. doi: 10.1038/ng.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X., Gierman H.J., Levy D., Plump A., Dobrin R., Goring H.H., Curran J.E., Johnson M.P., Blangero J., Kim S.K., et al. Synthesis of 53 tissue and cell line expression QTL datasets reveals master eQTLs. BMC Genomics. 2014;15:532. doi: 10.1186/1471-2164-15-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 18.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Encode Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 20.Encode Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kellis M., Wold B., Snyder M.P., Bernstein B.E., Kundaje A., Marinov G.K., Ward L.D., Birney E., Crawford G.E., Dekker J., et al. Defining functional DNA elements in the human genome. Proc. Natl Acad. Sci. U.S.A. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X. Rare variant association testing for sequencing data using the sequence kernel association test (SKAT) Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]