

Abstract
Fanconi anemia (FA) is a cancer predisposition syndrome characterized by cellular hypersensitivity to DNA interstrand cross-links (ICLs). Within the FA pathway, an upstream core complex monoubiquitinates and recruits the FANCD2 protein to ICLs on chromatin. Ensuing DNA repair involves the Fanconi-associated nuclease 1 (FAN1), which interacts selectively with monoubiquitinated FANCD2 (FANCD2Ub) at ICLs. Importantly, FANCD2 has additional independent functions: it binds chromatin and coordinates the restart of aphidicolin (APH)-stalled replication forks in concert with the BLM helicase, while protecting forks from nucleolytic degradation by MRE11. We identified FAN1 as a new crucial replication fork recovery factor. FAN1 joins the BLM-FANCD2 complex following APH-mediated fork stalling in a manner dependent on MRE11 and FANCD2, followed by FAN1 nuclease-mediated fork restart. Surprisingly, APH-induced activation and chromatin recruitment of FAN1 occur independently of the FA core complex or the FAN1 UBZ domain, indicating that the FANCD2Ub isoform is dispensable for functional FANCD2-FAN1 cross talk during stalled fork recovery. In the absence of FANCD2, MRE11 exonuclease-promoted access of FAN1 to stalled forks results in severe FAN1-mediated nucleolytic degradation of nascent DNA strands. Thus, FAN1 nuclease activity at stalled replication forks requires tight regulation: too little inhibits fork restart, whereas too much causes fork degradation.
INTRODUCTION
Inherited genomic instability diseases, such as Fanconi anemia (FA) and Bloom syndrome (BS), predispose affected individuals to cancer. FA is characterized by bone marrow failure and a high risk of developing leukemia and squamous cell carcinomas (1, 2). FA cells are sensitive to DNA interstrand cross-link (ICL)-inducing agents, such as mitomycin C (MMC), and exhibit spontaneous chromosomal aberrations that are further exacerbated upon treatment with replication-inhibiting agents, such as hydroxyurea (HU) or aphidicolin (APH) (3–5). The 16 currently known FA proteins act in a common pathway that is activated when the replication machinery encounters DNA ICLs. Upon activation, an upstream FA core complex (composed of eight known FA proteins) is recruited to chromatin by one of its members, FANCM (6–8). The core complex then monoubiquitinates two central FA pathway proteins, FANCD2 and FANCI, which subsequently localize to chromatin and into DNA repair foci (9, 10). Additional FA pathway members include the breast cancer-associated proteins FANCD1/BRCA2 (breast cancer-associated protein 2), FANCN/PALB2 (partner and localizer of BRCA2), and FANCJ/BRIP1 (BRCA1-interacting protein 1), which function in homologous recombination (HR) repair of DNA double-strand breaks (DSBs) (11, 12).
Recent work from our laboratory and others suggests that FANCD2—beyond its role in DNA ICL repair—has additional roles at sites of HU- or APH-stalled replication forks. We demonstrated that FANCD2 interacts constitutively with BLM and mediates assembly of the so-called BLM complex (13–16) (consisting of BLM, RMI1, RMI2, topoisomerase IIIα, and replication proteins A1 to A3 [RPA1 to RPA3]) upon APH-mediated replication fork stalling. Notably, FANCD2 fulfills this role independently of its interaction partner, FANCI (17). We further demonstrated a novel role for FANCD2—in concert with BLM—to promote the restart of replication forks following temporary, APH-triggered fork stalling. Another study showed that FANCD2 protects nascent DNA strands at stalled replication forks from nucleolytic degradation by the MRE11 exonuclease (5). Collectively, these findings indicate that FANCD2 has a dual role in replication fork restart and fork protection.
Intriguingly, recent studies identified a novel protein, FAN1 (Fanconi-associated nuclease 1), as an interactor of FANCD2 that promotes DNA ICL repair (18–22). Intriguingly, FAN1 contains a ubiquitin-binding (UBZ) domain that promotes its interaction with FANCD2 and mediates FAN1 relocalization into nuclear DNA repair foci in a manner dependent on FANCD2 monoubiquitination. Therefore, current models propose that FAN1 utilizes its UBZ domain to selectively interact with the monoubiquitinated FANCD2 (FANCD2Ub) isoform at sites of active DNA ICL repair. Interestingly, FAN1 focus formation is also triggered by cellular treatment with HU (18), hinting at putative additional roles for FAN1 in the cellular replication stress response; however, if and how FAN1 has additional functions at halted replication forks (in the absence of DNA ICLs) are not known.
We asked if FAN1 plays a role—possibly in concert with FANCD2 and BLM—during replication fork recovery. Our results demonstrate that FAN1 docks onto the BLM-FANCD2 complex on chromatin upon induction of APH-stalled forks. FAN1's recruitment to chromatin is regulated by combined efforts of FANCD2 and the upstream MRE11 nuclease. Once recruited, FAN1 utilizes its nuclease activity—in cooperation with the BLM-FANCD2 complex—to promote replication fork restart and simultaneous suppression of new origin firing. Unexpectedly, the interaction between FAN1 and FANCD2 in this context does not require FANCD2Ub formation or the FAN1 UBZ domain, supporting a model where nonubiquitinated FANCD2 promotes replication fork recovery. In addition, our results indicate that FANCD2 simultaneously restricts inappropriate access of FAN1 to stalled forks to prevent degradation of nascent DNA strands.
MATERIALS AND METHODS
Cell culture.
PD20 (FANCD2 deficient), PD20+D2 (complemented with wild-type human FANCD2), PD221 (FANCA deficient), and PD221+A (complemented with wild-type human FANCA) patient cells were obtained from the FA Cell Repository at the Oregon Health and Science University (OHSU). PD331 (FANCC-deficient FA patient cells) and PD331+C (complemented with wild-type human FANCC) cells were a kind gift from F. Rosselli. GM08505 (BLM deficient) and GM00637 (BLM proficient) cells were purchased from the Coriell Institute. A1170 (FAN1-deficient patient cells) and A1170+FAN1 (complemented with wild-type human FAN1) cells were a gift from A. Smogorzewska. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at 37°C in 5% CO2. MG132 was used at a concentration of 10 μM. The ATR inhibitor VE-821 (Selleck Chemicals) was used at a concentration of 10 μM.
FAN1 plasmid constructs.
Full-length, D960A point mutant, and ubiquitin-binding ZNF domain-deleted (UBZ) FAN1 constructs in pDONR201 (Invitrogen) were a kind gift from Junjie Chen (19). These constructs were mutated by changing 6 nucleotides in the FAN1 small interfering RNA (siRNA) target region (A1152G, C1155A, A1158G, T1161C, G1164A, and T1167C substitutions) to generate siRNA-resistant FAN1 constructs following the method of Liu et al. (19). In addition, a DNA sequence encoding a 3×FLAG tag was inserted via PCR at the 5′ end of each FAN1 gene construct, using the following primers: forward primer, 5′-GATCACGATATAGATTACAAGGATGACGACGATAAGATCAACAAGTTTGTACAAAAAAGC-3′; and reverse primer, 5′-CTTGTAATCTCCATCGTGATCCTTGTAATCCATGGTGGCAGCTTAACTAGCCAGCTTGGG-3′. The siRNA-resistant FAN1 constructs were then subcloned into the Gateway-compatible pcDNA3.2 expression vector (Invitrogen).
Stable expression of siRNA-resistant FAN1 constructs.
To create cell lines stably expressing siRNA-resistant 3×FLAG-tagged wild-type or mutant FAN1 proteins, pcDNA3.2-3xFLAG-FAN1-FL, pcDNA3.2-3xFLAG-D960A, and pcDNA3.2-3xFLAG-ΔUBZ constructs were transfected by use of Lipofectamine into PD20+D2 cells, followed by continuous selection in the presence of 400 μg/ml Geneticin (G418; Corning).
Transient expression of the RAD51-K133R mutant.
A1170 cells or PD20 cells were transiently transfected with 2 μg of RAD51-K133R-pCaggs expression plasmid by use of Lipofectamine (Invitrogen). Forty-eight hours after transfection, the cells were used for DNA fiber assay; in parallel, whole-cell extracts (WCEs) were prepared and analyzed for RAD51-K133R expression by Western blotting.
siRNA experiments.
siRNA duplexes were purchased from Dharmacon Research (Thermo Scientific). The sequence of FAN1 siRNA is AAACCGTACTTGAGAATGA (19). siGENOME nontargeting siRNA was used as a control. Transfections were performed using DharmaFECT1 transfection reagent according to the manufacturer's protocol. Optimal siRNA-mediated knockdown of FAN1 was observed at 72 h.
Preparation of WCEs and cell fractions.
For WCE preparation, cells were washed in phosphate-buffered saline (PBS), resuspended in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM dithiothreitol [DTT], 0.5 mg/ml Pefabloc protease inhibitor), and incubated on ice for 20 min. Cell extracts were centrifuged for 5 min at 10,000 rpm, and the supernatant was used for further analysis. Cytoplasmic and chromatin fractions were prepared using a subcellular protein fractionation kit (Thermo Scientific).
IP.
Untreated or aphidicolin-treated human cells were lysed in buffer containing 10 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM DTT, 0.5 mg/ml protease inhibitor (Boehringer). Lysates were precleared with rabbit IgG and subjected to immunoprecipitation (IP) with FANCD2, BLM, or IgG antibody at 4°C overnight. One hundred microliters of Sepharose 4B beads (50% slurry) was added and rotated for 30 min at 4°C. To rule out DNA-bridged interaction of the proteins, IP was performed in the presence of 10 μg/ml ethidium bromide (EtBr) (a DNA-intercalating agent). Beads were pelleted from solution, washed in cell lysis buffer, boiled in 1× NuPAGE buffer (Invitrogen), and analyzed for the presence of proteins by SDS-PAGE and Western blotting.
Immunoblotting.
Protein samples were separated in gradient gels and transferred to Immobilon P membranes (Millipore). After blocking in 5% milk, membranes were incubated with the following primary antibodies: FANCD2 (1:1,000), FAN1 (1:100), BLM (1:1,500), TOP3A (1:1,000), RPA1 (1:1,000), histone H2AX (1:6,000), tubulin (1:10,000), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1,000), RAD51 (1:1,000), and pCHK1-S317 (1:1,000). Horseradish peroxidase-conjugated rabbit secondary antibody (Jackson Laboratory) and mouse secondary antibody (Bio-Rad) were used at dilutions of 1:10,000 and 1:4,000, respectively. Protein bands were visualized using an ECL Plus system (Amersham).
Antibodies.
Commercial antibodies against human FANCD2 (sc-20022; Santa Cruz), FLAG (F3165-0.2MG; Sigma), FAN1 (H00022909-B01P; Abnova), BLM (ab476; Abcam), TOP3a (14525-1-AP; Proteintech), RPA1 (NA-13; Calbiochem), MRE11 (sc-5859; Santa Cruz), tubulin (ab7291; Abcam), histone H2AX (ab1791; Abcam), GAPDH (GTX627408; Genetex), RAD51 (05-530; Millipore), and pCHK1-S317 (12302; Cell Signaling Technologies) were used.
DNA fiber assay.
We used the DNA fiber assay protocol as described previously (17). Briefly, moving replication forks were labeled with digoxigenin-dUTPs (DigU) for 25 min and then with biotin-dUTPs (BioU) for 40 min. To allow efficient incorporation of the dUTPs, a hypotonic buffer treatment (10 mM HEPES, 30 mM KCl, pH 7.4) preceded each dUTP labeling step. To visualize labeled fibers, cells were mixed with a 10-fold excess of unlabeled cells, fixed, and dropped onto slides. After cell lysis, DNA fibers were released and extended by tilting the slides. Incorporated dUTPs were visualized by immunofluorescence detection using antidigoxigenin-rhodamine (Roche) and streptavidin-Alexa Fluor 488 (Invitrogen). Images were captured using a Deltavision microscope (Applied Precision) and analyzed using Deltavision softWoRx 5.5 software. All reported DNA fiber results are means of results from three independent experiments (300 DNA fibers/experiment) and were analyzed as described previously (17). Statistics were calculated using Prism software. Error bars show standard errors of the means (SEM). P values were determined using the Mann-Whitney test. P values of <0.001 were considered significant.
Cytokinesis block micronucleus assay.
The cytokinesis block micronucleus (MN) assay was performed as described by Fenech et al. (23). Cells were cultured on coverslips, fixed, and stained with DAPI (4′,6-diamidino-2-phenylindole) for fluorescence microscopy. Cytochalasin B (0.72 μg/ml) was added to block cytokinesis 16 h before harvest. The resulting binucleated cells were counted for the presence of micronuclei. Two hundred binucleated cells were scored per experiment.
Colony formation assay.
Cellular survival was measured by a colony formation assay as described previously (24). Briefly, 500 cells were seeded in 6-cm dishes and treated with 30 μM APH for 6 h. Cells were then replenished with fresh medium and allowed to grow for 8 days. Colonies were stained with crystal violet solution and counted.
RESULTS
FAN1 joins the BLM-FANCD2 protein complex in response to cellular APH treatment.
Previous studies identified FAN1 as a FANCD2 interactor that is recruited by FANCD2 to nuclear DNA ICL repair foci (18–22). Our laboratory recently demonstrated that FANCD2 also fulfills a separate role, in complex with the BLM helicase, to promote the restart of replication forks following APH-mediated fork stalling (17). Here we asked if FAN1 is involved in the recovery of stalled replication forks, possibly in concert with BLM and FANCD2.
To test if FAN1 is part of the BLM-FANCD2 protein complex, we utilized patient-derived FANCD2-deficient (PD20) cells and their retrovirally complemented, FANCD2-proficient counterpart (PD20+D2 cells [wild-type-like cells]). The cells were either left untreated or treated with APH for 6 h to induce transient replication fork stalling, followed by immunoprecipitation with a BLM-specific antibody. In agreement with our previous findings, FANCD2 coimmunoprecipitated with BLM and another BLM complex member, RPA1, from untreated or APH-treated wild-type cells (Fig. 1A), confirming that the BLM-FANCD2 complex exists independently of replication stress induction (17). Intriguingly, FAN1 also coimmunoprecipitated with BLM, but only following cellular treatment with APH, indicating that complex formation between BLM and FAN1 is induced upon replication fork stalling. Importantly, coimmunoprecipitation of FAN1 with BLM was abrogated in FANCD2-deficient cells (Fig. 1A), indicating that FANCD2 acts as a physical bridge between BLM and FAN1 to allow the formation of a BLM-FANCD2-FAN1 complex at sites of stalled replication forks.
FIG 1.

Replication fork stalling stimulates formation of a BLM-FANCD2-FAN1 complex and triggers FAN1 binding to chromatin in an MRE11- and FANCD2-dependent manner. (A) APH treatment induces formation of a BLM-FANCD2-FAN1 complex in a FANCD2-dependent manner. FANCD2-proficient cells (PD20+D2) (lanes 1 and 2) and FANCD2-deficient cells (PD20) (lanes 3 and 4) were either left untreated (lanes 1 and 3) or treated with 30 μM APH for 6 h (lanes 2 and 4). Whole-cell extracts from these cells (lanes 1 to 4) were further subjected to immunoprecipitation (IP) with rabbit IgG (lanes 5 and 8; negative control) or an anti-BLM antibody (lanes 6, 7, 9, and 10). WCEs and IP samples were analyzed by Western blot (WB) detection using antibodies against BLM, FANCD2, FAN1, and RPA1. (B) Recruitment and stabilization of FAN1 at APH-stalled forks depend on MRE11 exonuclease activity and FANCD2. FANCD2-proficient (PD20+D2) and -deficient (PD20) cells were either left untreated or incubated with the MRE11 exonuclease inhibitor mirin; additionally, cells were left untreated or treated with 30 μM APH for 6 h, as indicated. Chromatin fractions were isolated from the cells and analyzed by WB for the presence of FANCD2, FAN1, and RPA1. Histone H2AX was used as a loading control. (C) Inhibition of MRE11 exonuclease activity triggers proteasomal degradation of FAN1. (i) Cellular mirin treatment reduces FAN1 protein levels. Cytoplasmic extracts isolated simultaneously with the chromatin fractions shown in panel B were analyzed by WB for the presence of FANCD2, FAN1, and RPA1. Tubulin was used as a loading control. (ii) Cellular treatment with MG132 restores FAN1 chromatin binding. FANCD2-proficient (PD20+D2) cells were left untreated or incubated with mirin; additionally, cells were left untreated or treated with 30 μM APH for 6 h, as indicated. Three hours after the addition of APH, cells were additionally either left untreated or treated with 10 μM MG132. Chromatin fractions were isolated from the cells and analyzed by WB for the presence of FANCD2, FAN1, and RPA1. Histone H2AX was used as a loading control. (D) Recruitment of FAN1 to APH-stalled forks occurs independently of BLM. BLM-proficient (GM00637) and BLM-deficient (GM08505) cells were left untreated or treated with 30 μM APH for 6 h, as indicated. Cytoplasmic and chromatin fractions were isolated from the cells and analyzed by WB for the presence of BLM, FANCD2, FAN1, and MRE11. GAPDH and histone H2AX were used loading controls for the cytoplasmic and chromatin fractions, respectively. (E) Recruitment of BLM to APH-stalled forks occurs independently of FAN1. FAN1-proficient and -deficient cells were left untreated or treated with 30 μM APH for 6 h, as indicated. Cytoplasmic and chromatin fractions were isolated and analyzed by WB for the presence of BLM, FANCD2, FAN1, and MRE11. GAPDH and histone H2AX were used as loading controls for the cytoplasmic and chromatin fractions, respectively. (Note that RPA1 was used in panels B to E as a positive control for the cellular response to APH.)
FAN1 recruitment to APH-stalled replication forks is regulated by MRE11 and FANCD2 but occurs independently of BLM.
We recently showed that the MRE11 exonuclease, a crucial factor during replication fork recovery (25), is an early responder at stalled replication forks and promotes the recruitment of FANCD2, which in turn supports the recruitment of BLM (17, 26). To determine where FAN1 is positioned within this hierarchy, we first tested if FANCD2 and/or MRE11 contributed to FAN1 chromatin recruitment following cellular APH treatment. We utilized wild-type (PD20+D2) and FANCD2-deficient (PD20) cells that were left untreated or incubated with the MRE11 exonuclease inhibitor mirin. Additionally, replication fork stalling was triggered via cellular treatment with APH for 6 h followed by isolation of the cellular chromatin fractions.
Basal levels of chromatin-bound FAN1 (FAN1-chr) were reduced in FANCD2-deficient cells compared to wild-type cells (Fig. 1B, compare lanes 1 and 5). Moreover, APH treatment robustly increased FAN1 binding to chromatin in wild-type cells but not in FANCD2-deficient cells (Fig. 1B, compare lanes 3 and 7). Similarly, mirin treatment of wild-type cells strongly reduced the basal FAN1-chr levels (Fig. 1B, compare lanes 1 and 2) and blocked any increase in FAN1 chromatin recruitment in response to APH (Fig. 1B, compare lanes 3 and 4). Importantly, mirin treatment completely abolished the residual FAN1-chr levels in FANCD2-deficient cells, even in the presence of APH (Fig. 1B, lanes 6 and 8). These results suggested that FANCD2 and MRE11 promote FAN1 binding to chromatin both under unperturbed conditions and in response to APH-induced replication fork stalling. Unexpectedly, however, we noticed that mirin treatment caused a reduction of cellular FAN1 protein levels (Fig. 1C, panel i, lanes 2, 4, 6, and 8). Cellular treatment with the proteasome inhibitor MG132 restored FAN1 protein stability and FAN1 chromatin recruitment in untreated and APH-treated cells (Fig. 1C, panel ii, compare lanes 2 and 4 with lanes 6 and 8). Thus, cells degrade FAN1 when MRE11 exonuclease activity is blocked.
Next, we tested the hierarchy of chromatin recruitment between BLM and FAN1, utilizing the following cell pairs: (i) BLM-proficient (GM00637) and BLM-deficient (GM08505) cells and (ii) PD20+D2 cells treated with control siRNA (siControl) or FAN1 siRNA (siFAN1). BLM-deficient cells exhibited normal FAN1-chr levels under unperturbed or APH-treated conditions, demonstrating that FAN1 binds chromatin independently of BLM (Fig. 1D). In addition, FAN1-depleted cells were fully supportive of BLM binding to chromatin in the absence or presence of APH, indicating that BLM binds chromatin independently of FAN1 (Fig. 1E). Simultaneously, cells lacking BLM or FAN1 did not exhibit any defects in recruiting FANCD2 or MRE11 to unperturbed or APH-treated chromatin (Fig. 1D and E). Taken together, these results indicate that FAN1 is recruited to chromatin and sites of replication fork stalling by combined actions of MRE11 and FANCD2, but independently of BLM.
FAN1 acts in concert with FANCD2 and BLM to restart stalled replication forks.
We recently showed that FANCD2 and BLM act in concert to promote the restart of APH-stalled replication forks and to simultaneously suppress new origin firing (17). Our findings above suggested that FAN1 joins the BLM-FANCD2 protein complex upon replication fork stalling and is recruited to chromatin in a FANCD2-dependent manner. Thus, we asked if FAN1 functions during the restart of stalled replication forks, possibly in concert with FANCD2 and BLM. To this end, we monitored cellular replication events on individual chromosomes with a dual-labeling DNA fiber assay. In this assay, cells are first pulse labeled with DigU (red label) for 25 min and then either left untreated or treated with 30 μM APH for 6 h, followed by a pulse treatment with BioU (green label) for 40 min (Fig. 2A). To test if FAN1 promotes fork restart in concert with FANCD2, we used FANCD2-proficient (PD20+D2) and FANCD2-deficient (PD20) cells and also generated FAN1-deficient and FANCD2-FAN1-double-deficient cells via siRNA-mediated FAN1 knockdown in PD20+D2 and PD20 cells, respectively (Fig. 2B). Strikingly different from the efficient fork restart in the wild-type cells, FAN1-deficient cells exhibited a severe defect in the restart of APH-stalled replication forks, similar to FANCD2-deficient cells (29.0% and 26.0%, respectively; P < 0.0001). In addition, FANCD2-FAN1 double deficiency did not exacerbate this phenotype (28.0%; P < 0.0001) (Fig. 2C), indicating that FAN1 works in concert with FANCD2 to mediate replication fork restart. In parallel, the proportion of newly originated replication tracts (BioU label only) increased significantly and equally (approximately 5-fold; P < 0.0001) in FANCD2-, FAN1-, and FANCD2-FAN1-deficient cells compared to wild-type cells (Fig. 2D), suggesting that FAN1 and FANCD2 cooperate to suppress new origin firing during temporary replication fork stalling.
FIG 2.

FAN1 acts in the same pathway with FANCD2 and BLM to mediate restart of APH-stalled replication forks and suppression of new origin firing. (A) Schematic of DNA fibers depicting sites of replication. Red tracts, DigU; green tracts, BioU. (B) Cell types used for DNA fiber analysis in panels C and D. Cells included wild-type (PD20+D2, siControl), FAN1-deficient (PD20+D2, siFAN1), FANCD2-deficient (PD20, siControl), and FANCD2-FAN1-double-deficient (PD20, siFAN1) cells. (C) FAN1 and FANCD2 act in a common pathway to mediate replication fork restart after APH-induced fork blockade. The efficiencies of replication restart in wild-type, FAN1-deficient, FANCD2-deficient, and FANCD2-FAN1-double-deficient cells were measured as the number of restarted replication forks (DigU-BioU tracts) compared with the total number of DigU-labeled tracts (DigU plus DigU-BioU). (D) FAN1 and FANCD2 act in concert to suppress new origin firing during replication blockade. The numbers of new sites of replication originating during the 40-min recovery period after APH treatment were compared between wild-type, FAN1-deficient, FANCD2-deficient, and FANCD2-FAN1-double-deficient cells. New origins of replication were measured as the number of green-only (BioU) tracts per unit length. (E) Cell types used for DNA fiber analysis in panels F and G. Cells included wild-type (GM00637, siControl), FAN1-deficient (GM00637, siFAN1), BLM-deficient (GM08505, siControl), and BLM-FAN1-double-deficient (GM08505, siFAN1) cells. (F) FAN1 and BLM act in a common pathway to mediate replication fork restart after APH-induced fork blockade. The efficiencies of replication restart in wild-type, BLM-deficient, FAN1-deficient, and BLM-FAN1-double-deficient cells were measured as described for panel C. (G) FAN1 and BLM act in concert to suppress new origin firing during replication blockade. The numbers of new sites of replication originating during the 40-min recovery period after APH treatment were compared between wild-type, BLM-deficient, FAN1-deficient, and BLM-FAN1-double-deficient cells. New origins of replication were measured as the number of green-only (BioU) tracts per unit length.
Next, to investigate whether FAN1 has a similar functional relationship with BLM, we depleted FAN1 in BLM-proficient (GM00637) and BLM-deficient (GM08505) cells (Fig. 2E). FAN1- and BLM-deficient cells showed comparable, severe defects in the restart of APH-stalled replication forks compared to wild-type cells (27.3% and 26.2%, respectively; P < 0.0001), and BLM-FAN1 double deficiency did not exacerbate the phenotype (27.7%; P < 0.0001) (Fig. 2F). In parallel, the proportion of newly originated replication tracts (BioU label only) increased equally (approximately 5-fold; P < 0.0001) in FAN1-, BLM-, and BLM-FAN1-deficient cells compared to wild-type cells (Fig. 2G). Taken together, these results demonstrate that FAN1 has a novel role—in concert with FANCD2 and BLM—to promote replication fork restart and suppression of new origin firing.
FAN1 is dispensable for ATR activation following replication fork stalling.
The fork restart and origin firing defects in FAN1-deficient cells are similar to those caused by inhibition of the DNA damage response kinase ATR (ataxia telangiectasia and Rad3 related) (27). To test if FAN1 is required for ATR activation following fork stalling, we compared ATR-dependent phosphorylation of CHK1 following APH treatment in FAN1-proficient and -deficient cells. FAN1-deficient cells exhibited normal APH-triggered CHK1 phosphorylation that was comparable to that in wild-type cells (Fig. 3A and B). Thus, FAN1 is not required to activate ATR following APH-mediated replication fork stalling.
FIG 3.
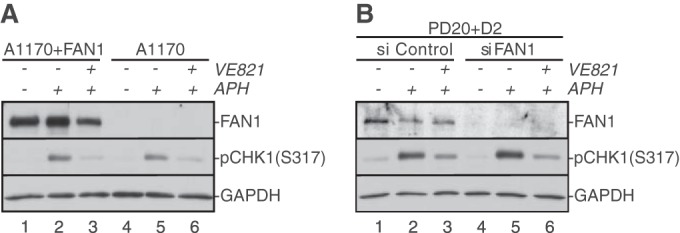
FAN1 does not regulate ATR activation in response to APH-triggered replication fork stalling. (A) Patient-derived FAN1-proficient (A1170+FAN1) or -deficient (A1170) cells were left untreated (lanes 1 and 4) or treated with 30 μM APH for 6 h (lanes 2, 3, 5, and 6). Additionally, cells were treated with dimethyl sulfoxide (DMSO) (lanes 2 and 5) or with the ATR inhibitor VE821 (lanes 3 and 6) 1 h prior to addition of APH. (B) Wild-type (PD20+D2, siControl) and FAN1-deficient (PD20+D2, siFAN1) cells were left untreated (lanes 1 and 4) or treated with 30 μM APH for 6 h (lanes 2, 3, 5, and 6) in the absence (lanes 2 and 5) or presence (lanes 3 and 6) of VE821. In panels A and B, WCE were analyzed for the presence of FAN1 and pCHK1-S317. GAPDH was used as a loading control.
FAN1 prevents the formation of APH-induced micronuclei.
Persistent replication fork stalling—if unresolved before M-phase entry—causes genomic instability and formation of micronuclei (MN) in the daughter cells (28–30). Previous studies showed that the FA and BLM pathways cooperate to prevent APH-induced formation of MN (3, 29). To test if FAN1 is involved in this protective mechanism, we compared APH-induced MN formation between siControl- and siFAN1-treated cells. As shown in Fig. 4A, FAN1-deficient cells exhibited significantly higher MN levels than those in wild-type cells following APH treatment (10.4% versus 4.4%; P < 0.01). APH-induced genomic instability, however, was not accompanied by acute cell death, since FAN1-deficient cells exhibited normal survival rates compared to FAN1-proficient cells upon APH treatment (Fig. 4B). These data suggest that FAN1—akin to the FA and BLM pathways—is required to avoid replication stress-induced chromosomal instability.
FIG 4.
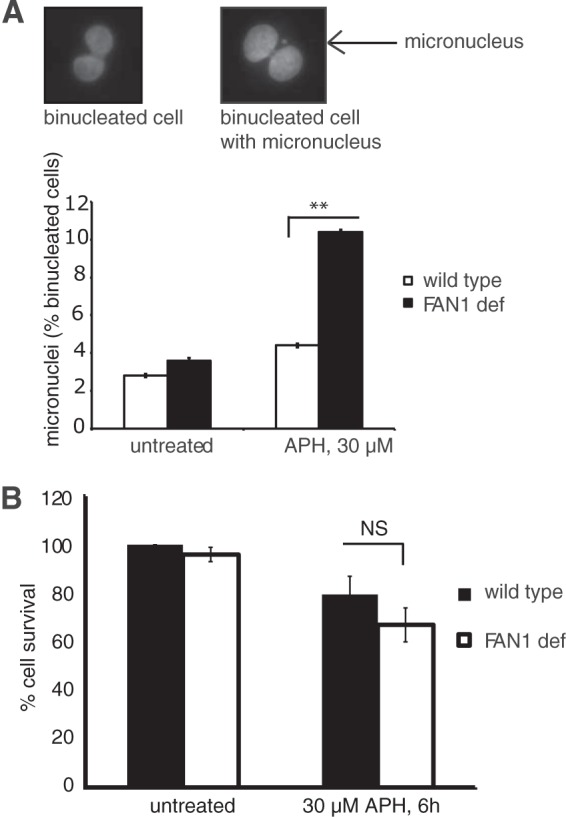
FAN1 prevents APH-induced MN formation. (A) FAN1-deficient cells show increased MN formation in response to APH. Wild-type (PD20+D2, siControl) and FAN1-deficient (PD20+D2, siFAN1) cells were either left untreated or treated with 30 μM APH for 6 h, followed by a recovery period of 16 h. Binucleated G1-phase cells were counted for the presence of MN by using the cytokinesis block micronucleus assay (23). (Top) Representative images of a binucleated cell without an MN (left) and with an MN (right). (Bottom) Average MN frequencies are shown for wild-type and FAN1-deficient cells under unperturbed conditions and following APH treatment. (B) FAN1 deficiency does not reduce cell viability in response to APH. Wild-type (PD20+D2, siControl) and FAN1-deficient (PD20+D2, siFAN1) cells were left untreated or treated with 30 μM APH for 6 h. Cellular survival was measured by colony formation assay. Results were plotted as averages of results from two independent experiments.
FANCD2 monoubiquitination is dispensable for APH-induced BLM-FANCD2-FAN1 complex formation or for the recruitment of FAN1 to chromatin following APH treatment.
Previous studies suggested that the interaction between FAN1 and FANCD2 following cellular induction of DNA ICLs or DNA DSBs is dependent on FANCD2 monoubiquitination (18–22). Thus, we asked if the APH-induced recruitment of FAN1 to the BLM-FANCD2 complex was mediated by the monoubiquitinated FANCD2 isoform (FANCD2Ub). To this end, we compared APH-triggered BLM-FANCD2-FAN1 complex formation between FANCC-proficient cells (PD331+FANCC) that are competent for FANCD2Ub formation and FANCC-deficient cells (PD331) that are unable to monoubiquitinate FANCD2. Surprisingly, the APH-induced coimmunoprecipitation of FAN1 with BLM was not interrupted in FANCC-deficient cells (Fig. 5A), suggesting that BLM-FANCD2-FAN1 complex formation occurred independently of a functional FA core complex, and thus independently of FANCD2Ub formation. Indeed, direct immunoprecipitation of FANCD2 from FANCC-proficient versus -deficient cells by use of a FANCD2-specific antibody revealed that the APH-stimulated FANCD2-FAN1 interaction occurred in the presence or absence of FANCC (Fig. 5B). Similarly, APH triggered a FANCD2-FAN1 interaction equally strongly in FANCA-proficient (PD220+FANCA) and FANCA-deficient (PD220) cells (Fig. 5C). These unexpected results indicate that the FANCD2-FAN1 interaction is inducible upon APH-triggered replication fork stalling but occurs independently of FANCD2Ub formation in this context. Moreover, they suggest that FANCD2 does not need to be monoubiquitinated in order to recruit FAN1 to APH-stalled replication forks. Thus, we compared FAN1 chromatin recruitment between FANCC-proficient and -deficient cells that were untreated or treated with APH for 6 h. As expected, FANCC-deficient cells exhibited greatly reduced levels of chromatin-bound FANCD2; however, we did detect very low residual levels of nonubiquitinated FANCD2 in FANCC-deficient cells in both the absence and presence of APH (Fig. 5D). Strikingly, chromatin-bound FAN1 levels were essentially unaffected in FANCC-deficient compared to FANCC-proficient cells in both the absence and presence of APH (Fig. 5D). We conclude that very low concentrations of chromatin-bound nonubiquitinated FANCD2 are sufficient to promote FAN1 recruitment to unperturbed chromatin and to APH-stalled replication forks.
FIG 5.

FAN1 joins the BLM-FANCD2 complex at APH-stalled replication forks independently of FANCD2 monoubiquitination. (A) APH-triggered formation of the BLM-FANCD2-FAN1 complex occurs independently of a functional FA core complex. FANCC-proficient cells (PD331+C) (lanes 1 and 2) or FANCC-deficient cells (PD331) (lanes 3 and 4) were left untreated (lanes 1 and 3) or treated with 30 μM APH for 6 h (lanes 2 and 4). Whole-cell extracts from these cells (lanes 1 to 4) were further subjected to IP with rabbit IgG (lanes 5 and 8; negative control) or an anti-BLM antibody (lanes 6, 7, 9, and 10). WCEs and IP samples were analyzed by WB using antibodies against BLM, FANCD2, FAN1, and RPA1. (B and C) APH-triggered interaction of FAN1 with FANCD2 does not depend on the FA core complex members FANCC and FANCA. (B) FANCC-proficient cells (PD331+C) (lanes 1 and 2) or FANCC-deficient cells (PD331) (lanes 3 and 4) were either left untreated (lanes 1 and 3) or treated with 30 μM APH for 6 h (lanes 2 and 4). Whole-cell extracts from these cells (lanes 1 to 4) were further subjected to IP with rabbit IgG (lanes 5 and 8; negative control) or an anti-FANCD2 antibody (lanes 6, 7, 9, and 10). WCEs and IP samples were analyzed by WB using antibodies against FANCD2 and FAN1. (C) The same IP experiment as that described for panel B was carried out in FANCA-proficient (PD220+A) versus FANCA-deficient (PD220) cells. (D) Recruitment of FAN1 to APH-stalled forks occurs independently of FANCD2 monoubiquitination. FANCC-proficient (PD331+C) and -deficient (PD331) cells were left untreated or treated with 30 μM APH for 6 h, as indicated. Cytoplasmic and chromatin fractions were isolated and analyzed by WB for the presence of FANCD2 and FAN1. RPA1 was used as a positive control for the cellular response to APH. Tubulin and histone H2AX were used as loading controls for the cytoplasmic and chromatin fractions, respectively. (E) The MMC- but not APH-triggered FANCD2-FAN1 interaction depends on a functional FA core complex. FANCC-proficient (PD331+C) (lanes 1 to 5) or -deficient (PD331) (lanes 6 to 10) cells were left untreated, treated with 1 μM MMC, or treated with 30 μM APH, as indicated. Cell extracts were subjected to IP with mouse IgG (lanes 1 and 6; negative control) or an anti-FANCD2 antibody (lanes 2 to 5 and 7 to 10). IP samples were analyzed for the presence of FANCD2 and FAN1. (F) The HU-triggered FANCD2-FAN1 interaction depends on a functional FA core complex. FANCC-proficient cells (PD331+C) (lanes 1 to 3) or FANCC-deficient cells (PD331) (lanes 4 to 6) were either left untreated (lanes 1, 2, 4, and 5) or treated with 2 mM HU (lanes 3 and 6) for 24 h. WCEs from these cells were subjected to IP with mouse IgG (lanes 1 and 4; negative control) or an anti-FANCD2 antibody (lanes 2, 3, 5, and 6). IP samples were analyzed for the presence of FANCD2 and FAN1.
Since our findings seemingly contradicted previous reports that FAN1 interacted selectively with FANCD2Ub following cellular treatment with MMC (24 h) or HU (24 h) (18–22), we compared the FANCD2-FAN1 interaction responses to APH (6 h and 24 h), HU (24 h), and MMC (24 h) in FANCC-proficient and -deficient cells. As expected, the FANCD2-FAN1 interaction was inducible by treatment with APH, MMC, or HU in FANCC-proficient cells. Strikingly, FANCC-deficient cells were able to promote the APH-triggered FANCD2-FAN1 interaction but exhibited strong defects in supporting the FANCD2-FAN1 interaction in response to MMC or HU (Fig. 5E and F). These results support the findings described above (Fig. 5B and C), as well as those of previous reports (18–22), and hint at distinct molecular requirements for FANCD2-FAN1 complex formation at APH-stalled forks compared to DNA lesions caused by MMC or HU treatment.
APH-induced BLM-FANCD2-FAN1 complex formation occurs independently of the FAN1 UBZ domain.
Based on our finding that FAN1 interacted with nonubiquitinated FANCD2 in response to APH (Fig. 5A to C), one would predict that the FAN1 ubiquitin-binding (UBZ) domain is dispensable for the FANCD2-FAN1 interaction and thus not required for the formation of the BLM-FANCD2-FAN1 complex. To test this, we expressed siRNA-resistant FLAG-tagged full-length FAN1 (FL-FAN1) and a FLAG-tagged FAN1 UBZ deletion mutant (ΔUBZ-FAN1) in wild-type cells (Fig. 6A and B). Following siRNA-mediated knockdown of endogenous FAN1, we performed immunoprecipitation studies with untreated or APH-treated cells, using specific FANCD2 and BLM antibodies. Strikingly, FL-FAN1 and ΔUBZ-FAN1 coimmunoprecipitated equally well with FANCD2 (Fig. 6C) or BLM (Fig. 6D) from APH-treated cells, demonstrating that FAN1 does not rely on its UBZ domain to join the BLM-FANCD2 complex upon APH induction of stalled forks.
FIG 6.

Functional cross talk between FAN1 and the BLM-FANCD2 complex during replication fork recovery requires FAN1 nuclease activity but not the FAN1 UBZ domain. (A) Schematic of wild-type and mutant FAN1 proteins used in the replication restart experiments. FL-FAN1, full-length wild-type FAN1; D960A-FAN1, full-length nuclease-dead FAN1 carrying a single amino acid substitution (D960A) in the nuclease domain (NUC); ΔUBZ-FAN1, truncated FAN1 lacking the N-terminal UBZ domain (21 amino acids). (B) Cell types used for DNA fiber analysis. Wild-type (PD20+D2) cells were stably transfected with either empty vector (EV) or a plasmid encoding siRNA-resistant, FLAG-tagged FL-FAN1, D960A-FAN1, or ΔUBZ-FAN1. The cells were then treated with control siRNA (siC) or FAN1 siRNA (siFAN1) for 72 h. WCEs were analyzed by WB for the presence of endogenous FAN1 (anti-FAN1 antibody) or FLAG-tagged FAN1 (anti-FLAG antibody). Tubulin was used as a loading control. (C) APH-triggered interaction of FAN1 with FANCD2 occurs independently of the FAN1 UBZ domain. Wild-type cells (PD20+D2) were transfected with either empty vector (EV; negative control) or a vector encoding siRNA-resistant, full-length FAN1 (FL-FAN1) or a FAN1 mutant lacking the UBZ domain (ΔUBZ-FAN1), followed by siFAN1 treatment to deplete endogenous FAN1 protein levels. Cells were either left untreated (lanes 1 to 4, 6, and 8) or treated with 30 μM APH for 6 h (lanes 5, 7, and 9) and then subjected to IP with rabbit IgG (lanes 1 to 3; negative control) or an anti-FANCD2 antibody (lanes 4 to 9). IP samples were analyzed by WB for the presence of FANCD2 and FAN1. (D) APH-triggered formation of the BLM-FANCD2-FAN1 complex occurs independently of the FAN1 UBZ domain. Wild-type cells were prepared as described for panel C and then subjected to IP with rabbit IgG (lanes 1 to 3; negative control) or an anti-BLM antibody (lanes 4 to 9). IP samples were analyzed by WB for the presence of FANCD2 and FAN1. (E) Analysis of replication fork restart following APH-triggered replication blockade in the cell types analyzed in panel B. The efficiencies of replication fork restart were compared between wild-type cells (siControl + EV), FAN1-deficient cells (siFAN1 + EV), and cells expressing exclusively FL-FAN1, D960A-FAN1, or ΔUBZ-FAN1. Fork restart efficiency was measured as the number of restarted replication forks (DigU-BioU tracts) compared with the total number of DigU-labeled tracts (DigU plus DigU-BioU tracts). (F) Mre11 exonuclease activity is required to mediate restart of APH-stalled replication forks. The efficiency of replication fork restart following APH-induced fork stalling was analyzed in wild-type (PD20+D2) cells in the presence or absence (NT) of the MRE11 exonuclease inhibitor mirin (50 μM). Restart efficiency was measured as the number of restarted replication forks (DigU-BioU tracts) compared with the total number of DigU-labeled tracts (DigU plus DigU-BioU tracts).
Replication fork restart requires FAN1 nuclease activity but not the FAN1 UBZ domain.
In addition to its N-terminal UBZ domain, FAN1 also contains a C-terminal nuclease domain. Previous studies indicated that FANCD2Ub formation and the FAN1 UBZ domain are crucial for the recruitment of FAN1 to sites of DNA ICL repair (18–22). Moreover, one study demonstrated that both the FAN1 UBZ and nuclease domains are crucial for cellular resistance to DNA ICLs (19).
In contrast, our results described above indicated that FANCD2Ub formation and the FAN1 UBZ domain are dispensable for the APH-induced FANCD2-FAN1 interaction and the FANCD2-dependent recruitment of FAN1 to chromatin (Fig. 5A to D and 6C and D). Thus, we set out to determine if the FAN1 UBZ and/or nuclease domain is required to promote replication fork restart. We stably expressed siRNA-resistant, FLAG-tagged wild-type FAN1 (FL-FAN1), a UBZ-deleted FAN1 mutant (ΔUBZ-FAN1), or a nuclease-dead FAN1 mutant (D960A-FAN1) in wild-type cells (PD20+D2) (Fig. 6A and B). Following siRNA-mediated knockdown of endogenous FAN1, cells expressing no FAN1 (empty vector [EV]), FL-FAN1, ΔUBZ-FAN1, or D960A-FAN1 were then either left untreated or treated with APH for 6 h, followed by analysis of replication fork restart efficiencies via the DNA fiber assay. Strikingly, expression of FL-FAN1 or ΔUBZ-FAN1 fully rescued replication fork restart in cells depleted of endogenous FAN1 (Fig. 6E). In contrast, cells expressing no FAN1 (EV) or the D960A-FAN1 mutant were unable to restart APH-stalled replication forks (28.2% and 30.1%, respectively; P < 0.0001) (Fig. 6E). These results suggest that the restart of APH-stalled replication forks requires the FAN1 nuclease activity but not the FAN1 UBZ domain. Since FAN1's function at APH-stalled replication forks depends on the MRE11 exonuclease (Fig. 1B), we also tested if MRE11 exonuclease activity was required for replication fork restart. DNA fiber analysis of untreated versus mirin-treated wild-type cells revealed that mirin-treated cells exhibited severe defects in the restart of APH-stalled replication forks that were comparable to those observed in cells lacking FAN1 or expressing the D960A-FAN1 mutant (Fig. 6F). We conclude that MRE11 and FAN1 nuclease activities are crucial to promote the restart of APH-stalled replication forks, whereas the FAN1 UBZ domain is dispensable for this function.
FAN1 degrades nascent DNA strands in FANCD2-deficient cells.
In addition to its role in replication fork restart, FANCD2 also functions to protect nascent DNA strands at APH- or HU-stalled replication forks from nucleolytic degradation by the MRE11 exonuclease (5, 17). This degradation can be measured via the DNA fiber assay: cells are treated with DigU-labeled dUTPs prior to APH treatment, and the lengths of the DigU tracts are measured before and after APH treatment. In FANCD2-deficient cells, DigU tract lengths shorten significantly during 6 h of HU or APH treatment (5, 17). Since FAN1—like MRE11—has nuclease activity (19–22) and shows residual APH-stimulated chromatin binding in FANCD2-deficient cells (Fig. 1B), we asked if FAN1 contributes to fork degradation in these cells. To this end, we compared nascent DNA strand degradation at APH-stalled forks in wild-type (PD20+D2), FANCD2-deficient (PD20), FAN1-deficient (PD20+D2; siFAN1), and FANCD2-FAN1-double-deficient (PD20; siFAN1) cells. APH-mediated fork stalling caused dramatic shortening of DigU-labeled replication tract lengths in FANCD2-deficient cells compared to wild-type cells (Fig. 7A) (4.13 μm and 8.16 μm, respectively; P < 0.0001), confirming our previous findings that FANCD2 prevents excessive nascent DNA strand degradation during APH-mediated fork stalling (17). In striking contrast, FANCD2-FAN1-double-deficient cells did not exhibit any reduction in DigU tract lengths during APH-mediated fork stalling compared to wild-type cells (Fig. 7B) (8.17 μm and 8.16 μm, respectively; P = 0.5288), indicating that FAN1 depletion completely restored replication fork stability in FANCD2-deficient cells. As expected, FAN1 depletion by itself did not cause any shortening of DigU tract lengths, which were similar to those of wild-type cells (Fig. 7C) (8.21 μm and 8.16 μm, respectively; P = 0.7659). Taken together, these data indicate that residual chromatin-bound FAN1 in FANCD2-deficient cells (also see Fig. 1B) is largely responsible for nucleolytic degradation of nascent DNA strands at APH-stalled replication forks.
FIG 7.

FAN1 degrades nascent DNA strands at APH-stalled replication forks in the absence of FANCD2. Lengths of nascent replication fork tracts indicating fork stability (labeled with DigU only) were measured before (NT) and after 6 h of APH treatment. Preformed DigU tract lengths shortened during APH treatment in FANCD2-deficient (PD20) cells compared to wild-type (PD20+D2) cells (A) but not in FANCD2-FAN1-double-deficient (PD20, siFAN1) cells compared to wild-type (PD20+D2) cells (B). (C) Preformed DigU tract lengths do not shorten in FAN1-deficient (PD20+D2, siFAN1) cells. (Insets) Plotted median tract lengths.
RAD51 filament stabilization does not compensate for the replication fork restart defects in FANCD2- or FAN1-deficient cells.
RAD51 forms nucleoprotein filaments and acts in concert with FANCD2 to protect nascent DNA strands at stalled replication forks from nucleolytic degradation (5). Moreover, overexpression of a RAD51 mutant (RAD51-K133R) that forms hyperstable DNA filaments can restore replication fork stability in FANCD2-deficient cells. We asked if RAD51-K133R overexpression can also restore replication fork restart in the absence of FAN1 or FANCD2. We expressed RAD51-K133R in FAN1-deficient cells (A1170) or FANCD2-deficient cells (PD20) (Fig. 8A and B) and analyzed fork restart in these cells. We found that RAD51-K133R expression in FAN1- or FANCD2-deficient cells did not increase the number of replication fork restart events following APH treatment. Importantly, in the same experiment, RAD51-K133R expression did restore replication fork stability in FANCD2-deficient cells, demonstrating that RAD51-K133R was expressed in sufficient amounts to protect stalled replication forks from degradation by MRE11 or FAN1 (Fig. 8C). Thus, defective replication fork restart caused by an absence of FANCD2 or FAN1 cannot be compensated for by RAD51 filament stabilization.
FIG 8.

RAD51-K133R overexpression does not compensate for replication restart defects in FAN1- or FANCD2-deficient cells. (A) RAD51-K133R does not rescue replication fork restart in FAN1-deficient cells. The efficiencies of replication restart in wild-type cells (A1170+FAN1) (lane 1), FAN1-deficient cells (A1170) (lane 2), and FAN1-deficient cells expressing RAD51-K133R (A1170+RAD51-K133R) (lane 3) were measured as the number of restarted replication forks (DigU-BioU tracts) compared with the total number of DigU-labeled tracts (DigU plus DigU-BioU tracts). (B) RAD51-K133R does not rescue replication fork restart in FANCD2-deficient cells. The efficiencies of replication restart in wild-type cells (PD20+D2) (lane 1), FANCD2-deficient cells (PD20) (lane 2), and FANCD2-deficient cells expressing RAD51-K133R (PD20+RAD51-K133R) (lane 3) were measured as the number of restarted replication forks (DigU-BioU tracts) compared with the total number of DigU-labeled tracts (DigU plus DigU-BioU tracts). (C) RAD51-K133R promotes replication fork stability in FANCD2-deficient cells. Lengths of nascent replication fork tracts indicating fork stability (labeled with DigU only) were measured after 6 h of APH treatment. Preformed DigU tract lengths shortened during APH treatment in FANCD2-deficient cells (median length = 4.71 μm) compared to wild-type cells (median length = 8.62 μm). DigU tract length shortening was counteracted by expression of RAD51-K133R in FANCD2-deficient cells (median length = 7.56 μm).
DISCUSSION
Recent work from our laboratory and others showed that FANCD2—beyond its role in DNA ICL repair—localizes directly to stalled replication forks, where it promotes fork restart in concert with BLM and protects nascent DNA strands at the stalled forks from degradation by MRE11 (5, 17, 31). Here we identified FAN1—a FANCD2 interactor and ICL repair protein (18–22)—as a crucial factor during the restart of APH-stalled replication forks. Moreover, our findings demonstrate a critical role for FANCD2 in regulating FAN1's nuclease activity to protect nascent DNA strands at stalled forks (Fig. 9).
FIG 9.

Model describing the role of FAN1 at a stalled replication fork. (A) Role of FAN1 in the presence of FANCD2. MRE11 is recruited to APH-stalled replication forks first, followed by FANCD2 and its constitutive interaction partner, BLM. Nonubiquitinated FANCD2 and MRE11 then both support the recruitment of FAN1. Subsequently, all four proteins (likely in concert with other BLM complex members) act in concert to promote replication fork restart in a manner dependent on the MRE11 and FAN1 nuclease activities. Concurrent with efficient fork restart, the firing of new origins is suppressed. In addition, FANCD2 protects the nascent DNA strands from nucleolytic attack by MRE11 and FAN1 during replication fork stalling. (B) Role of FAN1 in the absence of FANCD2. MRE11 is recruited to APH-stalled forks and enables partial recruitment of FAN1 independently of FANCD2. Despite the presence of MRE11 and FAN1, replication forks cannot restart efficiently, which in turn triggers firing of new replication origins. At the same time, the absence of FANCD2 allows for uncontrolled access of MRE11 and FAN1 to DNA at the stalled fork, leading to nucleolytic degradation of nascent DNA strands behind the fork.
Our results indicate that while BLM and FANCD2 form a constitutive complex, FAN1 is not associated with either protein under unperturbed conditions. Instead, FAN1 docks onto FANCD2 within the BLM-FANCD2 complex in response to APH, indicating that the BLM-FANCD2-FAN1 complex forms at specific DNA structures associated with stalled replication forks. The idea that an interaction of FAN1 with FANCD2 is stimulated by genotoxic influences is supported by previous reports. MacKay et al. found FANCD2-FAN1 interactions to be undetectable under unperturbed cellular conditions (22); moreover, Liu et al. showed that the FANCD2-FAN1 interaction was strongly stimulated in response to DNA ICLs, which ultimately cause replication fork stalling in S phase (19).
Rather unexpectedly, however, our results further demonstrate that the interaction between FAN1 and FANCD2 upon APH-induced replication fork stalling occurs in the absence of a functional FA core complex and does not require the FAN1 UBZ domain, indicating that nonubiquitinated FANCD2 recruits FAN1 to temporarily stalled forks. At first glance, these findings contradict previous reports indicating that FAN1 utilizes its UBZ domain to interact selectively with FANCD2Ub and relies on FANCD2Ub for its own recruitment to DNA ICLs caused by cellular treatment with MMC (24 h), and even to replication forks stalled in the presence of HU (24 h) (18–22). Moreover, our side-by-side comparison of the FANCD2-FAN1 interaction responses to APH, MMC, and HU confirms our findings and those of previous studies. How can these seeming discrepancies be reconciled? Importantly, the molecular mechanisms underlying the cellular responses to MMC, HU, and APH are different. DNA ICLs are incised to generate DNA DSBs, followed by homologous recombination (HR)-mediated break repair (32, 33). DNA DSBs can also stem from replication fork collapse after prolonged fork stalling (24 h) in the presence of HU (34). In contrast, replication forks stalled for a few hours in the presence of APH (6 h of APH treatment in the current study) remain stable and restart proficient (17, 26). Thus, we predict that the FANCD2Ub isoform is dispensable for FAN1 recruitment to APH-stalled replication forks for subsequent fork restart but crucial for promoting FANCD2-FAN1-dependent molecular actions during the repair of DNA DSBs generated at ICLs or collapsed replication forks. Importantly, our new findings strongly correlate with recent discoveries from our laboratory that interactions of FANCD2 with other DNA repair factors involved in replication fork restart, such as BLM or CtIP, occur independently of FANCD2Ub formation (17, 26), and thus provide further support for a novel model where nonubiquitinated FANCD2 coordinates several DNA repair factors to promote the restart of APH-stalled replication forks.
Of note, the FA literature typically describes only FANCD2Ub as chromatin bound, whereas nonubiquitinated FANCD2 is considered to be soluble. However, numerous studies have detected nonubiquitinated FANCD2 in chromatin fractions from human cells, including patient-derived fibroblasts (35, 36), HEK 293T cells (37), and HCT116 cells (38), as well as in chromatin fractions from chicken cells (39–41) and Xenopus egg extracts (17, 42). Undoubtedly, the ratio of chromatin-bound FANCD2Ub to FANCD2 is always greatly in favor of FANCD2Ub, regardless of the cell type, the species, or the chromatin isolation protocol, hinting that FANCD2Ub has a stronger affinity for chromatin. Intriguingly, a recent study found that replication fork stalling triggers a transient interaction of nonubiquitinated FANCD2 with the replicative MCM helicase (37). Thus, a short-lived presence of nonubiquitinated FANCD2 at stalled replication forks may be sufficient (or necessary?) to allow positioning of downstream factors, such as FAN1, and timely fork restart.
FAN1 now joins the growing group of fork restart proteins, including FANCD2, BRCA1, MRE11, XRCC3, RAD51, CtIP, and MUS81 (17, 25, 26, 34, 43). All proteins in this group have been implicated in HR repair of DNA DSBs, indicating that the recovery of APH-stalled replication forks involves HR mechanisms.
FANCD2 also has a second role at stalled forks: to promote RAD51 filament formation on nascent DNA strands as a protective mechanism against nucleolytic degradation. Interestingly, this role of FANCD2 is shared by the FA core complex (5), tempting us to speculate that FANCD2Ub may specifically function to protect nascent DNA strands at stalled forks. A previous study showed that overstabilizing RAD51 filaments protects nascent DNA even in the absence of FANCD2 (5). In contrast, we showed that overstabilization of RAD51 filaments cannot promote replication fork restart in the absence of FANCD2 or FAN1. These results hint that either (i) fork stabilization is necessary but not sufficient for fork restart or (ii) fork stabilization and fork restart are uncoupled events that may require distinct molecular machineries.
Concerning the order in which restart factors are recruited to APH-stalled replication forks, our current and previous results (17, 26) predict that MRE11 binds stalled forks as an early responder and—via its exonuclease activity—shapes a DNA intermediate substrate to recruit FANCD2. Interestingly, both MRE11 and FANCD2 contribute to the recruitment and stabilization of FAN1 on chromatin, although it remains unclear whether MRE11 recruits FAN1 directly—perhaps as part of the FANCD2-MRE11 complex (44)—or whether it provides a DNA substrate that is later processed by FAN1. Clearly, the recruitment hierarchy among these proteins is not strictly linear, possibly to allow for additional cellular adaptability during the replication stress response. In general, FANCD2's role in recruiting other DNA repair and replication fork recovery factors, such as FAN1 or BLM, to chromatin appears to be a partial one, indicating additional FANCD2-independent roles for these proteins. Indeed, FANCD2-independent functions of FAN1 and BLM are clearly reflected in the clinically distinct, disease-specific phenotypes associated with inherited mutations in the FANCD2, FAN1, and BLM genes (45–49).
On the other hand, single versus double deficiencies in FANCD2, BLM, and/or FAN1 expression cause identical replication restart defects, demonstrating that these three proteins act as a functional unit during fork restart. The FAN1 UBZ domain is completely dispensable during this process, further highlighting the idea that functional cross talk between FAN1 and FANCD2 during fork recovery does not require FANCD2Ub formation.
In contrast, the FAN1 nuclease domain appears to be crucial for promoting replication fork restart. Interestingly, FAN1 has dual nuclease activities: it is a structure-specific endonuclease and a 5′ → 3′ exonuclease (20–22). Unfortunately, we cannot determine which of these activities promotes fork restart, since mutations in the FAN1 nuclease domain (including the D960A mutation used in our study) disrupt both endo- and exonuclease activities of FAN1 (19–22).
Intriguingly, MRE11—the early responder at stalled replication forks—also harbors endo- and exonuclease activities; however, the latter has directionality opposite (3′ → 5′) that of FAN1 (50, 51). Our findings identify the MRE11 exonuclease activity as crucial for stabilization of FAN1 on chromatin. Since FAN1-deficient cells have normal chromatin-bound MRE11 levels but fail to promote replication fork restart, we speculate that the exonucleolytic activity of MRE11 reshapes the stalled replication fork into a substrate for FAN1 (as part of the FANCD2-FAN1 complex), which subsequently promotes fork restart. If MRE11 exonuclease activity is blocked, then cells degrade FAN1—perhaps to prevent FAN1 from inappropriately accessing DNA or because FAN1 is nonfunctional on chromatin without prior MRE11 activity. The stepwise action of MRE11 and FAN1 also extends to the situation in FANCD2-deficient cells. These cells have normal chromatin-bound MRE11 levels that suffice to directly recruit a subset of FAN1 molecules, eventually causing severe DNA strand degradation at stalled replication forks. Inhibition of MRE11 exonuclease activity or FAN1 depletion completely restores fork stability in these cells, indicating that MRE11 cannot perform significant DNA strand degradation without FAN1. The progressive shortening of nascent DNA strands in FANCD2-deficient cells is hence likely due to combined actions of MRE11 and FAN1. The idea of an early versus late function of MRE11 and FAN1 is mirrored by their predicted roles during HR-mediated repair of DNA breaks: MRE11 acts at an early stage of HR by promoting DNA end resection (reviewed in reference 52), whereas FAN1 is predicted to function downstream of DNA end resection and RAD51-mediated strand invasion (20, 22).
In summary, our results show that the FAN1 nuclease is a crucial replication fork recovery factor that works in concert with nonubiquitinated FANCD2 and the BLM helicase. Importantly, FAN1's access to stalled forks requires tight regulation by FANCD2; if this mechanism fails, forks become unprotected and FAN1's nuclease activity causes long-range degradation of nascent DNA strands. Our findings support accumulating evidence for a dual role of FANCD2 at stalled forks: coordinating access of DNA nucleases to promote replication fork restart while simultaneously restricting inappropriate activity of the very same nucleases to prevent fork instability (Fig. 9).
ACKNOWLEDGMENTS
We are grateful to F. Rosselli for sharing the PD331 and PD331+C cell lines with us. We thank A. Smogorzewska for sharing the A1170 and A1170+FAN1 cell lines. We also thank M. Jasin for sharing the RAD51-K133R-pCaggs expression plasmid. We thank N. Shima for helpful discussions regarding the manuscript.
A.S. was supported by the American Cancer Society (grant RSG-13-039-01-DMC). I.C. was supported by the American Heart Association.
Footnotes
Published ahead of print 18 August 2014
REFERENCES
- 1.Kee Y, D'Andrea AD. 2012. Molecular pathogenesis and clinical management of Fanconi anemia. J. Clin. Invest. 122:3799–3806. 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kupfer GM. 2013. Fanconi anemia: a signal transduction and DNA repair pathway. Yale J. Biol. Med. 86:491–497. [PMC free article] [PubMed] [Google Scholar]
- 3.Naim V, Rosselli F. 2009. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 11:761–768. 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- 4.Kee Y, D'Andrea AD. 2010. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 24:1680–1694. 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–116. 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deans AJ, West SC. 2009. FANCM connects the genome instability disorders Bloom's syndrome and Fanconi anemia. Mol. Cell 36:943–953. 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Mosedale G, Niedzwiedz W, Alpi A, Perrina F, Pereira-Leal JB, Johnson M, Langevin F, Pace P, Patel KJ. 2005. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat. Struct. Mol. Biol. 12:763–771. 10.1038/nsmb981. [DOI] [PubMed] [Google Scholar]
- 8.Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W. 2005. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 37:958–963. 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ. 2007. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129:289–301. 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. 2001. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 7:249–262. 10.1016/S1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 11.Wang W. 2007. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 8:735–748. 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 12.D'Andrea AD. 2010. Susceptibility pathways in Fanconi's anemia and breast cancer. N. Engl. J. Med. 362:1909–1919. 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh TR, Ali AM, Busygina V, Raynard S, Fan Q, Du CH, Andreassen PR, Sung P, Meetei AR. 2008. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 22:2856–2868. 10.1101/gad.1725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu D, Guo R, Sobeck A, Bachrati CZ, Yang J, Enomoto T, Brown GW, Hoatlin ME, Hickson ID, Wang W. 2008. RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 22:2843–2855. 10.1101/gad.1708608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin J, Sobeck A, Xu C, Meetei AR, Hoatlin M, Li L, Wang W. 2005. BLAP75, an essential component of Bloom's syndrome protein complexes that maintain genome integrity. EMBO J. 24:1465–1476. 10.1038/sj.emboj.7600622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W. 2003. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol. Cell. Biol. 23:3417–3426. 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaudhury I, Sareen A, Raghunandan M, Sobeck A. 2013. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 41:6444–6459. 10.1093/nar/gkt348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shereda RD, Machida Y, Machida YJ. 2010. Human KIAA1018/FAN1 localizes to stalled replication forks via its ubiquitin-binding domain. Cell Cycle 9:3977–3983. 10.4161/cc.9.19.13207. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Ghosal G, Yuan J, Chen J, Huang J. 2010. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329:693–696. 10.1126/science.1192656. [DOI] [PubMed] [Google Scholar]
- 20.Kratz K, Schopf B, Kaden S, Sendoel A, Eberhard R, Lademann C, Cannavo E, Sartori AA, Hengartner MO, Jiricny J. 2010. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142:77–88. 10.1016/j.cell.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 21.Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP, Elledge SJ. 2010. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 39:36–47. 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, Lilley DM, Rouse J. 2010. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142:65–76. 10.1016/j.cell.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fenech M. 2007. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2:1084–1104. 10.1038/nprot.2007.77. [DOI] [PubMed] [Google Scholar]
- 24.Luebben SW, Kawabata T, Akre MK, Lee WL, Johnson CS, O'Sullivan MG, Shima N. 2013. Helq acts in parallel to Fancc to suppress replication-associated genome instability. Nucleic Acids Res. 41:10283–10297. 10.1093/nar/gkt676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. 2009. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28:2601–2615. 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeo JE, Lee EH, Hendrickson E, Sobeck A. 2014. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum. Mol. Genet. 23:3695–3705. 10.1093/hmg/ddu078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D. 2013. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27:1610–1623. 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luebben SW, Kawabata T, Johnson CS, O'Sullivan MG, Shima N. 2014. A concomitant loss of dormant origins and FANCC exacerbates genome instability by impairing DNA replication fork progression. Nucleic Acids Res. 42:5605–5615. 10.1093/nar/gku170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. 2009. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11:753–760. 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 30.Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N. 2011. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell 41:543–553. 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. 2013. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 288:31458–31467. 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC. 2009. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326:1698–1701. 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. 2008. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134:969–980. 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37:492–502. 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang F, Fan Q, Ren K, Auerbach AD, Andreassen PR. 2010. FANCJ/BRIP1 recruitment and regulation of FANCD2 in DNA damage responses. Chromosoma 119:637–649. 10.1007/s00412-010-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howlett NG, Harney JA, Rego MA, Kolling FW, IV, Glover TW. 2009. Functional interaction between the Fanconi anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. J. Biol. Chem. 284:28935–28942. 10.1074/jbc.M109.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A. 2013. FANCD2 binds MCM proteins and controls replisome function upon activation of S phase checkpoint signaling. Mol. Cell 51:678–690. 10.1016/j.molcel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, Li L. 2014. Modularized functions of the Fanconi anemia core complex. Cell Rep. 7:1849–1857. 10.1016/j.celrep.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, Passmore LA. 2014. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol. Cell 54:858–869. 10.1016/j.molcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alpi A, Langevin F, Mosedale G, Machida YJ, Dutta A, Patel KJ. 2007. UBE2T, the Fanconi anemia core complex, and FANCD2 are recruited independently to chromatin: a basis for the regulation of FANCD2 monoubiquitination. Mol. Cell. Biol. 27:8421–8430. 10.1128/MCB.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, Tashiro S, Elledge SJ, Takata M. 2008. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 15:1138–1146. 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sareen A, Chaudhury I, Adams N, Sobeck A. 2012. Fanconi anemia proteins FANCD2 and FANCI exhibit different DNA damage responses during S-phase. Nucleic Acids Res. 40:8425–8439. 10.1093/nar/gks638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, Hickson ID. 2013. MUS81 promotes common fragile site expression. Nat. Cell Biol. 15:1001–1007. 10.1038/ncb2773. [DOI] [PubMed] [Google Scholar]
- 44.Roques C, Coulombe Y, Delannoy M, Vignard J, Grossi S, Brodeur I, Rodrigue A, Gautier J, Stasiak AZ, Stasiak A, Constantinou A, Masson JY. 2009. MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO J. 28:2400–2413. 10.1038/emboj.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, Diaz K, Lach FP, Bennett GR, Gee HY, Ghosh AK, Natarajan S, Thongthip S, Veturi U, Allen SJ, Janssen S, Ramaswami G, Dixon J, Burkhalter F, Spoendlin M, Moch H, Mihatsch MJ, Verine J, Reade R, Soliman H, Godin M, Kiss D, Monga G, Mazzucco G, Amann K, Artunc F, Newland RC, Wiech T, Zschiedrich S, Huber TB, Friedl A, Slaats GG, Joles JA, Goldschmeding R, Washburn J, Giles RH, Levy S, Smogorzewska A, Hildebrandt F. 2012. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 44:910–915. 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trujillo JP, Mina LB, Pujol R, Bogliolo M, Andrieux J, Holder M, Schuster B, Schindler D, Surralles J. 2012. On the role of FAN1 in Fanconi anemia. Blood 120:86–89. 10.1182/blood-2012-04-420604. [DOI] [PubMed] [Google Scholar]
- 47.Kalb R, Neveling K, Hoehn H, Schneider H, Linka Y, Batish SD, Hunt C, Berwick M, Callen E, Surralles J, Casado JA, Bueren J, Dasi A, Soulier J, Gluckman E, Zwaan CM, van Spaendonk R, Pals G, de Winter JP, Joenje H, Grompe M, Auerbach AD, Hanenberg H, Schindler D. 2007. Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype. Am. J. Hum. Genet. 80:895–910. 10.1086/517616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalb R, Neveling K, Nanda I, Schindler D, Hoehn H. 2006. Fanconi anemia: causes and consequences of genetic instability. Genome Dyn. 1:218–242. 10.1159/000092510. [DOI] [PubMed] [Google Scholar]
- 49.Singh DK, Ahn B, Bohr VA. 2009. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology 10:235–252. 10.1007/s10522-008-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paull TT, Gellert M. 1998. The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol. Cell 1:969–979. 10.1016/S1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 51.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA. 2014. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53:7–18. 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stracker TH, Petrini JH. 2011. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12:90–103. 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]