

Abstract
Pediatric acquired aplastic anemia (AA) is a bone marrow disorder that is difficult to distinguish from inherited bone marrow failure syndromes and hypocellular refractory cytopenia of childhood (RCC). Historically, patients with hypocellular RCC have been given the diagnosis of AA. To assess the clinical and histologic distinction between RCC and AA, we performed a retrospective analysis of 149 patients previously diagnosed with AA between 1976-2010. We evaluated event free survival (EFS), overall survival (OS), response rates to immunosuppressive therapy, treatment-related toxicities and clonal evolution. The 5-year EFS and OS were 50.8%±5.5% and 73.1%±4.7%, respectively. Patients with very severe AA had worse OS compared to patients with severe and moderately severe AA. Seventy-two patients had diagnostic pathology specimens available for review. Three pediatric hematopathologists reviewed and reclassified these specimens as AA, RCC or Other based on 2008 WHO Criteria. The concordance between pathologists in the diagnosis of AA or RCC was modest. RCC was associated with a trend towards improved OS and EFS and was not prognostic of immunosuppression therapy treatment failure. There was a low rate of clonal evolution exclusively associated with moderately severe AA. Our findings indicate that a diagnosis of RCC is difficult to establish with certainty and does not predict outcomes, calling into question the reproducibility and clinical significance of the RCC classification and warranting further studies.
Keywords: Aplastic Anemia, Pediatric Hematology/Oncology, Bone Marrow Failure, Myelodysplastic Syndrome, Hematopathology
Introduction
Bone marrow failure (BMF) disorders in children comprise a heterogeneous group of inherited and acquired conditions. Aplastic anemia (AA) is the most common form of acquired BMF. The diagnosis of AA requires a hypocellular bone marrow and two of the following criteria: i. hemoglobin (Hgb) < 10g/dL; ii. Platelets <50,000 cells/μL; iii. Absolute neutrophil count (ANC) <1500 cells/μL 1. AA is typically a diagnosis of exclusion; inherited bone marrow failure syndromes (IBMFS), and other causes of a hypoplastic bone marrow, such as infections, metabolic diseases, nutritional deficiencies, malignancies, and hypocellular myelodysplastic syndromes (MDS) must be ruled out. The majority of cases (~70-80%) of pediatric AA are idiopathic in origin1.
The pathophysiology of idiopathic AA in both children and adults is thought to be a Th1/CD8+ T-cell driven autoimmune process2. Immunosuppressive therapy (IST) – with triple therapy consisting of anti-thymocyte globulin (ATG), cyclosporine and prednisone - is the mainstay of therapy in patients who do not have a histocompatible matched related donor (MRD) for hematopoietic stem cell transplantation (HSCT). Pediatric AA patients treated with IST have an 3-5-year OS survival ranging from 81-93%3-7, depending upon the severity of disease. Response rates to IST range from 44-80%, with the majority of responses occurring within 4-6 months of therapy3,4,6,7. Relapse rates following IST in pediatric patients range between 13-15%3-5,8-10. A recent study focusing on treatment of adolescent AA patients with IST showed a 3-year EFS of 37%8, with an overall treatment failure rate of greater than 50%8. Most patients who fail to achieve a durable response to IST undergo a second round of IST or matched unrelated donor (MUD) HSCT as rescue therapy.
The frequency of clonal evolution in pediatric patients treated with IST, including cytogenetic changes, development of dysplastic hematopoiesis, or an increased blast count appears to be less than in adults, ranging from zero to 21.9%3-8. This wide range of response to IST and frequency of clonal evolution suggests that acquired AA may actually be a heterogeneous group of diseases, in which a subset of patients may have an underlying stem cell defect linked to clonal progression and the lack of a response to IST. Indeed, recent genomic studies have identified somatic genetic alterations in a small subset of adult AA patients, although the biological relevance of these changes remain unclear11,12.
In order to address the biological variability observed in the pediatric AA population, Baumann and colleagues defined and then studied the predictive value of a pathological classification of hypocellular bone marrows that included a new pediatric-specific subtype of MDS that they termed refractory cytopenia of childhood (RCC). They found that a diagnosis of RCC predicts a less favorable response to IST and an increased risk of clonal evolution, suggesting that initial use of HSCT might be an alternative therapeutic approach13,14. RCC was subsequently included as a provisional entity in the 2008 World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues13,15. The WHO defines RCC as: i. dysplasia in one or more hematopoietic lineages or in at least 10% of cells in one cell line; ii. less than 5% blasts in the marrow15. The MCV in RCC is usually elevated16. The majority of RCC patients present with a hypocellular bone marrow16; therefore, the histologic distinction between AA and RCC is challenging and requires the collaboration of experienced hematologists and hematopathologists, as well as cytogenetic and molecular analyses. RCC has been a controversial entity and has not been universally accepted, especially because the therapeutic and prognostic implications of the diagnosis have only been validated in a limited number of studies13,14,17. The distinction between AA and RCC can be somewhat confusing especially given the controversy around different taxonomies used to describe the BMF disorders. For the purposes of this manuscript, “previously diagnosed AA” or “clinical AA” comprises AA based on traditional diagnostic criteria1. In contrast, “AA” refers to histopathologically diagnosed AA based on 2008 WHO criteria. “RCC” here refers to histopathologically diagnosed RCC, a subset of pediatric MDS, based on 2008 WHO criteria, which is by definition a separate diagnosis from AA15.
Based on the existing data, we hypothesized that differences in the underlying biology of AA might explain variable responses to IST, clonal evolution, and clinical outcomes. Understanding these biological differences might suggest alternative initial therapies that could improve outcomes. In this single institution retrospective study, we reviewed and re-classified diagnostic material from 72 children previously diagnosed with AA according to the WHO classification - specifically distinguishing AA from hypocellular RCC - in order to determine the reproducibility of the proposed criteria as well as the clinical significance of the diagnosis of RCC. We also evaluated EFS, OS, and response rate to IST, outcomes after MRD and MUD HSCT following failed IST as well as toxicities and the rate of clonal disease progression. We found response rates to IST consistent with published literature and a low rate of clonal disease progression in classical pediatric AA patients. Furthermore a diagnosis of RCC does not predict disease outcome, or response rate to IST.
Methods
Patients
We conducted a retrospective chart review and analysis of pathology specimens from patients treated for classic AA at Boston Children's Hospital (BCH) and Dana-Farber Cancer Institute (DFCI) between 1976-2010. Patients were identified using ICD-9 code searches, local clinical databases and registries. Inclusion criteria were: a clinical and pathologic diagnosis of AA, age <22 years at diagnosis, a CBC obtained at diagnosis, and an available description of treatment and clinical course in the medical record. Exclusion criteria were: failure to meet the above inclusion criteria,concurrent diagnosis of Paroxysmal Nocturnal Hemoglobinuria (PNH), IBMFS by clinical criteria or molecular testing, pancytopenia secondary to other identified causes, advanced MDS such as refractory cytopenia with acess blasts (RAEB), and clonal cytogenetic changes. The institutional review board at BCH approved this study. A total of 149 eligible patients were identified.
Pathology Review
May-Gruenwald-Giemsa stained bone marrow (BM) aspirate and paraffin-embedded, H&E stained biopsy slides obtained prior to any therapeutic intervention were considered for review. Diagnostic material from 72 patients was available. Pre-therapy slides for the remaining 77 patients were not available,most often because the sample was obtained at another institution. Three board certified hematopathologists specializing in pediatrics (MHH, OW and IH) performed a double-blinded analysis and independently reviewed the entire study set of samples. Each pathologist assessed: 1) specimen adequacy, 2) biopsy cellularity, 3) confirmation of the diagnosis of AA employing conventional criteria 1, and 4) re-classification by 2008 WHO criteria (AA, RCC or other)15. In cases where one or more hematopathologists differed in their assessment, a consensus diagnosis was developed by reviewing and discussing each discrepant case with a senior hematopathologist (MDF). Together, the 4 pathologists determined a consensus diagnosis.
Definitions
Classic AA was defined using conventional criteria as a hypocellular bone marrow biopsy with <25% of the cellularity expected for age and the following published criteria: Hgb <10g/dL; Platelets <50,000 cells/μL; ANC <1500 cell/μL1. The severity of classic AA was categorized into moderate (MAA), severe (SAA) or very severe AA (vSAA) based on existing published criteria 1,18. Relapse and hematologic response (HR) was evaluated at 6 months and 1 year after initiation of therapy and defined as partial (PR) or complete response (CR) as previously published19. Failure to respond was defined as not achieving a PR or CR. Clonal evolution was determined by cloncal cytogenetics, increased blast count and progression to advanced MDS (RAEB). Medical records were reviewed for the following adverse events: renal toxicity, liver toxicity, fever and neutropenia, bacteremia, sepsis, hypertension, and transfusion dependence.
Data Collection
For each eligible patient data on clinical presentation, treatment and laboratory values (including CBC, reticulocyte count and HgF levels) were obtained at diagnosis, 6 months, 1 year, and yearly thereafter until the patient died or lost follow-up. Long-term outcome data also included HR to therapy, the provision of a MUD after completion of IST, development of a secondary malignancy, clonal disorder, and relapse of AA. Study data were collected and managed using REDCap electronic data capture tools hosted at DFCI20. REDCap (Research Electronic Data Capture) is a secure, web-based application designed to support data capture for research studies, providing: 1) an intuitive interface for validated data entry; 2) audit trails for tracking data manipulation and export procedures; 3) automated export procedures for seamless data downloads to common statistical packages; and 4) procedures for importing data from external sources.
Statistical Considerations
EFS time was calculated from diagnosis until the time of first occurrence of one of the following: HSCT, failure to respond to IST, diagnosis of advanced MDS, clonal evolution by cytogenetics, increased blast count, relapse, or death, or until last contact if none of these events occurred. A subanalysis was performed for patients treated after the year 2000 to asses outcomes over the last decade. OS time was calculated from diagnosis until death, or until last contact if the patient was alive. Survival curves were generated using the methods of Kaplan and Meier, with standard errors according to Peto21. Survival curves were compared using a log rank test. Tests of association were performed using Fisher's exact test. To describe the degree of concordance between pathologists, a kappa coefficient was calculated. A logistic regression model was used to identify factors prognostic of treatment failure. P-values less than 0.05 were considered statistically significant.
Results
Patient Characteristics
149 eligible classic AA patients were identified. Patient characteristics are outlined in Supplementary Table 1. The mean follow-up was 4.2 years (range, 0.02-21.5 years, median 2.6 years). The mean age at diagnosis was 9.3 years old (range, 0 to 21.1, median 9.0) and 48% of patients were female (n=71). There were 58, 50, and 41 patients with MAA, SAA, vSAA, respectively based on conventional criteria. Ninety-one patients received IST and were the focus of this study. The majority of patients treated with IST received triple therapy with cyclosporine (CSA), ATG and Prednisone (n=51). The remaining patients (n=40), largely treated prior to 1991, before triple IST became the standard of care22, received different combinations of immunosuppressive therapies, including steroids, in most cases.
Outcomes
Outcomes and treatment responses for patients receiving different IST modalities and outcomes by disease severity
The 5-year EFS and OS of the entire cohort of classic AA patients (n=149), including patients that eventually underwent a MRD or MUD HSCT, were 50.8%±5.5% and 73.1%±4.7%, respectively (Table 1). Assessing outcomes by disease severity, the overall 5-year EFS were 58.0%±8.9%, 54.3%±8.9% and 36.8%±8.8% (p=0.004 across groups) for patients diagnosed with MAA, SAA and vSAA, respectively. The 5-year OS in MAA, SAA and vSAA was 78.1%±6.6%,82.2%±7.4% and 55.2%±9.0%, respectively (p=0.03 across groups Table 1).
Table 1.
Event-free survival (EFS) and overall survival (OS) of study cohort (n=149)
| Predictors | |||||
|---|---|---|---|---|---|
| Event-free survival | Overall survival | ||||
| N | 5-yr EFS ± SE | P value | 5-yr OS ± SE | P value | |
| All patients | 149 | 50.8 ±5.5 | 73.1 ± 4.7 | ||
| Original diagnosis | |||||
| MAA | 58 | 58.0 ± 8.9 | 0.004 | 78.1 ± 6.6 | 0.003 |
| SAA | 50 | 54.3 ± 8.9 | 82.2 ± 7.4 | ||
| vSAA | 41 | 36.8 ± 8.8 | 55.2 ± 9.0 | ||
| Consensus diagnosis | |||||
| RCC | 30 | 71.3 ± 11.0 | 0.3~ | 84.9 ± 8.8 | 0.3~ |
| AA | 28 | 52.5 ± 13.7 | 72.5 ± 11.5 | ||
| Other | 10 | 58.3 ± 21.7 | 80.0 ± 20.7 | ||
| Not sufficient to evaluate | 6 | 75.0 ± 21.6 | 100 | ||
| Unknown | 75 | 40.3 ± 6.2 | 65.6 ± 6.2 | ||
| IST only patients | 91 | 34.6 ± 7.0 | 66 ± 6.7 | ||
| IST therapy | |||||
| Triple therapy | 51 | 51.5 ± 8.5 | 0.006 | 87.8 ± 5.5 | <0.0001 |
| Other | 40 | 16.3 ± 7.4 | 38.2 ± 10.0 | ||
| Triple IST | |||||
| Yr diagnosed: <2000 | 21 | 43.0 ± 12.2 | 0.4 | 82.1 ± 9.6 | 0.3 |
| Yr diagnosed: ≥2000 | 30 | 58.1 ± 10.0 | 91.5 ± 6.3 | ||
log rank test comparison of RCC vs. AA
SE = standard (error)
Classic AA patients that were administered triple IST had a better 5-year EFS of 51.5%±8.5%, compared to those who received other forms of IST (16.3%±7.4%; p=0.006). The 5-year OS for patients with triple IST was also significantly better (87.8%±5.5% compared to 38.2%±10.0%; p<0.0001) (Table 1). The outcome for patients diagnosed before and after the year 2000 and treated with triple IST was not significantly different (5-year EFS: 58.1%±10.0% versus 43.0%±12.2% respectively; 5-year OS: 82.1%±9.6% versus 91.5%±6.3%respectively) (p=0.4, Table 1). Sixty-one of the 91 patients that received initial IST ultimately failed or had an adverse outcome, including death (n=10), failure to respond (n=35), progression to MDS (n=4), relapse (n=7) or referral to HSCT (n=5). In classical AA diagnosed and treated with triple IST in the year 2000 and afterword, we observed a trend towards improved EFS for patients diagnosed with MAA/SAA compared to vSAA (p=0.08, Figure 1A). However, the 5-year OS was statistically significantly higher in MAA/SAA patients than in vSAA patients (p=0.02, Figure 1B). For classical AA patients that eventually underwent HSCT, the 5-year OS post-transplant was comparable in patients receiving a MUD or MRD HSCT following IST (74.1±9.1% and 85.4±5.2%, respectively; p=0.4).
Figure 1.
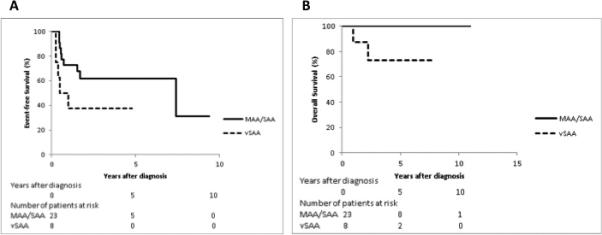
Figure 1A. EFS of AA patients who received IST and were diagnosed ≥2000, by disease subgroup (MAA/SAA, n=23; vSAA, n=8; p=0.08)
Figure 1B. OS of AA patients who received IST and were diagnosed ≥2000, by disease subgroup (MAA/SAA, n=23; vSAA, n=8; p=0.02)
Hematological response rates and treatment related toxicities for patients receiving IST
The HR after IST by disease severity was 54%, 46% and 30% for MAA, SAA and vSAA, respectively (p=0.2, across groups) (Supplementary Table 2). Forty-eight percent of patients treated with IST experienced treatment-related toxicities (TRT). The most commonly occurring toxicity was bacterial infection (25/89, 28%). Within the MAA and vSAA subgroups, the most common toxicities were renal toxicity (6/33, 18%) and bacterial infection (14/27, 52%), respectively; for the SAA subgroup, the most common toxicities included renal (6/28, 21%), and hepatic toxicities (6/28, 21%) and bacterial infections (6/28, 21%).
Reclassification of AA using the 2008 WHO criteria and concordance between hematopathologists
To determine the reproducibility of the 2008 WHO classification of RCC, three experienced board-certified pediatric hematopathologists independently reviewed diagnostic BM aspirates and biopsies and reclassifed specimens based on these criteria as “AA”, “RCC” or “Other”. Of 149 patients, 72 had material available for review. Interpretations were compared pairwise between all three pathologists. In 38 out of 72 cases (52.78%) all three independently arrived at the same diagnosis. For the 34 cases in which there was discordance, a consensus diagnosis of either AA (n=10) or RCC (n=14) was reached in conjunction with the assistance of a fourth pathologist. In 10 cases, no consensus could be reached, as the morphological features did not fit the diagnostic criteria for RCC or AA (Other, n=10). Pair-wise comparison between the 3 hematopathologists (A, B and C) yielded the following results: 1) pair A-B kappa coefficient 0.5 (95% CI: 0.4, 0.7) and concordance 74.6%, 2) pair B-C kappa coefficient 0.4 (95% CI: 0.1, 0.2) and concordance of 70.7%, and 3) pair A-C with kappa coefficient of 0.3 (95% CI: 0.1, 0.5) and concordance of 61.3% (Table 2).
Table 2.
Concordance between hematopathologists A, B and C of diagnostic classification of originally diagnosed AA patients into the diagnostic categories of AA, RCC or Other according to the WHO 2008 classification (n=74)
| Pathologist | B | Concordance* (%) | Kappa coefficient (95% CI) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RCC | AA | Other | Not sufficient to evaluate | Missing# | Total | ||||
| A | RCC | 25 | 10 | 2 | 1 | 0 | 38 | 74.6% | 0.5 (0.4, 0.7) |
| AA | 0 | 22 | 0 | 3 | 1 | 26 | |||
| Other | 1 | 3 | 0 | 2 | 0 | 6 | |||
| Not sufficient to evaluate | 0 | 0 | 0 | 4 | 0 | 4 | |||
| Total | 26 | 35 | 2 | 10 | 1 | 74 | |||
| Pathologist | C | Concordance* (%) | Kappa coefficient (95% CI) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RCC | AA | Other | Not sufficient to evaluate | Missing# | Total | ||||
| A | RCC | 19 | 15 | 2 | 1 | 1 | 38 | 61.3% | 0.3 (0.1, 0.5) |
| AA | 3 | 19 | 0 | 3 | 1 | 26 | |||
| Other | 1 | 3 | 0 | 2 | 0 | 6 | |||
| Not sufficient to evaluate | 0 | 0 | 0 | 4 | 0 | 4 | |||
| Total | 23 | 37 | 2 | 10 | 2 | 74 | |||
| Pathologist | C | Concordance* (%) | Kappa coefficient (95% CI) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RCC | AA | Other | Not sufficient to evaluate | Missing# | Total | ||||
| B | RCC | 15 | 8 | 1 | 1 | 1 | 26 | 70.7% | 0.4 (0.1, 0.2) |
| AA | 5 | 26 | 1 | 3 | 0 | 35 | |||
| Other | 1 | 1 | 0 | 0 | 0 | 2 | |||
| Not sufficient to evaluate | 1 | 2 | 0 | 6 | 1 | 10 | |||
| Missing# | 1 | 0 | 0 | 0 | 0 | 1 | |||
| Total | 22 | 37 | 2 | 10 | 2 | 74 | |||
Concordance was tested for the cells shaded in grey.
pt #88 was reviewed by A but was not reviewed by B, pt #3, 121 were reviewed by A but were not reviewed by C, # pt #88 was reviewed by Cbut was not reviewed by B, pt#3, 121 were reviewed by B but were not reviewed by C, as slides had to be returned to clinical pathology/referring institution.
Correlation of histopathology with clinical features
More patients in the MAA/SAA group were diagnosed with RCC than in the vSAA group (Table 3, p=0.0497) suggesting a correlation between lesser disease severity and the diagnosis of RCC. To evaluate this further, we determined if specific initial laboratory values were associated with AA or RCC. We found that, compared to RCC, AA patients had significantly lower hemoglobin (Hgb) values (AA: mean 6.4g/dL, median 6.6g/dL versus RCC: mean 8.1g/dL, median 8.1g/dL; p=0.03) and lower absolute reticulocyte count values (AA: mean 0.011 cells/uL, median 0.009 cells/uL versus RCC: 0.038 cells/uL, median 0.035 cells/uL; p=0.005). A trend towards a higher platelet count, higher mean corpuscular volume (MCV) and higher HgbF level was seen in RCC patients but did not reach statistical significance (Table 4). Since the MCV and HgbF has been previously reported to be elevated in RCC16 we assessed if a consensus diagnosis of RCC was associated with an MCV >100 and HgF% >4%. We did not confirm these associations in our study (Supplementary Table 3).
Table 3.
Consensus diagnosis by disease subgroup (MAA, SAA, vSAA) for patients whose consensus diagnosis was RCC or AA (n=58)
| Disease subgroup | RCC | AA | Total | p-value |
|---|---|---|---|---|
| MAA/SAA | 24 | 15 | 39 | 0.0497 |
| vSAA | 6 | 13 | 19 | |
| Total | 30 | 28 | 58 |
Table 4.
Patient age and laboratory values of the study cohort, by consensus diagnosis (n=149)
| n | Mean | Median | Min | Max | p-value* | |
|---|---|---|---|---|---|---|
| Age at diagnosis (in years) | ||||||
| All enrolled patients | 149 | 9.3 | 8.8 | 0 | 21.1 | |
| Patients with consensus diagnosis | 68 | 9.9 | 9.0 | 1.5 | 20.4 | |
| AA | 28 | 9.4 | 8.3 | 3.5 | 19.8 | 0.4 |
| RCC | 30 | 10.6 | 10.6 | 1.5 | 20.0 | |
| Other | 10 | 9.3 | 6.3 | 1.8 | 20.4 | |
| Follow up years | ||||||
| All enrolled patients | 149 | 4.2 | 2.6 | 0.02 | 21.5 | |
| Patients with consensus diagnosis | 68 | 4.2 | 3.1 | 0.2 | 17.7 | |
| AA | 28 | 3.8 | 2.4 | 0.2 | 16.1 | 0.3 |
| RCC | 30 | 4.7 | 3.7 | 0.2 | 17.7 | |
| Other | 10 | 4.1 | 2.5 | 0.3 | 16.0 | |
| Laboratory values | ||||||
| WBC | ||||||
| All enrolled patients | 144 | 2.8 | 2.6 | 0.5 | 12.0 | |
| Total | 68 | 2.7 | 2.5 | 0.5 | 9.1 | |
| AA | 28 | 2.4 | 2.1 | 0.5 | 5.4 | 0.1 |
| RCC | 30 | 2.7 | 2.8 | 0.9 | 5.4 | |
| Other | 10 | 3.5 | 2.6 | 0.5 | 9.1 | |
| ANC | ||||||
| All enrolled patients | 139 | 331.3 | 1.1 | 0 | 7333.0 | |
| Total | 64 | 338.6 | 1.1 | 0 | 7333.0 | |
| AA | 26 | 332.9 | 0.8 | 0 | 3255.0 | 0.7 |
| RCC | 29 | 193.7 | 1.2 | 0 | 2100.0 | |
| Other | 9 | 821.9 | 0.6 | 0 | 7333.0 | |
| Hgb | ||||||
| All enrolled patients | 128 | 7.4 | 7.8 | 2.0 | 14.3 | |
| Total | 60 | 7.6 | 7.8 | 2.4 | 14.3 | |
| AA | 23 | 6.4 | 6.6 | 2.4 | 10.2 | 0.03 |
| RCC | 28 | 8.1 | 8.1 | 2.7 | 13.7 | |
| Other | 9 | 9.1 | 8.8 | 5.0 | 14.3 | |
| Platelet count | ||||||
| All enrolled patients | 148 | 15.8 | 9.0 | 1.0 | 103.0 | |
| Total | 68 | 16.8 | 9.0 | 1.0 | 103.0 | |
| AA | 28 | 11.4 | 7.5 | 1.0 | 52.0 | 0.2 |
| RCC | 30 | 19.9 | 9.5 | 1.0 | 103.0 | |
| Other | 10 | 22.9 | 11.5 | 3.0 | 90.0 | |
| MCV | ||||||
| All enrolled patients | 116 | 89.1 | 88.5 | 60.0 | 115.0 | |
| Total | 56 | 88.4 | 87.0 | 60.0 | 111.0 | |
| AA | 23 | 86.3 | 85.0 | 60.0 | 110.0 | 0.1 |
| RCC | 26 | 91.0 | 92.5 | 76.0 | 111.0 | |
| Other | 7 | 86.0 | 85.0 | 72.0 | 105.0 | |
| Absolute reticulocyte count | ||||||
| All enrolled patients | 68 | 0.030 | 0.019 | 0 | 0.215 | |
| Total | 38 | 0.030 | 0.022 | 0 | 0.092 | |
| AA | 14 | 0.011 | 0.009 | 0 | 0.030 | 0.005 |
| RCC | 19 | 0.038 | 0.035 | 0 | 0.092 | |
| Other | 5 | 0.05 | 0.048 | 0.022 | 0.089 | |
| Hemoglobin F | ||||||
| All enrolled patients | 55 | 4.0 | 2.4 | 0 | 20.4 | |
| Total | 29 | 3.4 | 2.4 | 0 | 13.2 | |
| AA | 11 | 2.9 | 1.2 | 0 | 10.6 | 0.4 |
| RCC | 14 | 4.1 | 3.0 | 0 | 13.2 | |
| Other | 4 | 2.4 | 2.2 | 0.7 | 4.4 | |
p-value of Wilcoxon rank sums test comparing AA and RCC
“Total” refers to all patients with consensus diagnosis and available data.
Furthermore, we determined if a consensus diagnosis of RCC, a MCV > 100, or HgF% > 4% was predictive of treatment failure defined by failure to response to IST, progression to MDS or clonal evolution, and determined that none were statistically significant predictors (Table 5). However, and in contrast to previous studies, there was a trend towards improved EFS and OS in RCC (n=30) compared to AA (n=28) (Table 1). The 5-year EFS was 71.3 ± 11.0% for RCC, 52.5 ± 13.7% for AA, and 58.3 ± 21.7% for Other (Figure 2A). The 5-year OS was 84.9 ± 8.8% for RCC, compared to 72.5 ± 11.5% for AA, respectively, and 80.0 ± 20.7% for Other (Figure 2B).
Table 5.
Univariate tests for factors potentially prognostic of failure to treatment (Consensus diagnosis, MCV, HgF%)
| Risk factors | n* | Odds Ratio@ (95%CI) | p-value |
|---|---|---|---|
| Consensus diagnosis | 68 | 2.1 (0.7-6.2) | 0.2 |
| RCC | 30 | ||
| AA/Other (Reference) | 38 | ||
| MCV value | 116 | 0.9 (0.3-2.5) | 0.8 |
| >100 | 17 | ||
| ≤100 (Reference) | 99 | ||
| HgF% | 55 | 1.4 (0.4-4.2) | 0.6 |
| >4% | 19 | ||
| ≤4% (Reference) | 36 | ||
n is the number of patients with a known value for the given risk factor
for increased risk of failure to treatment, compared to the Reference level
Figure 2.
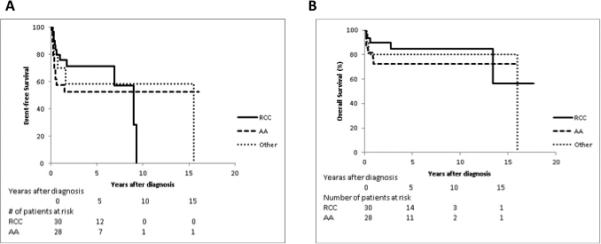
Figure 2A. EFS by consensus diagnosis (n=68, p=0.3 for log rank test comparison of RCC vs. AA )
Figure 2B. OS by consensus diagnosis (n=68, p=0.3 for log rank test comparison of RCC vs. AA)
Clonal evolution and disease progression to MDS
Five patients (3%) of the analytical cohort (n=149) experienced clonal evolution or disease progression to advanced MDS. All five patients (4 female, one male) had MAA. Four patients had received IST (triple IST, n=1; prednisone/CSA n=2; IVIG/prednisone/Rituximab n=1). Only one patient had a specimen available for review. While two of the three pathologists classified the marrow as RCC, the final consensus designation was “Other” given the concern for advanced MDS with a significant degree of multi-lineage dysplasia. All five patients underwent a HSCT for clonal evolution or development of MDS.
Discussion
Our study consists of a retrospective, single-center analysis of the outcomes of pediatric patients with classic AA, re-classification of these patients to AA vs RCC based on 2008 WHO Criteria and analysis the histomorphologic predictors of clinical outcomes using the newer classification.
First, we determined how our analysis compared to the published literature by assessing the OS and EFS for patients originally classified as AA. Focusing the analysis to patients diagnosed and treated with IST after the year 2000, our calculated OS and EFS of 88% and 52% respectively was comparable to the recent literature3-5,8. For patients receiving triple IST and diagnosed after 2000, there OS was significantly higher for patients diagnosed with MAA/SAA versus vSAA, which is also consistent with the literature4. The lower OS of patients with vSAA in this cohort is likely due to the higher rate of infectious complications in the vSAA group.
Considering the entire study cohort, the 5-year OS for all previously diagnosed AA patients was 72.5%±11.5% (n=149). The 5-year EFS and OS for AA patients treated with IST was 34.6%±7.0% and 66.0%±6.7% respectively (n=91). These outcomes are worse than the recently published literature in children8 and are likely a result of different treatment eras since our patient cohort encompassing pediatric patients from over 34 years with heterogeneous treatment approaches, limitations in molecular diagnostics to rule out IBMFS and improved supportive care utilized over the last decade.
We sought to assess the reproducibility and clinical relevance of the 2008 WHO classification distinguishing AA from hypocellular RCC. The WHO classification divides childhood MDS into three categories: RCC, RAEB, and RAEB in transformation (RAEB-T), which are largely distinguished by the peripheral blood (PB) and/or BM blast percentage15. While these guidelines allow for a highly accurate classification of most pediatric MDS patients,23 it is important to note that the provisional entity of RCC includes an array of phenotypes, ranging from hypo- to hypercellular RCC with limited unilineage to overt trilineage dysplasia. While the latter is more easily appreciated as unequivocal MDS, hypocellular RCC is a challenging diagnosis.
RCC remains a controversial classification that has not been widely adopted, largely due to limited data on the clinical implications of this diagnosis17,24. The European Working Group of Childhood MDS (EWOG-MDS) developed histomorphologic criteria to differentiate SAA from hypocellular RCC and demonstrated reproducibility of these criteria13. As proposed by the WHO, an accurate diagnosis of RCC requires both, evidence of dysplastic changes on the aspirate and characteristic features on the biopsy, including a hypocellular marrow with patchy clusters of left-shifted erythroid cells, increased mitosis, decreased granulopoiesis and megakaryocytes and optional micromegakaryocytes. In contrast, AA typically shows near complete hematopoietic aplasia, and the absence of left-shifted erythroid clusters and decreased but normal appearing megakaryocytes15.
In order to assess the reproducibility of the WHO diagnostic criteria, we had three pediatric hematopathologists evaluate both the BM aspirate and biopsy to determine if it was possible to distinguish AA from RCC. We included assessment of the BM aspirate wherever possible (42 out of 72 cases) given the current WHO criteria, and the generally accepted principle among morphologists that the BM biopsy is a reliable specimen to assess topography and cellularity, but it is insufficient to evaluate dysplasia. We found that the concordance of the assessment of diagnosis ranged from 61.3 to 74.6% with a kappa coefficient of 0.3-0.5. This is in contrast to the EWOG-MDS study where seven pathologists came to an agreement in 76% of cases with an inter-observer reliability of 79%13. This higher concordance may be due to a homogenously trained group of hematopathologists under the EWOG-MDS consortium, which was instrumental in developing the WHO diagnostic criteria for RCC. In contrast, the hematopathologists in our study had different training backgrounds. Thus, one interpretation of our data is that more specialized training of the interpreters of pathological specimens on the diagnostic criteria for RCC vs AA is required. However, our results may alternatively suggest that criteria for RCC and AA should be refined such that the classification is broadly reproducible by less homogeneously trained practitioners.
Since RCC is considered a subcategory of pediatric MDS treatment recommendations may differ significantly from classical AA and therefore accurate diagnosis has important clinical implications. In this study, we sought to determine if we could identify a subset of previously diagnosed AA patients that respond poorly to IST and might benefit from tailored therapy such as front-line HSCT. We assessed if a consensus diagnosis of RCC, MCV level greater than 100, or HgF% greater than 4%, were factors prognostic of treatment failure. While none of these, including the diagnosis of RCC, were associated with treatment failure, the small sample size may have limited the power to detect differences that would have been considered clinically significant. However, we did find an association between lower Hgb and absolute reticulocyte values in patients with AA compared to RCC.
The clinical relevance of the diagnosis of RCC remains controversial. Our data, and that of Yang et al17, showed an association between conventionally diagnosed MAA and RCC (Table 3). Since RCC is a subcategory of MDS, it has traditionally been treated with up front HSCT in an attempt to provide curative therapy prior to progression to clonal hematopoiesis or leukemia. A recent study suggested that IST may also be a reasonable treatment option for patients with hypoplastic RCC without monosomy 7,14 and showed a complete or partial response to IST in 71% of patients at 6 months. The 3-year OS and 3-year FFS of this study was 88% and 57% respectively, which is comparable to the OS and FFS in pediatric patients with classical AA treated with IST3-5,8. Interestingly, although our cohort was too small to show statistically significant results, our data indicate a trend towards improved OS and EFS in patients with RCC compared to AA (Figure 2A and 2B), potentially supporting the utility of this therapy. Larger prospective randomized clinical trials are needed to compare outcomes for pediatric patients with AA versus hypocellular RCC after treatment with IST.
Prior reports indicate that the classification of SAA versus hypocellular RCC was associated with a marked reduction in clonal evolution in patients with SAA13. These data suggest that precise morphological separation is important in predicting clonal disease. In our analysis, 5 patients all with MAA showed clonal progression or development of MDS after IST therapy. Unfortunately pathology was available in only 1 case. Therefore, a determination of RCC versus AA could not be made in the remaining cases. The only case available for review was diagnosed as RCC by 2 out of the 3 pathologists, but given the designation “Other” at consensus conference due to concern for significant dysplasia likely qualifying the patient for unequivocal MDS. These data suggest that perhaps some of the cases historically classified as MAA actually represent an underlying genetic defect leading to a clonal hematopoietic stem cell disorder and that correct differentiation between AA and RCC might allow clinicians to distinguish between a primarily immune mediated process and an underlying clonal stem cell defect or IBMFS.
While our study attempts to assess the reproducibility of the WHO criteria for AA and RCC, there are limitations to this analysis, which include a moderate sized cohort within which only a subset of pathology specimens were available for analysis and reclassification. Therefore, our study was not able to reach statistical significance with respect to the outcomes of OS and EFS after IST in patients with RCC. Furthermore, this is a single center study spanning a large time frame—34 years, with significant differences in treatment regimens, diagnostic work ups and availability and useage of molecular testing. Therefore, cryptic presentations of IBMFS that typically resemble the histological patterns of RCC16,25 could not be definitively excluded.
In conclusion, our findings indicate that a histologic diagnosis of RCC is difficult to establish with certainty and does not predict disease outcome, calling into question the reproducibility and clinical significance of the RCC classification and warranting further prospective studies. Our analysis suggests that a large multidisciplinary study is needed to examine the histopathologic validity of AA and hypoplastic RCC and prognostic impact of these diagnoses on treatment response and outcomes. In addition, given the limits of histopathological criteria and interpretation, efforts designed to further characterize the molecular underpinnings of AA and RCC would aid in differentiating these disorders and would inform the validity of any future classification systems.
Supplementary Material
Acknowledgements
The authors want to thank Nancy Potter for providing access to the institutional registry of AA patients complied and maintained by Nancy Potter and Dr. Frederick Pei Li between 1980 and 2000.
This work was supported by National Institutes of Health grants from the National Institute of Diabetes and Digestive and Kidney Diseases 1 R24 DK 094746-01 (I.H., M.D.F, D.A.W.). I.H. is also supported through a career development award from the National Cancer Institute 1K08CA140723.
Footnotes
Contribution: C.F., S.S., M.D.F., W.B.L., D.A.W. and I.H. designed the study and analyzed the data. C.F. and S.S. collected all clinical data. M.H.H., O.K.W., M.D.F. and I.H. reviewed pathology slides and collected pathology data. D.G. and W.B.L. performed the statistical analysis. C.F. and I.H. wrote and edtited the manuscript. All authors edited the manuscript.
Conflict-of-interest disclosures: The authors declare no competing financial interest.
References
- 1.Marsh JC, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70. doi: 10.1111/j.1365-2141.2009.07842.x. [DOI] [PubMed] [Google Scholar]
- 2.Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood. 2002;100(4):1185–1191. doi: 10.1182/blood-2002-01-0035. [DOI] [PubMed] [Google Scholar]
- 3.Fuhrer M, Burdach S, Ebell W, et al. Relapse and clonal disease in children with aplastic anemia (AA) after immunosuppressive therapy (IST): the SAA 94 experience. German/Austrian Pediatric Aplastic Anemia Working Group. Klin Padiatr. 1998;210(4):173–179. doi: 10.1055/s-2008-1043875. [DOI] [PubMed] [Google Scholar]
- 4.Fuhrer M, Rampf U, Baumann I, et al. Immunosuppressive therapy for aplastic anemia in children: a more severe disease predicts better survival. Blood. 2005;106(6):2102–2104. doi: 10.1182/blood-2005-03-0874. [DOI] [PubMed] [Google Scholar]
- 5.Goldenberg NA, Graham DK, Liang X, Hays T. Successful treatment of severe aplastic anemia in children using standardized immunosuppressive therapy with antithymocyte globulin and cyclosporine A. Pediatr Blood Cancer. 2004;43(7):718–722. doi: 10.1002/pbc.20114. [DOI] [PubMed] [Google Scholar]
- 6.Kojima S, Hibi S, Kosaka Y, et al. Immunosuppressive therapy using antithymocyte globulin, cyclosporine, and danazol with or without human granulocyte colony-stimulating factor in children with acquired aplastic anemia. Blood. 2000;96(6):2049–2054. [PubMed] [Google Scholar]
- 7.Kojima S, Ohara A, Tsuchida M, et al. Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children. Blood. 2002;100(3):786–790. doi: 10.1182/blood.v100.3.786. [DOI] [PubMed] [Google Scholar]
- 8.Dufour C, Pillon M, Passweg J, et al. Outcome of Aplastic Anemia in adolescence. A survey of the Severe Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation. Haematologica. 2014;99(10):1574–81. doi: 10.3324/haematol.2014.106096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamio T, Ito E, Ohara A, et al. Relapse of aplastic anemia in children after immunosuppressive therapy: a report from the Japan Childhood Aplastic Anemia Study Group. Haematologica. 2011;96(6):814–819. doi: 10.3324/haematol.2010.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003;289(9):1130–1135. doi: 10.1001/jama.289.9.1130. [DOI] [PubMed] [Google Scholar]
- 11.Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group of aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124(17):2698–704. doi: 10.1182/blood-2014-05-574889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lane AA, Odejide O, Kopp N, et al. Low frequency clonal mutations recoverable by deep sequencing in patients with aplastic anemia. Leukemia. 2013;27(4):968–971. doi: 10.1038/leu.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumann I, Fuhrer M, Behrendt S, et al. Morphological differentiation of severe aplastic anaemia from hypocellular refractory cytopenia of childhood: reproducibility of histopathological diagnostic criteria. Histopathology. 2012;61(1):10–17. doi: 10.1111/j.1365-2559.2011.04156.x. [DOI] [PubMed] [Google Scholar]
- 14.Yoshimi A, Baumann I, Fuhrer M, et al. Immunosuppressive therapy with anti-thymocyte globulin and cyclosporine A in selected children with hypoplastic refractory cytopenia. Haematologica. 2007;92(3):397–400. doi: 10.3324/haematol.10683. [DOI] [PubMed] [Google Scholar]
- 15.Cancer TIAfRo . Childhood Myelodysplastic Syndrome. In: Swerdlow SHCE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, vardimaan JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC; Lyon: 2008. pp. 104–107. [Google Scholar]
- 16.Niemeyer CM, Baumann I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program. 2011;2011:84–89. doi: 10.1182/asheducation-2011.1.84. [DOI] [PubMed] [Google Scholar]
- 17.Yang W, Zhang P, Hama A, Ito M, Kojima S, Zhu X. Diagnosis of acquired bone marrow failure syndrome during childhood using the 2008 World Health Organization classification system. Int J Hematol. 2012;96(1):34–38. doi: 10.1007/s12185-012-1092-z. [DOI] [PubMed] [Google Scholar]
- 18.Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70(2):177–182. doi: 10.1111/j.1365-2141.1988.tb02460.x. [DOI] [PubMed] [Google Scholar]
- 19.Kosaka Y, Yagasaki H, Sano K, et al. Prospective multicenter trial comparing repeated immunosuppressive therapy with stem-cell transplantation from an alternative donor as second-line treatment for children with severe and very severe aplastic anemia. Blood. 2008;111(3):1054–1059. doi: 10.1182/blood-2007-08-099168. [DOI] [PubMed] [Google Scholar]
- 20.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan ELM,P. Nonparametric estimation from incomplete observations. J Amer Statist Assn. 1958;53(282):457–481. [Google Scholar]
- 22.Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. The German Aplastic Anemia Study Group. N Engl J Med. 1991;324(19):1297–1304. doi: 10.1056/NEJM199105093241901. [DOI] [PubMed] [Google Scholar]
- 23.Elghetany MT. Myelodysplastic syndromes in children: a critical review of issues in the diagnosis and classification of 887 cases from 13 published series. Arch Pathol Lab Med. 2007;131(7):1110–1116. doi: 10.5858/2007-131-1110-MSICAC. [DOI] [PubMed] [Google Scholar]
- 24.Hasegawa D, Chen X, Hirabayashi S, et al. Clinical characteristics and treatment outcome in 65 cases with refractory cytopenia of childhood defined according to the WHO 2008 classification. Br J Haematol. 2014;166(5):758–766. doi: 10.1111/bjh.12955. [DOI] [PubMed] [Google Scholar]
- 25.Yoshimi A, Niemeyer C, Baumann I, et al. High incidence of Fanconi anaemia in patients with a morphological picture consistent with refractory cytopenia of childhood. Br J Haematol. 2013;160(1):109–111. doi: 10.1111/bjh.12083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.