
Figure 3. Divergent roles of class I PI3K catalytic isoforms in different signaling contexts.
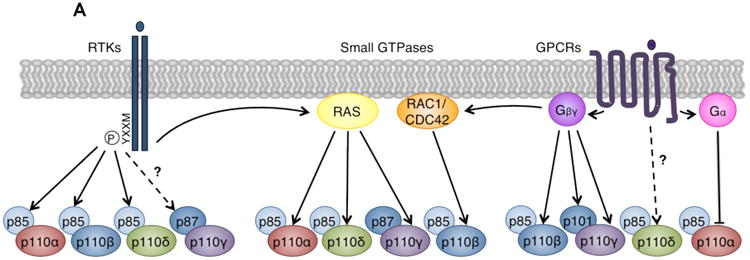
(A) Class I PI3Ks mediate signaling downstream of RTKs, GPCRs, and small GTPases. Left: p85 regulatory subunits bind phosphorylated YXXM motifs on activated RTKs. Because p110α, p110β, and p110δ bind p85, these isoforms mediate signaling downstream of RTKs. Recent evidence also suggests that p87-p110γ may be activated by certain RTKs110. Middle: Small GTPases synergize with RTK and GPCR signals to directly activate PI3Ks by interacting with their RAS-binding domains (RBDs). Isoforms p110α, p110δ, and p110γ bind RAS family GTPases, while p110β binds the RHO family GTPases RAC1 and CDC42143. Right: Gα and Gβγ proteins dissociate from activated GPCRs. Catalytic isoforms p110β and p110γ, and regulatory isoform p101, directly bind and are activated by Gβγ. p110δ may be activated downstream of GPCRs, but the mechanism is unknown126-128. Gα proteins have been reported to directly bind and inhibit p110α129-131. Modified with permission from Reference 3.
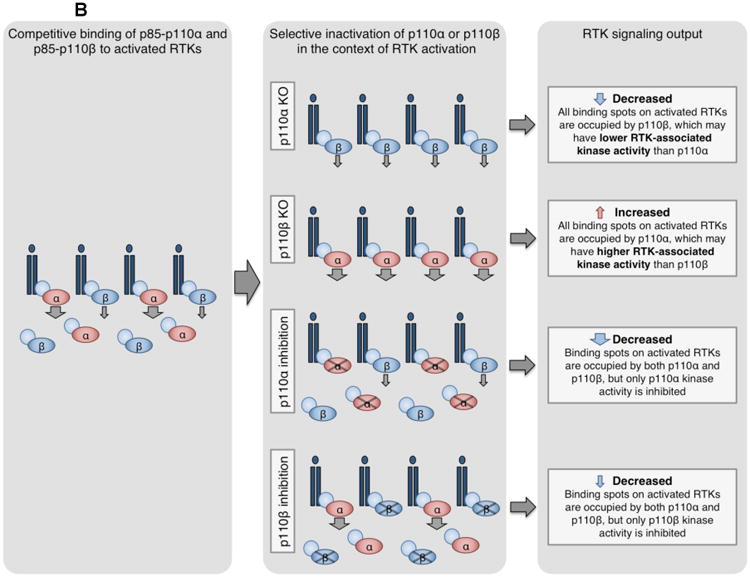
(B) Competition model for p110α and p110β regulation of RTK signaling96. Both p85-p110α and p85-p110β compete for phosphorylated YXXM sites on activated RTKs. However, the maximal specific activity and enzymatic rate of p110α are higher than that of p110β108, 109, and RTK-associated p110α may have higher lipid kinase activity than p110β96. By this model, loss or inactivation of p110α or p110β differentially modulates RTK signaling. Knockout of p110α allows all sites to be occupied by the less active p110β, decreasing RTK output. Conversely, knockout of p110β allows all sites to be bound by the more active p110α, increasing RTK output. Genetically or pharmacologically inactivated p110α or p110β can still bind RTKs but cannot signal, reducing RTK output.