

Abstract
Airway inflammation is a key aspect of diseases such as asthma. Several inflammatory cytokines (e.g., TNFα and IL-13) increase cytosolic Ca2+ ([Ca2+]cyt) responses to agonist stimulation and Ca2+ sensitivity of force generation, thereby enhancing airway smooth muscle (ASM) contractility (hyper-reactive state). Inflammation also induces ASM proliferation and remodeling (synthetic state). In normal ASM, the transient elevation of [Ca2+]cyt induced by agonists leads to a transient increase in mitochondrial Ca2+ ([Ca2+]mito) that may be important in matching ATP production with ATP consumption. In human ASM (hASM) exposed to TNFα and IL-13, the transient increase in [Ca2+]mito is blunted despite enhanced [Ca2+]cyt responses. We also found that TNFα and IL-13 induce reactive oxidant species (ROS) formation and endoplasmic/sarcoplasmic reticulum (ER/SR) stress (unfolded protein response) in hASM. ER/SR stress in hASM is associated with disruption of mitochondrial coupling with the ER/SR membrane, which relates to reduced mitofusin 2 (Mfn2) expression. Thus, in hASM it appears that TNFα and IL-13 result in ROS formation leading to ER/SR stress, reduced Mfn2 expression, disruption of mitochondrion–ER/SR coupling, decreased mitochondrial Ca2+ buffering, mitochondrial fragmentation, and increased cell proliferation.
Keywords: inflammation, ER/SR stress, airway, asthma, unfolded protein response
Introduction
Asthma affects more than 20 million people in the USA (~10% of the population), and more than 300 million people world-wide (GINA 2012). It is now recognized that inflammation is key to asthma pathophysiology, and involves mediators such as the cytokines tumor necrosis factor alpha (TNFα) and interleukin 13 (IL-13). Yet, therapies that blunt inflammation per se are not entirely effective. While a number of cell types such as airway epithelium and immune cells are likely to play some role in asthma, airway smooth muscle (ASM) is the key player by contributing to both exaggerated airway narrowing via enhanced cytosolic Ca2+ concentration ([Ca2+]cyt) and Ca2+ sensitivity of force responses to agonist (e.g., acetylcholine, ACh) stimulation (i.e., “hyper-reactive” state) and airway remodeling via increased ASM cell proliferation and the formation of extracellular matrix (ECM) (i.e., “synthetic” state) (Fig. 1) (James 2005; Joubert and Hamid 2005; Black et al. 2012; Prakash 2013; Wright et al. 2013a, 2013b). Thus, it appears that in asthmatics, ASM exists in both hyper-reactive and synthetic states, thus contributing to a thicker, more responsive airway. Accordingly, upstream mechanisms that promote both hyper-reactive and synthetic states of ASM, particularly under the influence of inflammation, represent an appealing target for therapeutic intervention.
Fig. 1.
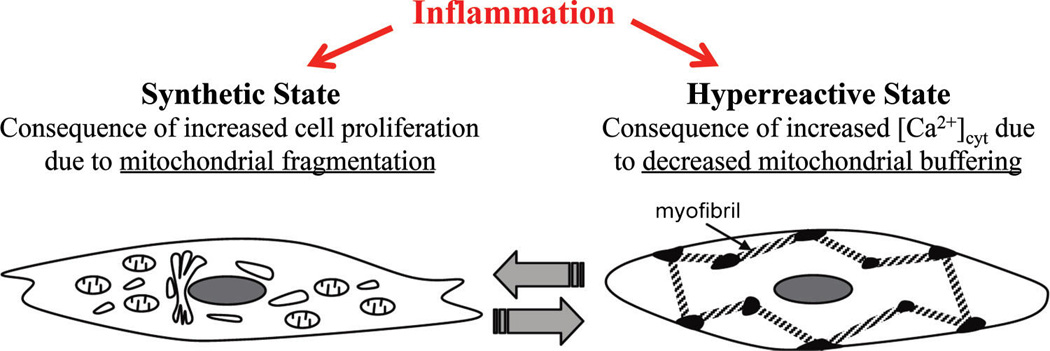
Inflammation plays a central role in asthma pathophysiology inducing both increased cell proliferation (synthetic state) and an enhanced contractile response to agonist stimulation (hyper-reactive state) in airway smooth muscle (ASM). We hypothesize that increased ASM cell proliferation is associated with decreased mitofusin-2 (Mfn2) expression and mitochondrial fragmentation, while ASM hyper-reactivity results from an increased [Ca2+]cyt due to an uncoupling of mitochondria from the endoplasmic reticulum/sarcoplasmic reticulum membrane (decreased Mfn2 expression) and decreased mitochondrial Ca2+ buffering (reduced [Ca2+]mito response).
Relationship between elevated [Ca2+]cyt and force in ASM
Force generation in ASM is initiated by a transient increase in [Ca2+]cyt in response to agonist (e.g., ACh) stimulation (Sieck et al. 2001). The mechanisms underlying the transient increase in [Ca2+]cyt are complex, but involve Ca2+ influx and (or) Ca2+ release from the endoplasmic/sarcoplasmic reticulum (ER/SR). Based on available evidence, it is unlikely that depolarization-dependent Ca2+ influx is required for the immediate [Ca2+]cyt response to agonist stimulation in ASM (Kannan et al. 1997; Prakash et al. 1997). However, Ca2+ influx in ASM does have an important role in establishing basal [Ca2+]cyt levels (Wylam et al. 2012) as well as in replenishing depleted SR Ca2+ stores via store-operated Ca2+ entry (SOCE) (Ay et al. 2004, 2006; White et al. 2006; Sieck et al. 2008; Sathish et al. 2012; Jia et al. 2013).
The transient elevation of [Ca2+]cyt in ASM cells is mediated in part by the production of inositol 1,4,5-triphosphate (IP3), which activates IP3 receptors to allow SR Ca2+ release (Foskett et al. 2007; Jude et al. 2008; Bai et al. 2009; Mei et al. 2014). The initial release of SR Ca2+ is amplified by the Ca2+-induced activation of additional IP3 receptors as well as ryanodine receptors (RyR); a process called Ca2+-induced Ca2+ release (CICR) (Mei et al. 2014). In ASM, the open probability of the RyR channels and CICR is also enhanced by agonist-induced production of cyclic ADP ribose (Kannan et al. 1996; Prakash et al. 1998; White et al. 2006).
The SR Ca2+ stores are replenished via the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) (Sathish et al. 2008, 2009; Prakash et al. 2009). In ASM cells, the interaction between SR Ca2+ release via IP3 receptor and ryanodine receptor (RyR) channels and SR Ca2+ uptake via SR calcium transport ATPase (SERCA) is dynamic, resulting in localized [Ca2+]cyt oscillations (~800–1000 nmol·L−1 amplitude at frequencies ranging from ~5–40 min−1) that propagate through the cell (15–25 µm−1) (Prakash et al. 1997, 2000; Sieck et al. 1997, 2001). Both the frequency and propagation velocity of [Ca2+]cyt oscillations in ASM cells depend on ACh concentration, whereas the amplitude of the localized [Ca2+]cyt response is independent of ACh concentration (Prakash et al. 1997, 2000; Sieck et al. 1997, 2001) Thus, the ACh concentration dependence of the global [Ca2+]cyt response in ASM cells is based on the frequency modulation of local responses and their fusion into larger responses.
In ASM cells, there is a delay of several hundred milliseconds between the agonist-induced elevation of [Ca2+]cyt and the resulting mechanical response (Sieck et al. 2001). This time delay reflects a cascade of events underlying excitation–contraction coupling in ASM that can be summarized into 4 main steps: (i) mobilization of calmodulin (CaM); (ii) activation of myosin light chain kinase (MLCK) following Ca2+ binding to CaM; (iii) MLCK-mediated phosphorylation of MLC20; and (iv) cross-bridge recruitment and cycling (Sieck et al. 2001). It has been shown that mobilization of CaM in ASM introduces the longest delay in excitation–contraction coupling in ASM, while delays owing to MLC20 phosphorylation, cross-bridge recruitment, and cycling are minimal (Sieck et al. 2001). The level of MLC20 phosphorylation also depends on myosin light chain phosphatase (MLCP), and the Ca2+ sensitivity of ASM force generation is modulated by the balance between MLCK and MLCP activities (Sieck et al. 2001). In ASM, agonist stimulation increases Rho-associated protein kinase (ROCK), which inhibits MLCP activity, thereby mediating a Ca2+ independent pathway for modulating ASM force generation (Ca2+ sensitivity). Agonist-induced increase in ROCK activity also indirectly mediates cyto-skeletal remodeling in ASM via inhibition of actin filament depolymerization.
Actin/myosin interactions in ASM
The essential molecular mechanism underlying force generation and contraction in ASM, like all muscle, is based on the Ca2+-regulated binding of myosin to actin to form cross-bridges. Force generation in ASM depends on the number of cross-bridges recruited in parallel, whereas the velocity of shortening (contraction) depends on the cycling of cross-bridges from bound to unbound states. In skeletal and cardiac (striated) muscle, myosin filaments form α-helical, bipolar configuration, whereas in smooth muscle, monomeric myosin is assembled into polymers or thick filaments in a nonhelical, side-polar configuration (Hodgkinson et al. 1995; Xu et al. 1996). The bipolar arrangement of myosin in skeletal and cardiac muscle together with the steric constraints of the sarcomeric structure impose limits on muscle length and shortening that do not exist in smooth muscle (Xu et al. 1996; Seow 2005). In striated muscle, actin filaments are anchored to the Z-disks of the sarcomere for longitudinal transmission of force. In contrast, actin filaments in smooth muscle are clustered into regions called dense bodies or membrane-associated dense plaques that allow force transmission to the ECM only when anchored to the plasma membrane (Hodgkinson et al. 1995; Gunst and Tang 2000; Kuo et al. 2003a; Sieck and Gransee 2012). Importantly, the internal arrangement of actin/myosin filaments, dense bodies, and dense plaques in smooth muscle is not highly ordered as in striated muscle, and this arrangement undergoes remodeling during the contractile response (Mehta and Gunst 1999; Gunst et al. 2003; Herrera et al. 2004; Seow 2005; Gunst and Zhang 2008; Zhang and Gunst 2008).
Actin exists in 2 distinct but interchangeable forms: polymeric, filamentous actin (F-actin) and monomeric actin (G-actin) (Jones et al. 1999b; Mehta and Gunst 1999; Gunst and Zhang 2008). During ASM activation, there is a decrease in G-actin content indicating that actin cytoskeletal remodeling and inhibition of actin polymerization reduces isometric force generation (Jones et al. 1999b; Mehta and Gunst 1999; Gunst et al. 2003). In addition to actin polymerization, there is also evidence that the contractile network is remodeled through myosin polymerization (Kuo et al. 2001, 2003a, 2003b; Seow 2005; Ali et al. 2007; Ijpma et al. 2011), such that the length of the myosin filaments increases, thereby increasing the number of myosin heads that can potentially form cross-bridges. In ASM, focal adhesion complexes (membrane-bound dense plaques) couple F-actin to the ECM (Gunst et al. 2003). The focal adhesion complexes comprise a scaffold of structural and signaling proteins that include mechano-sensitive transmembrane integrins (Gunst and Zhang 2008; Zhang and Gunst 2008). It is possible that in ASM, the location of focal adhesion complexes on the plasma membrane are dynamically regulated such that they recruit actin polymerization (increasing internal loading) according to local mechanical stimuli experienced during contraction (Gunst and Zhang 2008; Zhang and Gunst 2008). External loading of ASM is complex and dynamic, varying with inflation and deflation of the lung (Gunst et al. 2003; Gunst and Zhang 2008; Zhang and Gunst 2008). Thus, the internal organization of the actin/myosin filament lattice in ASM is likely to be complex and dynamic as well, changing in response to internal cell signaling and external loading (Gunst and Tang 2000; Gunst et al. 2003; Kuo et al. 2003a; Zhang and Gunst 2008).
Cytokine enhancement of [Ca2+]cyt and force responses in ASM: hyper-reactivity
As mentioned above, one hallmark of asthma is ASM hyper-reactivity in response to agonist (e.g., ACh) stimulation. This effect on ASM force generation and contractility is mimicked in animal models following airway sensitization (Stephens et al. 2003; Aravamudan et al. 2012), or following cytokine exposure in ASM muscle strips (Fig. 2), or in lung slice preparations (Fig. 3) (Amrani 2014). The increase in ASM force generation is the consequence of both an increased [Ca2+]cyt response to agonist stimulation as well as an increase in the Ca2+ sensitivity of force generation. We and others have demonstrated that inflammatory cytokines enhance agonist-induced [Ca2+]cyt responses in ASM (Parris et al. 1999; Amrani et al. 2000; Reynolds et al. 2000; Amrani and Panettieri 2002; Chen et al. 2003; Hunter et al. 2003; Sakai et al. 2004; White et al. 2006; Tirumurugaan et al. 2007; Sathish et al. 2008, 2009, 2011a, 2011b; Delmotte et al. 2012). Several major mechanisms involved in Ca2+ homeostasis and agonist-induced [Ca2+]cyt responses are modulated by cytokines (Jude et al. 2008; Sanderson et al. 2008; Prakash 2013; Wright et al. 2013c; Amrani 2014; Delmotte et al. 2014; Matsumoto 2014).
Fig. 2.
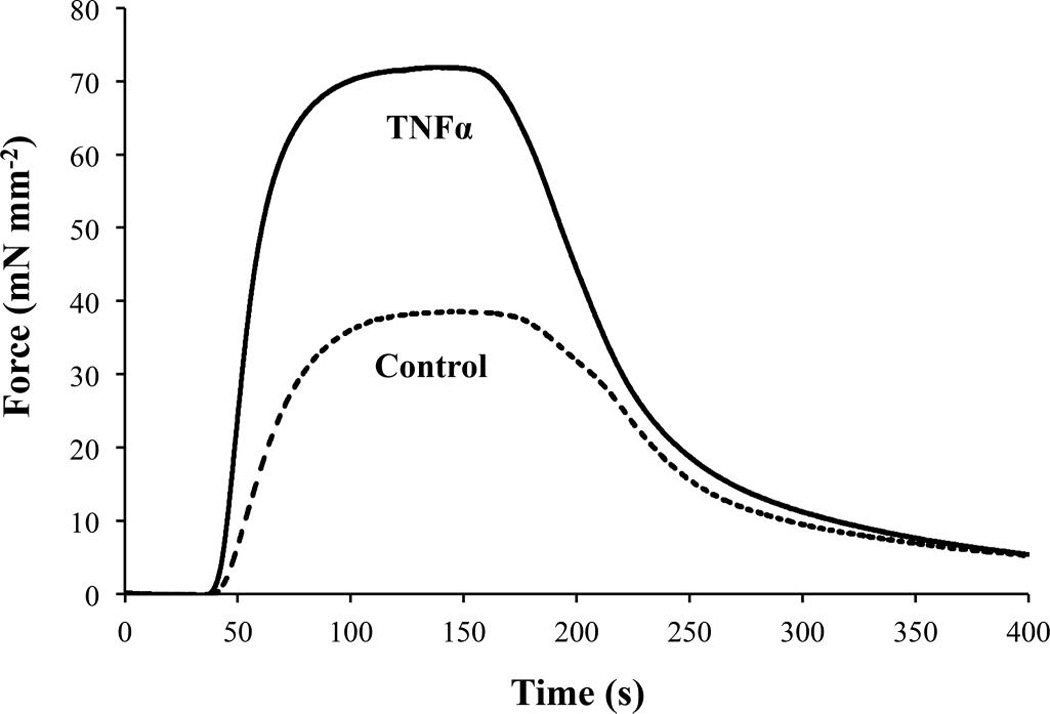
Exposure to tumor necrosis factor α (TNFα; 20 ng/mL for 24 h) increases the isometric force response of permeabilized (Triton X-100; 1%) porcine tracheal smooth muscle activated by 10 µmol·L−1 free Ca2+ (n = 4).
Fig. 3.
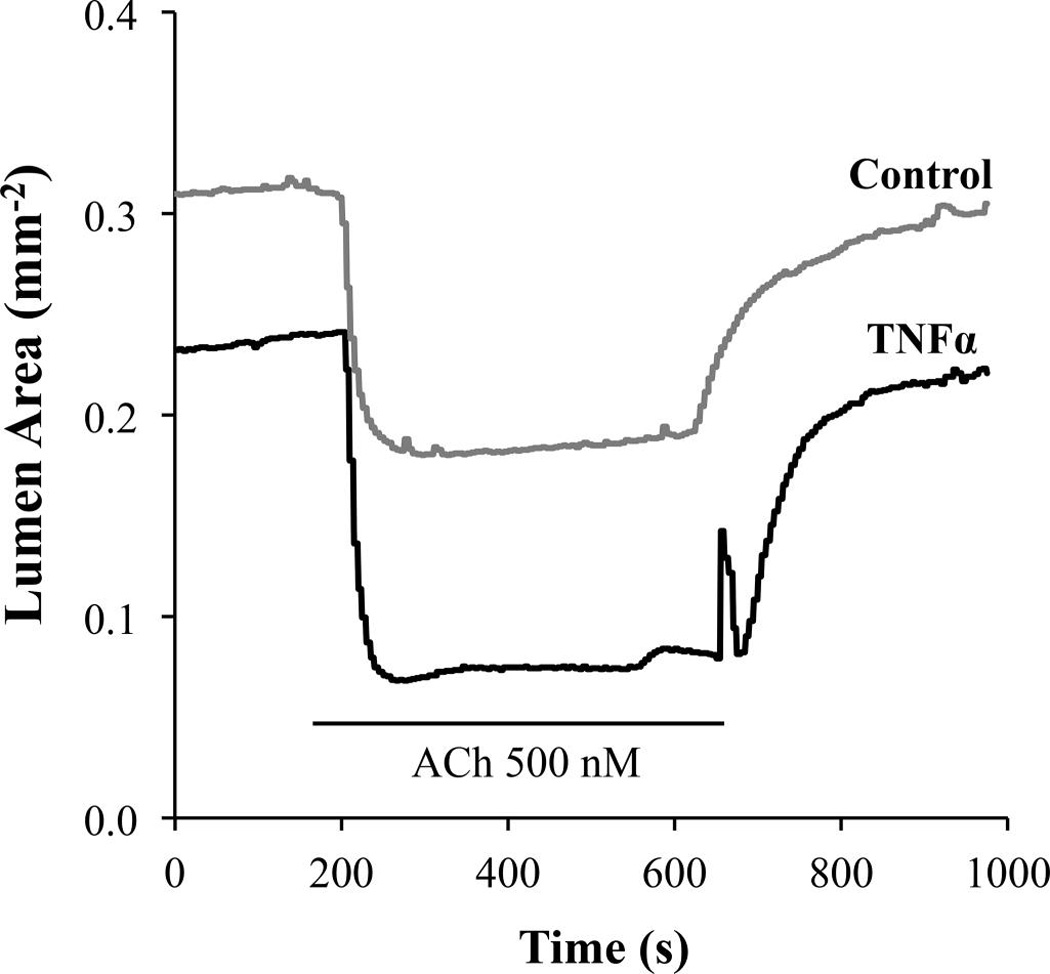
In a mouse lung slice, tumor necrosis factor α (TNFα) exposure (20 ng/mL for 12 h) increases constriction of small bronchioles induced by 500 nmol·L−1 acetylcholine (ACh) (n = 2 mice).
Mechanical work and ATP hydrolysis in ASM
In all muscle types, cross-bridge recruitment and cycling underlie the mechanical work performed through force generation and contraction. To perform this mechanical work, muscle requires chemical energy in the form of ATP hydrolysis (actin-activated myosin ATPase). The energy released by ATP hydrolysis is coupled to the intrinsic force development and load displacement that is mediated by cyclic attachment and detachment of myosin heads to actin (cross-bridge cycling). In skeletal muscle fibers, isometric force generation at any given level of Ca2+ activation is accompanied by a sustained rate of ATP hydrolysis; thus, tension cost (ATP consumed per unit of force) does not change (Sieck et al. 1998). In contrast, during sustained isometric force generation in ASM, the rate of ATP hydrolysis decreases and as a result tension cost decreases with time (Fig. 4) (Sieck et al. 1998; Jones et al. 1999a, 1999b). The time-dependent decrease in rate of ATP hydrolysis in ASM is also associated with a slowing of shortening velocity and has been termed the “latch state” (Hai and Murphy 1988; Murphy and Rembold 2005). According to the “latch bridge” hypothesis, a unique cross-bridge state occurs when the myosin head of an attached cross-bridge (due to prior MLC20 phosphorylation) is then dephosphorylated. These dephosphorylated latch-bridges cycle at a slower rate with an increased duty cycle (amount of time attached versus detached), and therefore maintaining force at a reduced energy cost. The main assumption of the latch-bridge model is that the extent of MLC20 phosphorylation determines the “latch” or energetic state of smooth muscle force generation (Hai and Murphy 1988; Murphy and Rembold 2005). However, in permeabilized canine ASM, we (Jones et al. 1999a) showed that with sustained maximum Ca2+ activation, isometric force and the level of MLC20 phosphorylation are sustained, while the rate of ATP hydrolysis decreases with time reflecting a reduction in energy cost, consistent with a latch state, but without a change in MLC20 phosphorylation (Fig. 4). Furthermore, when dephosphorylation of MLC20 was prevented by thiophosphorylation (using ATPγS), the time-dependent decrease in ATP hydrolysis rate following Ca2+ activation was unaffected. Thus, the slowing of cross-bridge cycling rate and ATP hydrolysis rate in ASM is independent of any change in the level of MLC20 phosphorylation. Alternatively, we proposed that the decline in the rate of ATP hydrolysis in ASM during sustained Ca2+ activation may be due to an increase in “internal loading” as a result of cytoskeletal remodeling (Jones et al. 1999b). This alternative “internal loading” hypothesis was tested by inhibition of actin remodeling with phalloidin (which binds F-actin, preventing its depolymerization), which essentially eliminated the time-dependent decrease ATP hydrolysis rate in ASM (Fig. 4) (Jones et al. 1999b). According to this internal loading hypothesis, cross-bridges in ASM cells are recruited by MLC20 phosphorylation, but a proportion of the cross-bridges are not fully attached to the internal cytoskeletal structure, resulting in lower internal loading, faster cross-bridge cycling, and a high rate of ATP hydrolysis. Subsequently, with cytoskeletal remodeling, there is an increase in cross-bridge attachment to the cytoskeleton imposing increased internal loading, a slowing of cross-bridge cycling rate, and a decrease in ATP hydrolysis rate.
Fig. 4.
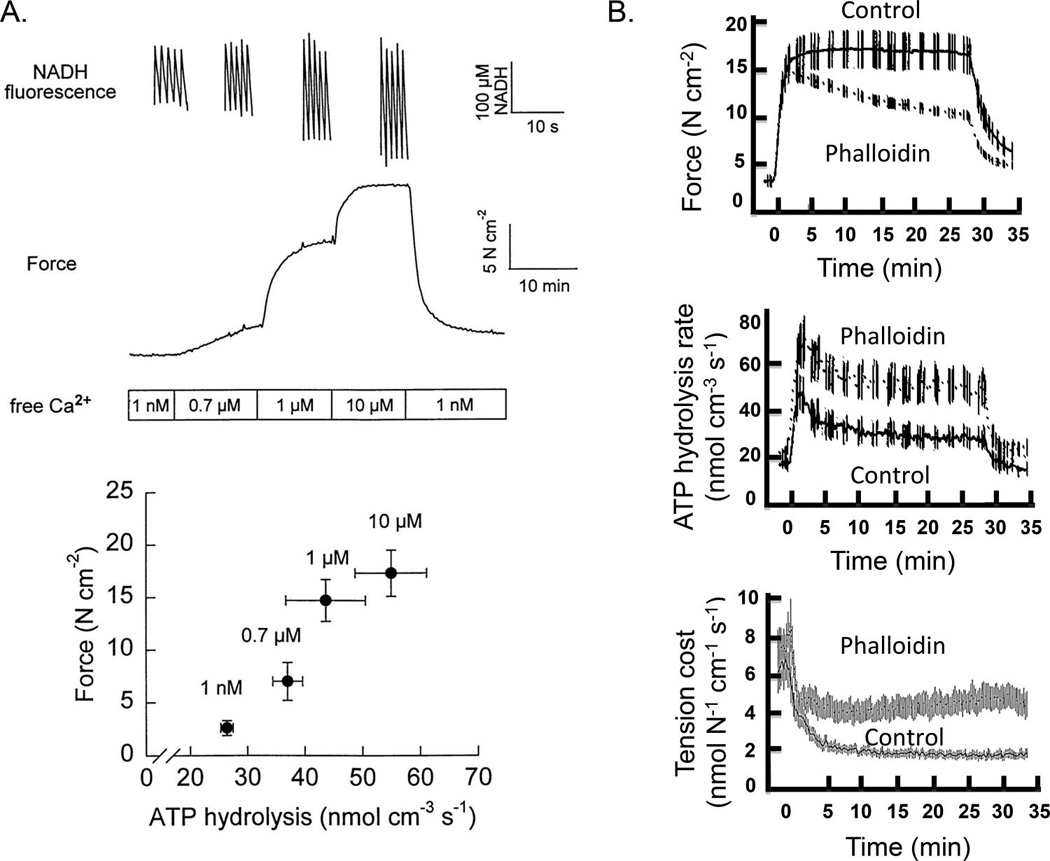
(A) Changes in isometric force and ATP hydrolysis rate (measured via extinction of NADH fluorescence) in Triton X-100 (0.1%)-permeabilized canine tracheal smooth muscle activated by various levels of free Ca2+ (1.0 nmol·L−1 to 10.0 µmol·L−1). Note the linear relationship between isometric force and ATP hydrolysis rate. (B) Time course of changes in isometric force, ATP hydrolysis rate, and tension cost of permeabilized canine tracheal smooth muscle activated by 10 µmol·L−1 free Ca2+. During normal isometric activation of airway smooth muscle (ASM), ATP hydrolysis rate is initially faster and then declines with time to a sustained level, even though isometric force is maintained. Tension cost (ATP consumption per unit force generation) of ASM activation also starts higher and then decreases with time. Phalloidin induced disruption of actin cytoskeletal remodeling results in force decline and blunted the time-dependent changes in ATP hydrolysis rate and tension cost. (Modified from Fig. 2 of Jones et al. 1999b and from Figs. 2 and 4 of Jones et al. 1999a).
Metabolic cost of cytokine-induced hyper-reactivity in ASM
In previous studies in ASM (Sieck et al. 1998; Jones et al. 1999a, 1999b), we demonstrated a direct relationship between ATP hydrolysis rate and force generation (Fig. 4), velocity of shortening, and power output. Thus, in ASM exposed to inflammatory cytokines, the increase in force generation is associated with an increase in ATP consumption. In porcine ASM, maximum ATP hydrolysis occurs at peak power output, attaining a rate of ~0.12 nmol ATP·mm−3·s−1 that is approximately 3-fold higher than the ATP hydrolysis rate during maximum isometric force (~0.04 nmol ATP·mm−3·s−1) (Sieck et al. 1998). By comparison, in fast-twitch diaphragm muscle fibers, the rate of ATP hydrolysis is ~1.2 nmol ATP·mm−3·s−1 during maximum isometric force and increases to ~2.7 nmol ATP·mm−3·s−1 during peak power output (Sieck et al. 1998). Tension cost ([ATP] consumed per force generated) in diaphragm muscle fibers ranges from ~16 000–35 000 nmol ATP·s−1·mN−1 depending on fiber type. In contrast, the tension cost of ASM is 6- to 14-fold lower at ~2500 nmol ATP·s−1·mN−1 (Sieck et al. 1998). Of course these differences reflect the mechanical/work performance of these different muscle types, e.g., the maximum power output of skeletal muscle is ~10 to 100 times greater than that of ASM (Sieck et al. 1998). In skeletal muscle fibers this greater energy demand is met by a relatively large pool of phosphocreatine (PCr) that is available to buffer the transient increase in ATP hydrolysis (Paul 1989). Smooth muscle contains a very small PCr pool, with a much greater relative dependence on ATP production rather than PCr buffering (Krisanda and Paul 1983; Spurway and Wray 1987; Ishida et al. 1994). Thus, skeletal muscle mechanical performance is optimized for maximum power output, while the mechanics of smooth muscle is optimized for maximum energy efficiency. Efficient matching of energy supply with energy demand may be an even more significant governing factor for the ASM contractile response.
In all muscle types, mitochondria provide ATP through O2 consumption and oxidative phosphorylation to meet the energy demands of force generation and contraction. In vascular smooth muscle, it has been shown that isometric force is strongly correlated with O2 consumption (Krisanda and Paul 1983; Paul 1989; Ishida et al. 1994). It is likely that the same relationship holds for ASM. Thus, with an inflammatory-cytokine-induced increase in force and contractile response and the associated increase in ATP consumption, it is likely that O2 consumption and oxidative phosphorylation also increase (Fig. 5).
Fig. 5.
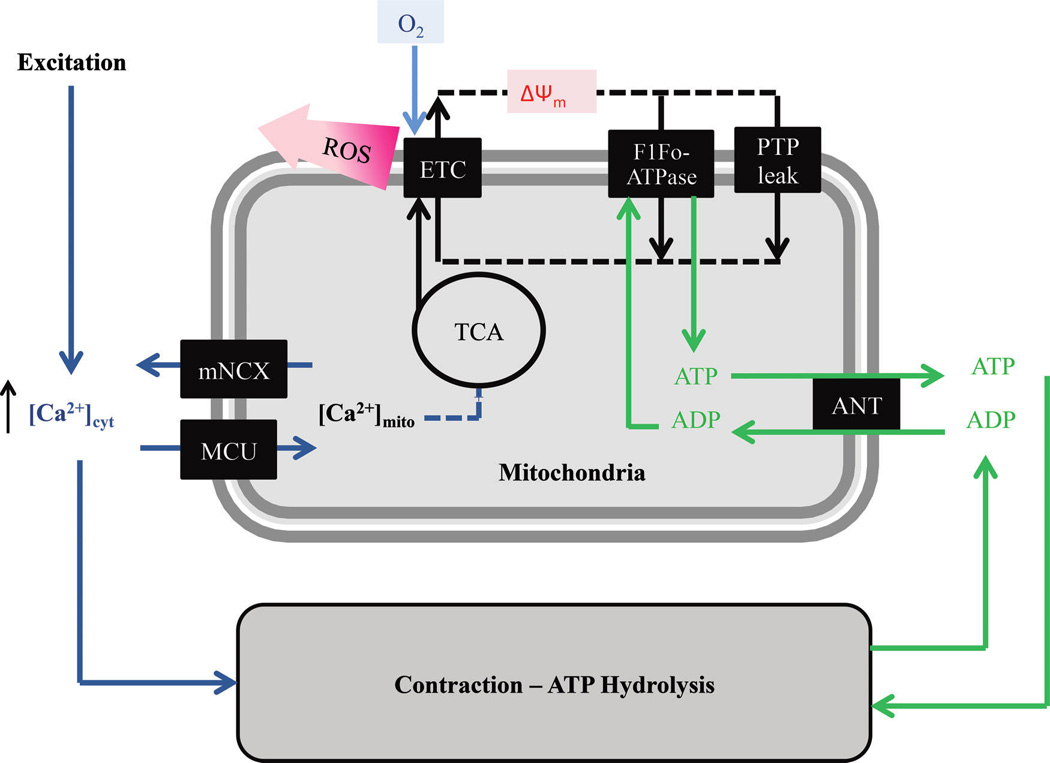
In airway smooth muscle cells, the agonist-induced transient increase of [Ca2+]cyt is accompanied by activation of the MCU and a transient increase in [Ca2+]mito thereby increasing activities of intra-mitochondrial dehydrogenases of the TCA cycle and the amount of NADH available to the electron transport chain. Mitochondrial O2 consumption and ATP synthesis are also linked to cross-bridge cycling and ATP hydrolysis via changes in the ATP/ADP ratio and the ANT. A portion of the O2 consumed in the ETC is incompletely reduced resulting in ROS. MCU, mitochondrial Ca2+ uniporter; TCA, tricarboxylic acid; ANT, adenine nucleotide transporter; ETC, electron transport chain; ROS, reactive oxidant species; mNCX, mitochondrial Na–Ca exchanger; F1Fo, F-type ATPases; PTP, permeability transition pore.
Mitochondrial ATP production is stimulated by an increase in mitochondrial Ca2+ concentration ([Ca2+]mito) via an increase in activity of Ca2+-sensitive intra-mitochondrial dehydrogenases associated with the tricarboxylic acid (TCA) cycle – pyruvate dehydrogenase (PDH), NAD-isocitrate dehydrogenase (ICDH), and oxoglutarate dehydrogenase (OGDH) (Denton and McCormack 1990; Denton 2009; Griffiths and Rutter 2009; Glancy and Balaban 2012; Tarasov et al. 2012). The relationship between dehydrogenase activity and [Ca2+]mito can also be reversed by an excess of end-product (for example, excess acetyl-CoA in the case of PDH), and can also be modulated by the ratio of ATP/ADP (which decreases during muscle contraction and increased ATP consumption) and substrate availability within the mitochondria (Denton 2009).
In the process of oxidative phosphorylation, NADH donates electrons to the electron transport chain (ETC) within the inner membrane of the mitochondria. As the ETC catalyzes electron flow, H+ is pumped against its concentration gradient out of the mitochondrial matrix. The final stage of the ETC requires O2 to accept electrons (at Complex IV). Therefore, electrons flowing through the ETC results in O2 consumption and, owing to H+ pumping, the formation of a proton motive force (pmf) that has electrical (ΔΨ) and chemical (ΔpH) features. The pmf favors the flow of protons into the mitochondrial matrix. The F1Fo-ATPase (also called Complex V), which is permeable to H+, uses the energy dissipated by pmf-driven proton flow into the matrix to synthesize ATP from ADP and inorganic phosphate (Pi). Synthesized ATP is then transported to the cytosol in exchange for cytosolic ADP by the adenine nucleotide transporter (ANT). Normally, the O2 consumed in the ETC is reduced to produce water. However, approximately 1% of O2 is incompletely reduced and yields the superoxide radical (O2−). Thus, reactive oxidant species (ROS) formation is a natural by product of oxidative phosphorylation. However, metabolic stress can increase ROS formation, triggering oxidative stress in cells (Fig. 5).
Cytokine-induced ER/SR stress and the unfolded protein response in ASM
A major feature of asthma is an increase in ROS formation (Katsumata et al. 1990; Comhair and Erzurum 2010; Zuo et al. 2013). In other cell systems, it has been shown that one consequence of excessive ROS formation is an accumulation of unfolded or misfolded proteins in the ER/SR lumen, which triggers a highly conserved restorative/corrective response (i.e., the ER/SR stress or unfolded protein response) (Adolph et al. 2012; Baban et al. 2012; Garg et al. 2012; Hasnain et al. 2012a, 2012b; Martino et al. 2012). As a normal physiological process, the ER/SR stress response serves to restore normal function by halting protein translation and activating protein degradation and expression of molecular chaperones involved in protein folding (Welihinda et al. 1999; Fulda et al. 2010; Adolph et al. 2012; Wang and Kaufman 2012). Three ER/SR stress protein markers located at the ER/SR membrane are involved in the unfolded protein response: protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1α) (Fig. 6). Normally, the natural molecular chaperone protein binding immunoglobulin protein (BiP) (also known as 78 kDa glucose-regulated protein (GRP)-78 or 70 kDa heat shock protein 5 (HSPA5)) is associated with PERK, ATF6, and IRE1 and maintains these proteins in an inactive state. However, as unfolded proteins accumulate in the ER/SR lumen, expression of BiP/GRP-78 increases and it dissociates from PERK, ATF6, and IRE1 leading to the activation of these ER/SR stress proteins and the downstream pathways they mediate. IRE1α catalyzes the alternative splicing of X-box binding protein 1 (XBP1) mRNA leading to expression of the active (spliced) XBP1 transcription factor. ATF6 translocates to the golgi and is cleaved by the proteases S1P and S2P, leading to an active ATF6 transcription factor ATF6 (i.e., ATF6p50). PERK phosphorylates the translation-initiator factor eukaryotic initiation factor 2 (eIF2α), leading to selective translation of the transcription factor ATF4. The IRE1, ATF6, and PERK pathways, and thereby XBP1, ATF6p50, and ATF4 act to: (i) block protein translation; (ii) increase chaperone expression (e.g., calnexin); and (iii) enhance SR-associated protein degradation pathways (Fig. 6) (Samali et al. 2010; Munoz and Zorzano 2011; Adolph et al. 2012; Baban et al. 2012; Garg et al. 2012; Dromparis et al. 2013).
Fig. 6.
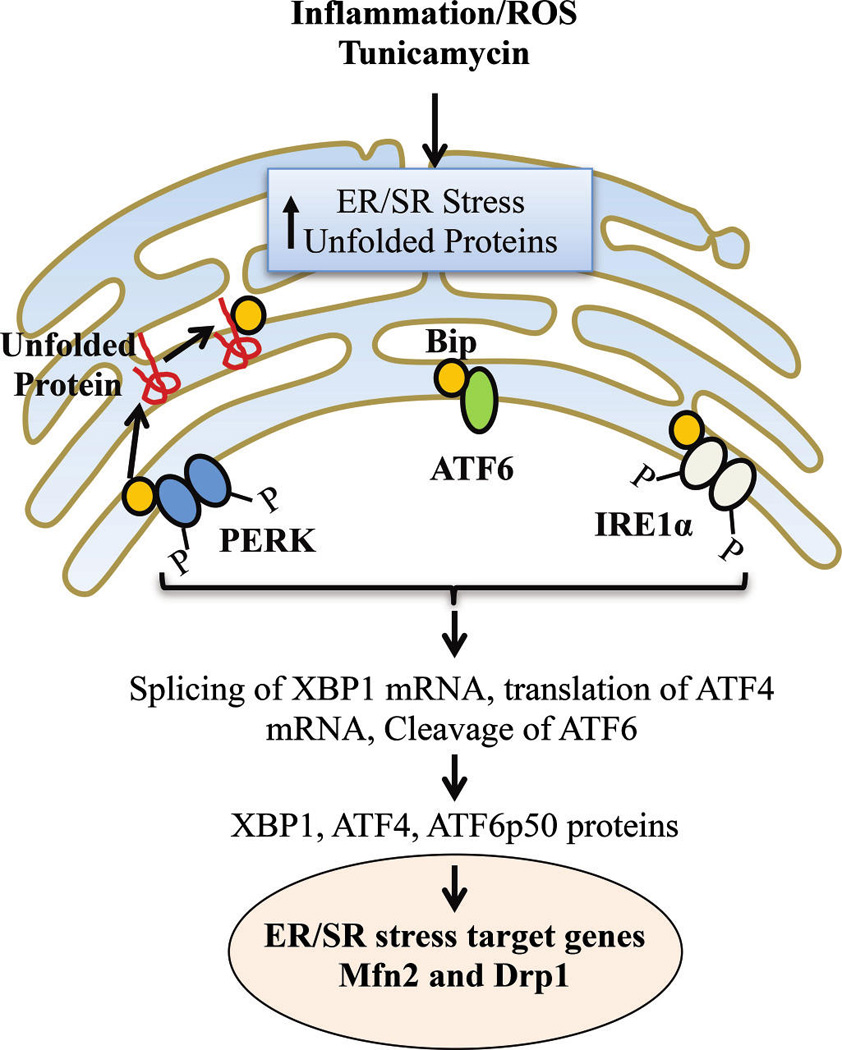
In hASM, inflammation, ROS, and (or) tunicamycin induce an accumulation of unfolded proteins and ER/SR stress (unfolded protein response). The molecular chaperone BiP (GRP78) is normally associated with PERK, ATF6, and IRE1α and maintains these proteins in an inactive state. As unfolded proteins accumulate in the ER/SR lumen, expression of BiP increases, and it dissociates from PERK, ATF6, and IRE1 leading to their activation and resulting in selective translation of the transcription factor ATF4; active transcription factor ATF6 (i.e., ATF6p50); and the active (spliced) XBP1 transcription factor. The IRE1, ATF6, and PERK pathways act to: (i) block protein translation, (ii) increase chaperone expression (e.g., calnexin), (iii) enhance SR-associated protein degradation pathways, and (iv) alter mitochondrial fusion (Mfn2) and fission (Drp1) protein expression. ROS, reactive oxidant species; hASM, human airway smooth muscle cells; ER/SR, endoplasmic reticulum/sarcoplasmic reticulum; BiP, binding immunoglobulin protein; PERK, protein kinase RNA-like endoplasmic reticulum kinase; ATF, active transcription factor; XBP1, X-box binding protein 1; IRE1, inositol-requiring enzyme 1; Mfn2, mitofusin-2; Drp1, dynamin related protein 1.
A recent study (Makhija et al. 2013) showed that there is an increased expression of ER/SR stress protein markers such as BiP/GRP-78 and ATF6 in lungs from an ovalbumin (OVA) sensitized mouse model of asthma, and that chemical chaperone treatment used to attenuate ER/SR stress is effective in reducing airway remodeling and inflammation. Others recent studies confirmed the presence of ER/SR stress markers in the OVA mouse model (Kim et al. 2013) as well as another mouse model of asthma: house dust mite sensitization (Hoffman et al. 2013). However, the involvement of ASM per se, and the downstream mechanisms by which ER/SR stress influences airway reactivity and remodeling were not explored in these studies.
In a recent unpublished study, we confirmed that exposure to inflammatory cytokines (TNFα or IL13) induces ROS formation and ER/SR stress in hASM cells. In these studies, hASM cells were acutely dissociated from >3rd order bronchi (~3 mm diameter) from normal human lung biopsy tissue obtained during thoracic surgery. The hASM cells were exposed to TNFα or IL-13 (20 ng/mL) for 24 h (n = 3 ASM samples per group). The response to tunicamycin (1.0 µg/mL) served as a positive control for ER/SR stress. The induction of ER/SR stress was verified by increased expression (determined by Western blot) of BiP/GRP78 (upstream marker), IRE1, ATF6, and PERK (ER/SR stress markers), as well as spliced XBP1 and calnexin (downstream markers). ER/SR stress was also confirmed by the increased ratio of spliced versus unspliced XBP1 mRNA measured using real-time reverse-transcription PCR, which peaked at 12 h of cytokine exposure.
Cytokine-induced disruption of mitochondrial coupling with ER/SR membrane in ASM
Within ASM cells, the ER/SR and mitochondria are discreet intracellular organelles. However, both the ER/SR and mitochondria are highly dynamic, moving and undergoing constant structural and spatial reorganization (see below). Physical contact between the ER/SR membrane and the outer mitochondrial membrane (e.g., mitochondria-associated ER/SR membrane, MAM) is especially important for efficient transfer of metabolites (lipids, Ca2+) between the 2 organelles and is essential for their normal function (de Brito and Scorrano 2010; Kornmann 2013; Rainbolt et al. 2014). Disruption of the normal coupling between mitochondria and the ER/SR membrane appears to be an important downstream consequence of ER/SR stress (Bravo et al. 2012; Verfaillie et al. 2012; Vannuvel et al. 2013; Rainbolt et al. 2014). In addition, PERK and another ER/SR protein, ER oxidoreduclin 1α (Ero1α), are associated with MAMs, and they appear to leave MAMs when cells experience ER/SR stress (Gilady et al. 2010; Raturi and Simmen 2013). While the role of PERK and Ero1α within MAMs is unclear, it suggests that uncoupling of mitochondria with the ER/SR membrane may be an important component of the overall ER/SR stress response. In this regard, mitofusin-2 (Mfn2), which is well known for its role in the regulation of mitochondria morphology (see below), is also an important component of MAMs, involved in the formation of bridges between mitochondria and the ER/SR membrane (de Brito and Scorrano 2008a, 2008b, 2010). Mfn2 protein expressed on the ER/SR membrane forms homotypic and heterotypic complexes with Mfn2 and Mfn1 proteins present on the outer mitochondrial layer (Fig. 7). Interestingly, the ER/SR stress marker PERK co-localizes with Mfn2 protein within MAMs (Verfaillie et al. 2012; Munoz et al. 2013). Ablation of the Mfn2 gene enhances ER/SR stress, while ablation of the PERK gene increases Mfn2 expression suggesting a homeostatic interaction between Mfn2 and PERK expression (Munoz et al. 2013). However, the exact mechanisms are unknown. In a recent study, we reported that Mfn2 expression is decreased in asthmatic ASM (Aravamudan et al. 2014); however, the relationship between reduced Mfn2 expression and mitochondrial association with the ER/SR membrane was not explored. More recently, we found that in hASM cells exposed to TNFα or IL13, Mfn2 expression was markedly reduced, whereas PERK expression increased, consistent with the ER/SR stress response. We also found that following 24 h exposure of hASM to TNFα or IL-13, the normal enhancement of mitochondria proximity to the ER/SR induced by ACh stimulation was blunted. These results suggested that the normal coupling between mitochondria and the ER/SR membrane was disrupted in hASM cells as a result of ER/SR stress.
Fig. 7.
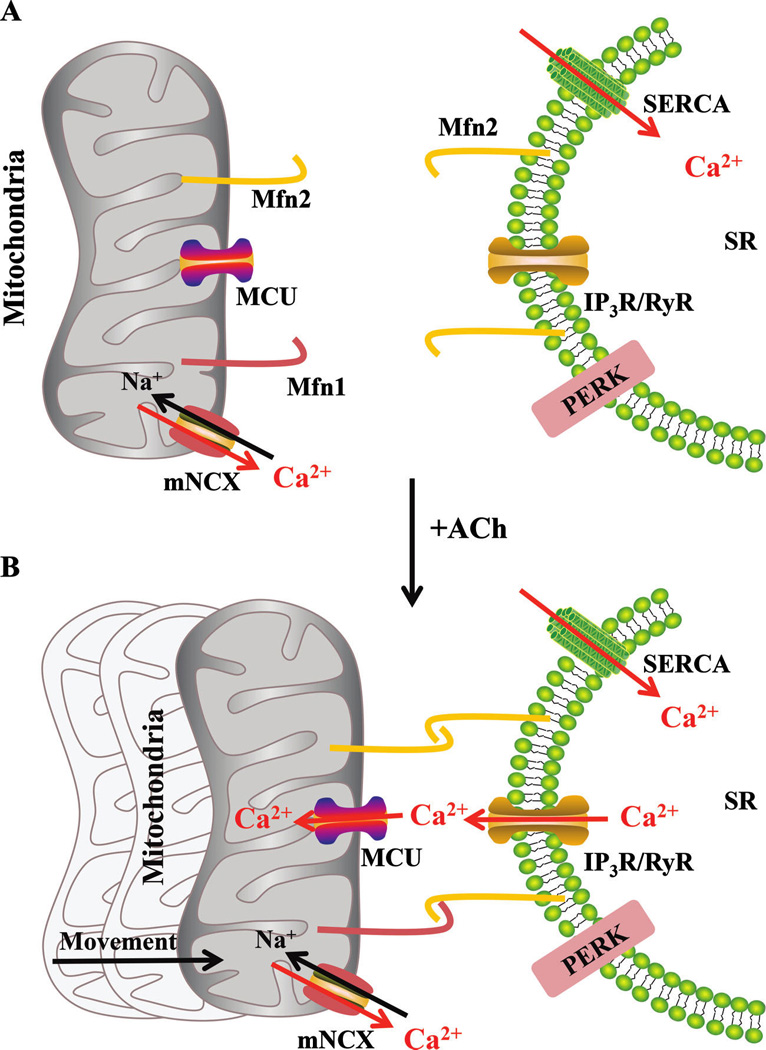
In hASM, agonist-induced elevations of [Ca2+]cyt are associated with corresponding elevations in [Ca2+]mito, reflecting mitochondrial Ca2+ buffering via activation of the MCU that requires close proximity of mitochondria to the ER/SR membrane. Activation of MCU is only achieved within localized microdomains (i.e., “Ca2+ hot spots”) in the vicinity of the IP3R or RyR. This mitochondrial coupling to the ER/SR membrane requires mitochondrial movement and tethering mediated by dimerization of Mfn2s (homodimer) or Mfn2 and Mfn1 (hetrodimer). Subsequent to agonist activation, [Ca2+]mito decreases via the mNCX. Refilling of the ER/SR is achieved by SERCA. hASM, human airway smooth muscle cells; MCU, mitochondrial Ca2+ uniporter; IP3R, inositol 1,4,5-triphosphate receptor; RyR, ryanodine receptor; ER/SR, endoplasmic reticulum/sarcoplasmic reticulum; mNCX, mitochondrial Na–Ca exchanger; SERCA, SR calcium transport ATPase.
Cytokine-induced reduction of mitochondrial movement/motility in ASM
Mitochondrial movement or motility is another potent regulator of the coupling between mitochondria and the ER/SR membrane. Movement of mitochondria toward the ER/SR should promote MAM formation. A redistribution of mitochondria to areas of high [Ca2+]cyt has been reported (Yi et al. 2004; Anesti and Scorrano 2006; Boldogh and Pon 2007), and agonist stimulation increases mitochondrial density near the ER/SR (Brough et al. 2005; Malaiyandi et al. 2005). Mitochondria movement is also critical to maintain normal mitochondrial morphology (see below), and therefore, mitochondrial function and integrity. It is not clear whether ER/SR stress affects mitochondrial movement. In hASM, there is no information regarding the effect of inflammation on mitochondrial movement. Similarly, there is no information whether mitochondrial movement is impaired in airway diseases such as asthma.
In a recent study (Zavaletta et al. 2011), we observed 2 general types of mitochondrial movement in hASM cells: (i) a continuous apparent random, non-directed movement that occurs over relatively short distances, and (ii) an infrequent directed trajectory movement that is most apparent in the distal compartment of the cell. In hASM cells we found that agonist (ACh, histamine) stimulation reduced the random, non-directed movement of mitochondria, whereas the directed trajectory movement of mitochondria in distal compartments of the cell appeared to increase. We also found that 24 h exposure of hASM cells to TNFα decreases both types of mitochondrial movement. It is possible that the movement of mitochondria toward the ER/SR in hASM cells is essential for MAM formation and coupling of mitochondria to the ER/SR membrane (Fig. 7).
Cytokine-induced disruption of coupling between [Ca2+]cyt and [Ca2+]mito in ASM
In many cell types, including hASM, a transient elevation of [Ca2+]cyt leads to a coupled transient increase in mitochondrial Ca2+ concentration ([Ca2+]mito) mediated via Ca2+([Ca2+]cyt)-induced activation of the mitochondrial Ca2+ uniporter (MCU) (Fig. 8) (Baughman et al. 2011; De Stefani et al. 2011; Delmotte et al. 2012, 2014; Raffaello et al. 2012). However, the MCU is relatively insensitive to Ca2+, requiring [Ca2+]cyt levels that are much higher (>10 µM Ca2+) than normally encountered in the bulk cytosol during agonist stimulation ([Ca2+]cyt is typically <2.0 µmol·L−1 Ca2+) (Gunter and Sheu 2009). This high threshold for MCU activation (>10 µmol·L−1 Ca2+) compared with the lower subthreshold bulk [Ca2+]cyt is reconciled by the “hotspot hypothesis”, which presumes that the cytosol in the immediate vicinity of an open Ca2+ channel (either inositol triphosphate (IP3) or RyR) can transiently experience intense increases in [Ca2+]cyt (Fig. 7) (Rizzuto et al. 2004). Interestingly, both IP3Rs and RyRs have been identified within MAMs (Hajnoczky et al. 2002; Kiviluoto et al. 2013). A Ca2+ hotspot thereby exposes the MCU to a microdomain of high [Ca2+]cyt sufficient for MCU activation and a coupled increase in [Ca2+]mito (Fig. 8) (Rizzuto et al. 1993, 2004, 2009; Rizzuto and Pozzan 2006; Pizzo et al. 2012) Thus, mitochondrial coupling with the ER/SR membrane is essential for MCU activation and the transient elevations of [Ca2+]mito that normally are coupled to transient [Ca2+]cyt responses (Fig. 8). Recent studies reported that ER/SR stress is associated with reduced mitochondrial coupling with the ER/SR membrane, resulting in changes in [Ca2+]mito (Csordas et al. 2006; Raturi and Simmen 2013). In various models of ER/SR stress, expression of Mfn2 is altered and this is correlated with uncoupling of mitochondria with the ER/SR membrane and blunting of the transient [Ca2+]mito response. Ablation of the Mfn2 gene in mouse embryonic fibroblasts and HeLa cells reduces [Ca2+]mito in a manner similar to direct MCU inhibition of mitochondrial Ca2+ uptake (de Brito and Scorrano 2008a). We also found that 24 h exposure of hASM cells to TNFα and (or) IL-13 resulted in reduced Mfn2 expression, reduced proximity of mitochondria to the ER/SR, and a marked reduction in the transient [Ca2+]mito response to ACh stimulation, even though the transient [Ca2+]cyt response was enhanced (Delmotte et al. 2012).
Fig. 8.
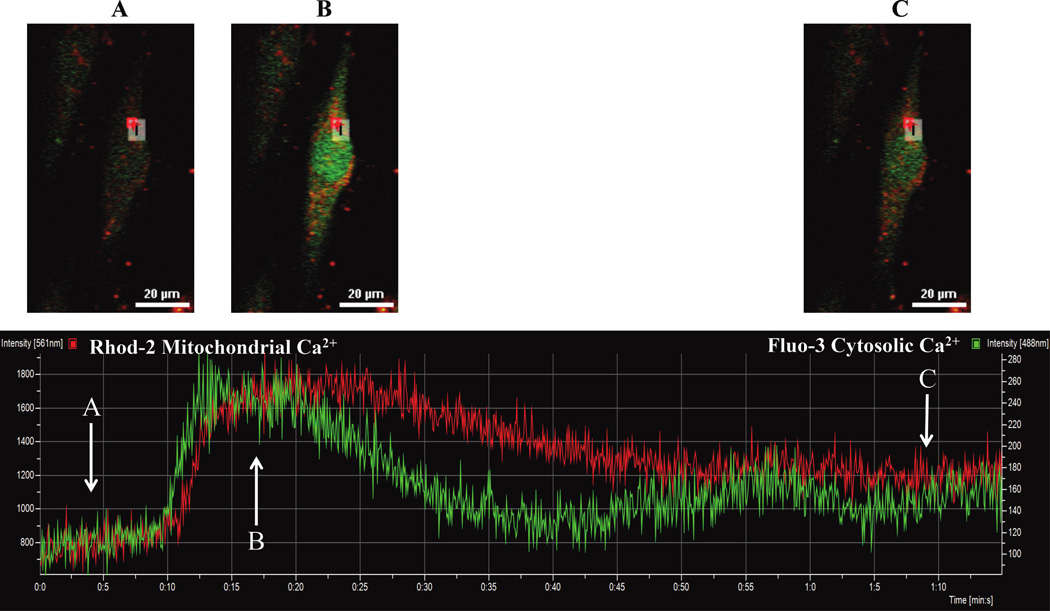
Human ASM cells were loaded with 2.5 µmol·L−1 fluo3-AM (green) and 2.5 µmol·L−1 rhod-2-AM (red) to measure changes in [Ca2+]cyt and [Ca2+]mito, respectively (top: images at 3 different time points (A, B, and C) relative to ACh stimulation). Fluo3-AM and rhod-2-AM fluorescence increased in response to 1.0 µmol·L−1 ACh stimulation (bottom). Note that the transient [Ca2+]cyt response slightly preceded the [Ca2+]mito response, whereas the [Ca2+]mito response was more prolonged. ASM, airway smooth muscle cells; ACh, acetylcholine.
The concept that mitochondria could compete with the SR to buffer [Ca2+]cyt is controversial (Collins et al. 2000; Cheranov and Jaggar 2004; Ishii et al. 2006; Chalmers and McCarron 2008, 2009). However, mitochondria have a huge Ca2+ buffering capacity and, importantly, are often strategically located near IP3 or RyR Ca2+ channels (Hajnoczky et al. 2002; Kiviluoto et al. 2013). It is presumed that mitochondrial Ca2+ uptake and (or) release in proximity to IP3 or RyR Ca2+ channels may affect local [Ca2+]cyt and thereby their probability of opening. In addition, changes in [Ca2+]mito affect mitochondrial function (i.e., ATP synthesis, ROS generation), which in turn could affect nearby Ca2+ channels (Perocchi et al. 2010; Delmotte et al. 2014). It has been suggested that ROS increase the “open probability” of RyR channels (Favero et al. 1995). Several studies showed that endogenous ROS lead to an elevation in [Ca2+]cyt (Lin et al. 2007; Chen et al. 2010; Perez-Vizcaino et al. 2010), which is blunted by the inhibition of RyR channels (Lin et al. 2007). When mitochondrial Ca2+ uptake (via MCU) is inhibited in ASM cells, the [Ca2+]cyt response to agonist stimulation is increased, similar to the effect of inflammatory cytokine exposure. Importantly, exposure to TNFα or IL-13 blunted mitochondrial Ca2+ uptake during agonist stimulation, suggesting an uncoupling of the SR/ER and mitochondria and a reduction in mitochondrial buffering of [Ca2+]cyt. The effect of ER/SR stress on [Ca2+]cyt and [Ca2+]mito in ASM has not been previously reported, but in other cell types it induces an increase in [Ca2+]cyt. Similarly, ablation of the Mfn2 gene increases basal [Ca2+]cyt and reduces [Ca2+]mito, similar to the effect of inflammatory cytokine exposure in ASM.
Elevation of [Ca2+]mito and excitation/energy coupling
Elevation of [Ca2+]mito stimulates enzymes of the tricarboxylic acid (TCA) cycle, and thus, the downstream electron transport chain (Brookes et al. 2004; Denton 2009; Gunter and Sheu 2009; Glancy and Balaban 2012). Therefore, mitochondrial coupling with the ER/SR membrane to create a Ca2+ hotspot and cause mitochondrial Ca2+ uptake (increased [Ca2+]mito) is critical for matching the increased energy demands that result from cross-bridge recruitment and cycling via excitation–contraction coupling (triggered by transient elevation of [Ca2+]cyt) and increased mitochondrial ATP production (i.e., excitation–energy coupling) (Fig. 5) (Schaible et al. 2014). An abnormal increase or decrease in [Ca2+]mito (relative to the transient [Ca2+]cyt response) is associated with mitochondrial dysfunction and increased ROS generation (Malli et al. 2003; Brookes et al. 2004; Romagnoli et al. 2007; Dada and Sznajder 2011; Csordas et al. 2012; Dorn and Maack 2013).
Cytokine-induced mitochondrial fragmentation
Apart from its role in ER/SR-mitochondrial coupling, Mfn2 is also essential in the fusion of mitochondrial filamentous networks within ASM. Mitochondria normally exhibit tubular, reticular, or networked morphology, which is regulated by dynamic remodeling, represented by a balance between fission vs. fusion that shifts in response to the environment (Chan 2006a, 2006b, 2012; Liesa et al. 2009; Youle and van der Bliek 2012). The balance between fission and fusion is now thought to be important for mitochondrial DNA stability, respiratory function, cell fate determination, and adaptation to cellular stress (Chan 2012). In mammalian cells, mitochondrial fusion involves 2 steps: fusion of the outer membrane, which is dependent on 2 GTPase proteins, mitofusin 1 (Mfn1) and 2 (Mfn2); and fusion of the inner membrane, which requires optic atrophy protein 1 (OPA1) (Fig. 9). The fusion of the outer mitochondrial membrane is initiated by dimerization of mitofusins and tethering of outer membranes of adjacent mitochondria. The fusion of the inner membranes of the mitochondria requires a motor-like process driven by OPA1 (Song et al. 2009; Palmer et al. 2011; Ranieri et al. 2013) Mitochondrial fission depends largely on dynamin related protein 1 (Drp1), which promotes fission by tethering to mitochondria at specific positions known as constriction sites and then forming multimeric spirals around mitochondria further constricting mitochondrial tubules leading to fission/fragmentation. In human cells, it has been reported that fission 1 protein (hFis1), present in the outer membrane, is involved in recruiting cytosolic Drp1 to constriction sites (Smirnova et al. 2001; James et al. 2003; Lee et al. 2004; Sheridan and Martin 2010). We recently reported that Mfn2 expression is reduced, whereas Drp1 is increased in hASM from patients with moderate asthma (Aravamudan et al. 2014). As a result, we also found that mitochondrial fragmentation is increased in asthmatic hASM. Recent evidence has suggested that Mfn2 and dynamic mitochondrial remodeling (balance between fusion and fission) are involved in pro-proliferative pathways (Liesa et al. 2009; Antico Arciuch et al. 2012).
Fig. 9.
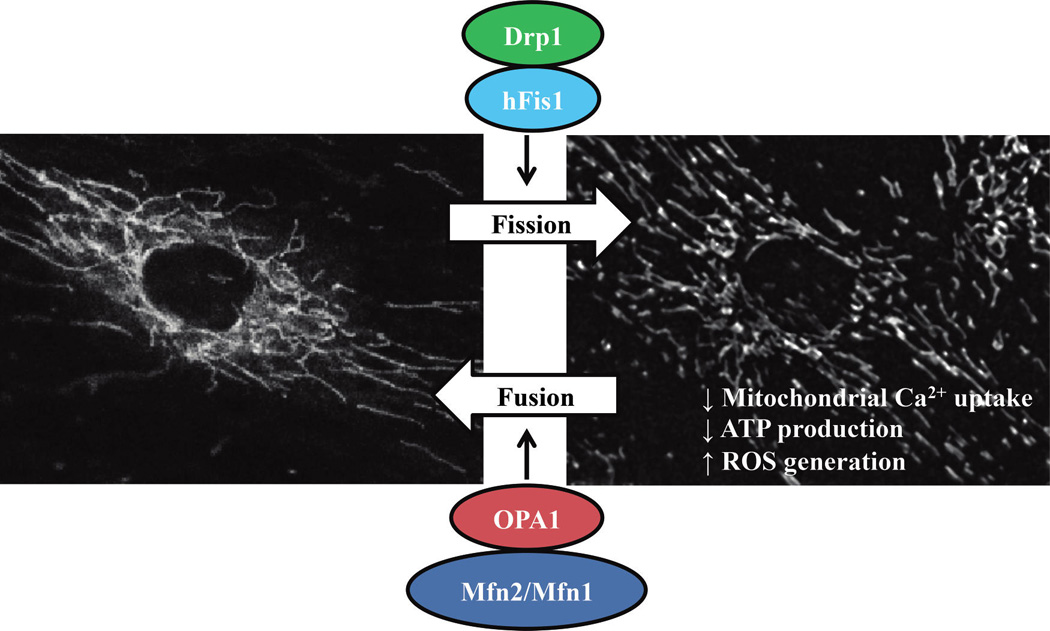
Human ASM cells were loaded with 500 nmol·L−1 MitoTraker Green AM to visualize the mitochondrial network. Control hASM cells displayed a filamentous/elongated mitochondrial network (fusion), which is promoted by Mfn1, Mfn2, and OPA1 proteins. In contrast, after 24 h exposure to TNFα (20 ng/mL for 12 h), mitochondria in hASM cells were more fragmented (fission). Mitochondria fragmentation (fission) is promoted by Drp1 and hFis1 proteins, and is associated with reduced mitochondrial Ca2+ uptake, ATP production, and increased ROS generation. ASM, airway smooth muscle; ROS, reactive oxidant species; Mfn, mitofusin; Drp1, dynamin related protein 1; OPA1, optic atrophy protein 1.
Cytokine-induced mitochondrial fragmentation and cell proliferation in ASM
In the most fundamental sense, mitochondrial fragmentation and elongation is required for mitochondrial biogenesis to ensure that mitochondrial number is maintained subsequent to cell division (Antico Arciuch et al. 2012). Interestingly, studies have shown that mitochondrial biogenesis is enhanced in asthmatic ASM (Trian et al. 2007; Girodet et al. 2011); however, these studies did not examine mitochondrial network morphology. As a first step in mitochondrial biogenesis, fragmentation may reflect or induce an increased proliferative state of the cell. Indeed, inducing mitochondrial fragmentation leads to cell proliferation (Liesa et al. 2009; Antico Arciuch et al. 2012). Recent studies have suggested that in vascular smooth muscle, Mfn2 plays a regulatory role in cell division (Liesa et al. 2009; Antico Arciuch et al. 2012). In vascular smooth muscle, Mfn2 overexpression leads to cell growth arrest (Liesa et al. 2009). In addition, Mfn2 may inhibit the activation of pro-proliferative kinases such as extracellular signal-regulated kinase (ERK1/2) (Liesa et al. 2009). Importantly, many studies suggest that ERK1/2 activation plays a critical role in mediating ASM proliferation (Lee et al. 2001; Yu et al. 2013; Dragon et al. 2014;Movassagh et al. 2014).
ER/SR stress in ASM provides potential therapeutic targets for asthma
There is a growing amount of evidence to indicate that inflammation/ROS-induced ER/SR stress is a major contributor in the pathophysiology of various diseases, now including asthma. Recently, ER/SR stress markers have been identified in lungs from mouse models of airway diseases and chemical chaperones. Inhibition of ER/SR stress responses successfully reduced airway inflammation and airway hyper-reactivity observed in those models (Hoffman et al. 2013; Kim et al. 2013; Makhija et al. 2013). The results of our studies on hASM cells support a central role of inflammatory cytokine-induced ER/SR stress in both the enhanced contractile response of ASM (hyper-reactive state) and the increase in ASM cell proliferation (synthetic state) (Fig. 10). The functional impact of ER/SR stress in ASM cells is mediated in large part by the reduced expression of Mfn2. Thus, these results suggest potential therapeutic target in asthma that include: (i) ROS scavengers, (ii) chemical chaperones, and (iii) gene therapy to increase Mfn2 expression.
Fig. 10.
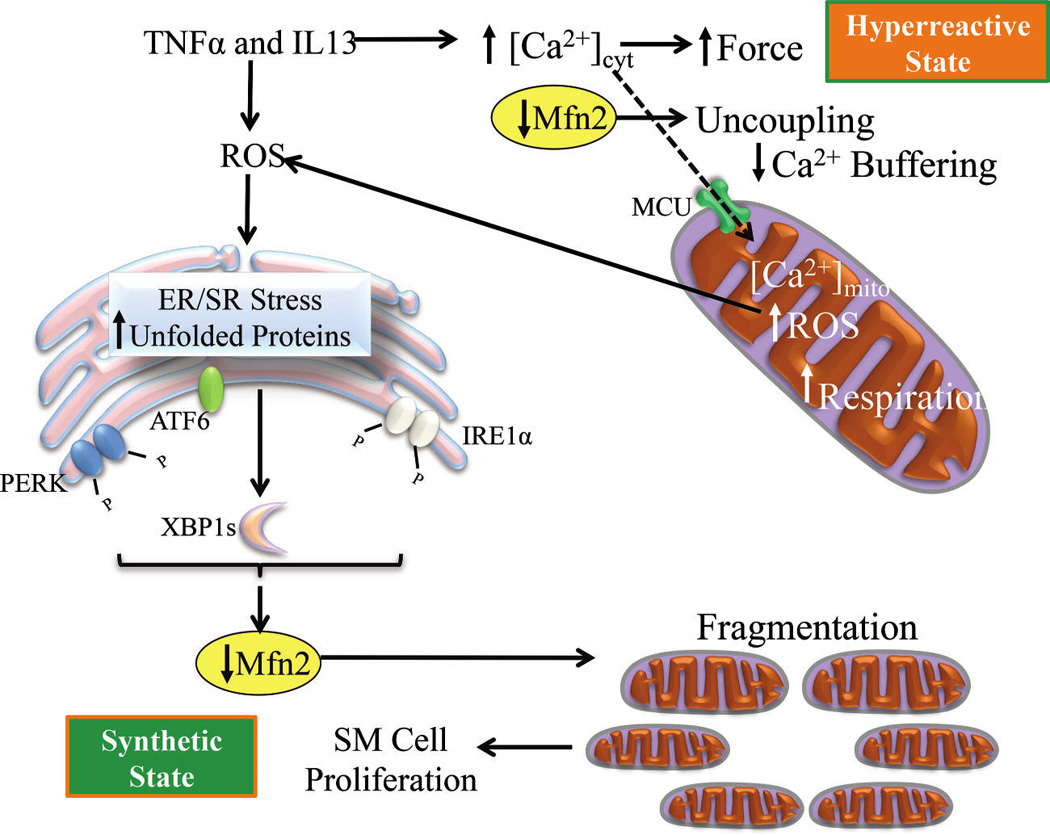
Inflammatory cytokines induce ROS generation in hASM that leads to ER/SR stress with downstream impact of reduced Mfn2 expression (increased Drp1), uncoupling of mitochondria to the ER/SR membrane, decreased [Ca2+]mito buffering, which contributes to increased [Ca2+]cyt and force responses to ACh stimulation (hyper-reactive state). A decrease in [Ca2+]mito buffering also leads to mitochondrial dysfunction and a further increase in ROS generation. Reduced Mfn2 expression also leads to mitochondrial fragmentation (fission) and increased cell proliferation and remodeling (synthetic state). ROS, reactive oxidant species; hASM, human airway smooth muscle cells; ER/SR, endoplasmic reticulum/sarcoplasmic reticulum; ACh, acetycholine; Mfn2, mitofusin-2; Drp1, dynamin related protein 1.
Acknowledgements
This work was supported by a grant from the US National Institutes of Health (No. HL74309, to G.C. Sieck).
Footnotes
This Invited Review is part of a Special Issue entitled “Smooth muscle and asthma: a tribute to Newman Stephens.”
References
- Adolph TE, Niederreiter L, Blumberg RS, Kaser A. Endoplasmic reticulum stress and inflammation. Dig. Dis. 2012;30(4):341–346. doi: 10.1159/000338121. PMID: 22796794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali F, Chin L, Pare PD, Seow CY. Mechanism of partial adaptation in airway smooth muscle after a step change in length. J. Appl. Physiol. (1985) 2007;103(2):569–577. doi: 10.1152/japplphysiol.00216.2007. PMID: 17495118. [DOI] [PubMed] [Google Scholar]
- Amrani Y. Modulation of airway smooth muscle contractile function by TNFα and IL-13 and airway hyperresponsiveness in asthma. In: Wang Y-X, editor. Calcium signaling in airway smooth muscle cells. Springer International Publishing; 2014. pp. 423–439. [Google Scholar]
- Amrani Y, Panettieri RA., Jr Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr. Opin. Allergy Clin. Immunol. 2002;2(1):39–45. doi: 10.1097/00130832-200202000-00007. PMID: 11964749. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Chen H, Panettieri RA., Jr Activation of tumor necrosis factor receptor 1 in airway smooth muscle: a potential pathway that modulates bronchial hyper-responsiveness in asthma? Respir. Res. 2000;1(1):49–53. doi: 10.1186/rr12. PMID: 11667965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta. 2006;1757(5–6):692–699. doi: 10.1016/j.bbabio.2006.04.013. PMID: 16729962. [DOI] [PubMed] [Google Scholar]
- Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 2012;16(10):1150–1180. doi: 10.1089/ars.2011.4085. PMID: 21967640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravamudan B, VanOosten SK, Meuchel LW, Vohra P, Thompson M, Sieck GC, et al. Caveolin-1 knockout mice exhibit airway hyperreactivity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012;303(8):L669–L681. doi: 10.1152/ajplung.00018.2012. PMID: 22923642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, et al. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;306(9):L840–L854. doi: 10.1152/ajplung.00155.2013. PMID: 24610934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay B, Prakash YS, Pabelick CM, Sieck GC. Store-operated Ca2+ entry in porcine airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286(5):L909–L917. doi: 10.1152/ajplung.00317.2003. PMID: 14617522. [DOI] [PubMed] [Google Scholar]
- Ay B, Iyanoye A, Sieck GC, Prakash YS, Pabelick CM. Cyclic nucleotide regulation of store-operated Ca2+ influx in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290(2):L278–L283. doi: 10.1152/ajplung.00188.2005. PMID: 16155088. [DOI] [PubMed] [Google Scholar]
- Baban B, Liu JY, Mozaffari MS. Endoplasmic reticulum stress response and inflammatory cytokines in type 2 diabetic nephropathy: Role of indoleamine 2,3-dioxygenase and programmed death-1. Exp. Mol. Pathol. 2012;94(2):343–351. doi: 10.1016/j.yexmp.2012.11.004. PMID: 23219834. [DOI] [PubMed] [Google Scholar]
- Bai Y, Edelmann M, Sanderson MJ. The contribution of inositol 1,4,5-trisphosphate and ryanodine receptors to agonist-induced Ca2+ signaling of airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297(2):L347–L361. doi: 10.1152/ajplung.90559.2008. PMID: 19465516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345. doi: 10.1038/nature10234. PMID: 21685886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JL, Panettieri RA, Jr, Banerjee A, Berger P. Airway smooth muscle in asthma: just a target for bronchodilation? Clin. Chest Med. 2012;33(3):543–558. doi: 10.1016/j.ccm.2012.05.002. PMID: 22929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldogh IR, Pon LA. Mitochondria on the move. Trends Cell Biol. 2007;17(10):502–510. doi: 10.1016/j.tcb.2007.07.008. PMID: 17804238. [DOI] [PubMed] [Google Scholar]
- Bravo R, Gutierrez T, Paredes F, Gatica D, Rodriguez AE, Pedrozo Z, et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int. J. Biochem. Cell Biol. 2012;44(1):16–20. doi: 10.1016/j.biocel.2011.10.012. PMID: 22064245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love–hate triangle. Am. J. Physiol. Cell Physiol. 2004;287(4):C817–C833. doi: 10.1152/ajpcell.00139.2004. PMID: 15355853. [DOI] [PubMed] [Google Scholar]
- Brough D, Schell MJ, Irvine RF. Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem J. 2005;392(2):291–297. doi: 10.1042/BJ20050738. PMID: 15982187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers S, McCarron JG. The mitochondrial membrane potential and Ca2+ oscillations in smooth muscle. J. Cell Sci. 2008;121(1):75–85. doi: 10.1242/jcs.014522. PMID: 18073239. [DOI] [PubMed] [Google Scholar]
- Chalmers S, McCarron JG. Inhibition of mitochondrial calcium uptake rather than efflux impedes calcium release by inositol-1,4,5-trisphosphatesensitive receptors. Cell Calcium. 2009;46(2):107–113. doi: 10.1016/j.ceca.2009.05.007. PMID: 19577805. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006a;125(7):1241–1252. doi: 10.1016/j.cell.2006.06.010. PMID: 16814712. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006a;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. PMID: 16704336. [DOI] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. PMID: 22934639. [DOI] [PubMed] [Google Scholar]
- Chen H, Tliba O, Van Besien CR, Panettieri RA, Jr, Amrani Y. TNF-α modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J. Appl. Physiol. 2003;95(2):864–872. doi: 10.1152/japplphysiol.00140.2003. PMID: 12730147. [DOI] [PubMed] [Google Scholar]
- Chen T, Zhu L, Wang T, Ye H, Huang K, Hu Q. Mitochondria depletion abolishes agonist-induced Ca2+ plateau in airway smooth muscle cells: potential role of H2O2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;298(2):L178–L188. doi: 10.1152/ajplung.00134.2009. PMID: 19933400. [DOI] [PubMed] [Google Scholar]
- Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J. Physiol. 2004;556(3):755–771. doi: 10.1113/jphysiol.2003.059568. PMID: 14766935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ, Lipp P, Berridge MJ, Li W, Bootman MD. Inositol 1,4,5-trisphosphate-induced Ca2+ release is inhibited by mitochondrial depolarization. Biochem J. 2000;347(2):593–600. doi: 10.1042/0264-6021:3470593. PMID: 10749691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comhair SA, Erzurum SC. Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2010;12(1):93–124. doi: 10.1089/ars.2008.2425. PMID: 19634987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006;174(7):915–921. doi: 10.1083/jcb.200604016. PMID: 16982799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Varnai P, Golenar T, Sheu SS, Hajnoczky G. Calcium transport across the inner mitochondrial membrane: Molecular mechanisms and pharmacology. Mol. Cell. Endocrinol. 2012;353(1–2):109–113. doi: 10.1016/j.mce.2011.11.011. PMID: 22123069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dada LA, Sznajder JI. Mitochondrial Ca2+ and ROS take center stage to orchestrate TNF-alpha-mediated inflammatory responses. J. Clin. Invest. 2011;121(5):1683–1685. doi: 10.1172/JCI57748. PMID: 21519140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008a;456(7222):605–610. doi: 10.1038/nature07534. PMID: 19052620. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid. Redox Signal. 2008b;10(3):621–633. doi: 10.1089/ars.2007.1934. PMID: 18092941. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. An intimate liaison: spatial organization of the endoplasmic reticulum–mitochondria relationship. EMBO J. 2010;29(16):2715–2723. doi: 10.1038/emboj.2010.177. PMID: 20717141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A fortykilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. doi: 10.1038/nature10230. PMID: 21685888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmotte P, Yang B, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. Inflammation alters regional mitochondrial Ca2+ in human airway smooth muscle cells. Am. J. Physiol. Cell Physiol. 2012;303(3):C244–C256. doi: 10.1152/ajpcell.00414.2011. PMID: 22673614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmotte P, Jia L, Sieck G. The role of mitochondria in calcium regulation in airway smooth muscle. In: Wang Y-X, editor. Calcium signaling in airway smooth muscle cells. Springer International Publishing; 2014. pp. 211–234. [Google Scholar]
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta. 2009;1787(11):1309–1316. doi: 10.1016/j.bbabio.2009.01.005. PMID: 19413950. [DOI] [PubMed] [Google Scholar]
- Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu. Rev. Physiol. 1990;52:451–466. doi: 10.1146/annurev.ph.52.030190.002315. PMID: 2184763. [DOI] [PubMed] [Google Scholar]
- Dorn GW, II, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 2013;55:42–49. doi: 10.1016/j.yjmcc.2012.07.015. PMID: 22902320. [DOI] [PubMed] [Google Scholar]
- Dragon S, Hirst SJ, Lee TH, Gounni AS. Interleukin-17A mediates a selective gene expression profile in asthmatic human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2014;50(6):1053–1063. doi: 10.1165/rcmb.2012-0267OC. PMID: 24393021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dromparis P, Paulin R, Stenson TH, Haromy A, Sutendra G, Michelakis ED. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation. 2013;127(1):115–125. doi: 10.1161/CIRCULATIONAHA.112.133413. PMID: 23149668. [DOI] [PubMed] [Google Scholar]
- Favero TG, Zable AC, Abramson JJ. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1995;270(43):25557–25563. doi: 10.1074/jbc.270.43.25557. PMID: 7592726. [DOI] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007;87(2):593–658. doi: 10.1152/physrev.00035.2006. PMID: 17429043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int. J. Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. PMID: 20182529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol. Med. 2012;18(10):589–598. doi: 10.1016/j.molmed.2012.06.010. PMID: 22883813. [DOI] [PubMed] [Google Scholar]
- Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, et al. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM) Cell Stress Chaperones. 2010;15(5):619–629. doi: 10.1007/s12192-010-0174-1. PMID: 20186508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GINA (Global Strategy for Asthma Management and Prevention) GINA Report, Global Strategy for Asthma Management and Prevention. [Accessed 9 January 2014];2012 Available from www.ginasthma.org. [Google Scholar]
- Girodet PO, Ozier A, Bara I, Tunon de Lara JM, Marthan R, Berger P. Airway remodeling in asthma: New mechanisms and potential for pharmacological intervention. Pharmacol. Ther. 2011;130(3):325–337. doi: 10.1016/j.pharmthera.2011.02.001. PMID: 21334378. [DOI] [PubMed] [Google Scholar]
- Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51(14):2959–2973. doi: 10.1021/bi2018909. PMID: 22443365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta. 2009;1787(11):1324–1333. doi: 10.1016/j.bbabio.2009.01.019. PMID: 19366607. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Tang DD. The contractile apparatus and mechanical properties of airway smooth muscle. Eur. Respir. J. 2000;15(3):600–616. doi: 10.1034/j.1399-3003.2000.15.29.x. PMID: 10759460. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am. J. Physiol. Cell Physiol. 2008;295(3):C576–C587. doi: 10.1152/ajpcell.00253.2008. PMID: 18596210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunst SJ, Tang DD, Opazo Saez A. Cytoskeletal remodeling of the airway smooth muscle cell: a mechanism for adaptation to mechanical forces in the lung. Respir. Physiol. Neurobiol. 2003;137(2–3):151–168. doi: 10.1016/s1569-9048(03)00144-7. PMID: 14516723. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim. Biophys. Acta. 2009;1787(11):1291–1308. doi: 10.1016/j.bbabio.2008.12.011. PMID: 19161975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai CM, Murphy RA. Cross-bridge phosphorylation and regulation of latch state in smooth muscle. Am. J. Physiol. 1988;254(1):C99–C106. doi: 10.1152/ajpcell.1988.254.1.C99. PMID: 3337223. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Yi M. Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium. 2002;32(5–6):363–377. doi: 10.1016/s0143416002001872. PMID: 12543096. [DOI] [PubMed] [Google Scholar]
- Hasnain SZ, Lourie R, Das I, Chen AC, McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 2012a;90(3):260–270. doi: 10.1038/icb.2011.112. PMID: 22249202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasnain SZ, Tauro S, Das I, Tong H, Chen AC, Jeffery PL, et al. IL-10 promotes production of intestinal mucus by suppressing protein misfolding and endoplasmic reticulum stress in goblet cells. Gastroenterology. 2012b;144(2):357–368. doi: 10.1053/j.gastro.2012.10.043. PMID: 23123183. [DOI] [PubMed] [Google Scholar]
- Herrera AM, Martinez EC, Seow CY. Electron microscopic study of actin polymerization in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286(6):L1161–L1168. doi: 10.1152/ajplung.00298.2003. PMID: 14751850. [DOI] [PubMed] [Google Scholar]
- Hodgkinson JL, Newman TM, Marston SB, Severs NJ. The structure of the contractile apparatus in ultrarapidly frozen smooth muscle: freeze-fracture, deep-etch, and freeze-substitution studies. J. Struct. Biol. 1995;114(2):93–104. doi: 10.1006/jsbi.1995.1009. PMID: 7612400. [DOI] [PubMed] [Google Scholar]
- Hoffman SM, Tully JE, Nolin JD, Lahue KG, Goldman DH, Daphtary N, et al. Endoplasmic reticulum stress mediates house dust mite-induced airway epithelial apoptosis and fibrosis. Respir. Res. 2013;14:141. doi: 10.1186/1465-9921-14-141. PMID: 24364984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter I, Cobban HJ, Vandenabeele P, MacEwan DJ, Nixon GF. Tumor necrosis factor-alpha-induced activation of RhoA in airway smooth muscle cells: role in the Ca2+ sensitization of myosin light chain20 phosphorylation. Mol. Pharmacol. 2003;63(3):714–721. doi: 10.1124/mol.63.3.714. PMID: 12606782. [DOI] [PubMed] [Google Scholar]
- Ijpma G, Al-Jumaily AM, Cairns SP, Sieck GC. Myosin filament polymerization and depolymerization in a model of partial length adaptation in airway smooth muscle. J. Appl. Physiol. 2011;111(3):735–742. doi: 10.1152/japplphysiol.00114.2011. PMID: 21659490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Riesinger I, Wallimann T, Paul RJ. Compartmentation of ATP synthesis and utilization in smooth muscle: roles of aerobic glycolysis and creatine kinase. Mol. Cell. Biochem. 1994;133–134:39–50. doi: 10.1007/BF01267946. PMID: 7808464. [DOI] [PubMed] [Google Scholar]
- Ishii K, Hirose K, Iino M. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep. 2006;7(4):390–396. doi: 10.1038/sj.embor.7400620. PMID: 16415789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James A. Airway remodeling in asthma. Curr. Opin. Pulm. Med. 2005;11(1):1–6. doi: 10.1097/01.mcp.0000146779.26339.d8. PMID: 15591881. [DOI] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003;278(38):36373–36379. doi: 10.1074/jbc.M303758200. PMID: 12783892. [DOI] [PubMed] [Google Scholar]
- Jia L, Delmotte P, Aravamudan B, Pabelick CM, Prakash YS, Sieck GC. Effects of the inflammatory cytokines TNF-alpha and IL-13 on stromal interaction molecule-1 aggregation in human airway smooth muscle intracellular Ca2+ regulation. Am. J. Respir. Cell Mol. Biol. 2013;49(4):601–608. doi: 10.1165/rcmb.2013-0040OC. PMID: 23713409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Lorenz RR, Prakash YS, Sieck GC, Warner DO. ATP hydrolysis during contraction of permeabilized airway smooth muscle. Am. J. Physiol. 1999a;277(2):L334–L342. doi: 10.1152/ajplung.1999.277.2.L334. PMID: 10444528. [DOI] [PubMed] [Google Scholar]
- Jones KA, Perkins WJ, Lorenz RR, Prakash YS, Sieck GC, Warner DO. F-actin stabilization increases tension cost during contraction of permeabilized airway smooth muscle in dogs. J. Physiol. (Lond.) 1999b;519(2):527–538. doi: 10.1111/j.1469-7793.1999.0527m.x. PMID: 10457068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert P, Hamid Q. Role of airway smooth muscle in airway remodeling. J. Allergy Clin. Immunol. 2005;116(3):713–716. doi: 10.1016/j.jaci.2005.05.042. PMID: 16159653. [DOI] [PubMed] [Google Scholar]
- Jude JA, Wylam ME, Walseth TF, Kannan MS. Calcium signaling in airway smooth muscle. Proc. Am. Thorac. Soc. 2008;5(1):15–22. doi: 10.1513/pats.200704-047VS. PMID: 18094080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan MS, Fenton AM, Prakash YS, Sieck GC. Cyclic ADP-ribose stimulates sarcoplasmic reticulum calcium release in porcine coronary artery smooth muscle. Am. J. Physiol. 1996;270(2):H801–H806. doi: 10.1152/ajpheart.1996.270.2.H801. PMID: 8779859. [DOI] [PubMed] [Google Scholar]
- Kannan MS, Prakash YS, Brenner T, Mickelson JR, Sieck GC. Role of ryanodine receptor channels in Ca2+ oscillations of porcine tracheal smooth muscle. Am. J. Physiol. 1997;272(4):L659–L664. doi: 10.1152/ajplung.1997.272.4.L659. PMID: 9142939. [DOI] [PubMed] [Google Scholar]
- Katsumata U, Miura M, Ichinose M, Kimura K, Takahashi T, Inoue H, et al. Oxygen radicals produce airway constriction and hyperresponsiveness in anesthetized cats. Am.Rev. Respir. Dis. 1990;141(5):1158–1161. doi: 10.1164/ajrccm/141.5_Pt_1.1158. PMID: 2131784. [DOI] [PubMed] [Google Scholar]
- Kim SR, Kim DI, Kang MR, Lee KS, Park SY, Jeong JS, et al. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor kappaB activation. J. Allergy Clin. Immunol. 2013;132(6):1397–1408. doi: 10.1016/j.jaci.2013.08.041. PMID: 24161747. [DOI] [PubMed] [Google Scholar]
- Kiviluoto S, Vervliet T, Ivanova H, Decuypere JP, De Smedt H, Missiaen L, et al. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta. 2013;1833(7):1612–1624. doi: 10.1016/j.bbamcr.2013.01.026. PMID: 23380704. [DOI] [PubMed] [Google Scholar]
- Kornmann B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013;25(4):443–448. doi: 10.1016/j.ceb.2013.02.010. PMID: 23478213. [DOI] [PubMed] [Google Scholar]
- Krisanda JM, Paul RJ. Phosphagen and metabolite content during contraction in porcine carotid artery. Am. J. Physiol. 1983;244(5):C385–C390. doi: 10.1152/ajpcell.1983.244.5.C385. PMID: 6846527. [DOI] [PubMed] [Google Scholar]
- Kuo KH, Wang L, Pare PD, Ford LE, Seow CY. Myosin thick filament lability induced by mechanical strain in airway smooth muscle. J. Appl. Physiol. 2001;90(5):1811–1816. doi: 10.1152/jappl.2001.90.5.1811. (1985), PMID: 11299271. [DOI] [PubMed] [Google Scholar]
- Kuo KH, Herrera AM, Seow CY. Ultrastructure of airway smooth muscle. Respir. Physiol. Neurobiol. 2003a;137(2–3):197–208. doi: 10.1016/s1569-9048(03)00147-2. PMID: 14516726. [DOI] [PubMed] [Google Scholar]
- Kuo KH, Herrera AM, Wang L, Pare PD, Ford LE, Stephens NL, et al. Structure–function correlation in airway smooth muscle adapted to different lengths. Am. J. Physiol. Cell Physiol. 2003b;285(2):C384–C390. doi: 10.1152/ajpcell.00095.2003. PMID: 12700138. [DOI] [PubMed] [Google Scholar]
- Lee JH, Johnson PR, Roth M, Hunt NH, Black JL. ERK activation and mitogenesis in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;280(5):L1019–L1029. doi: 10.1152/ajplung.2001.280.5.L1019. PMID: 11290527. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell. 2004;15(11):5001–5011. doi: 10.1091/mbc.E04-04-0294. PMID: 15356267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009;89(3):799–845. doi: 10.1152/physrev.00030.2008. PMID: 19584314. [DOI] [PubMed] [Google Scholar]
- Lin MJ, Yang XR, Cao YN, Sham JS. Hydrogen peroxide-induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292(6):L1598–L1608. doi: 10.1152/ajplung.00323.2006. PMID: 17369291. [DOI] [PubMed] [Google Scholar]
- Makhija L, Krishnan V, Rehman R, Chakraborty S, Maity S, Mabalirajan U, et al. Chemical chaperones mitigate experimental asthma by attenuating endoplasmic reticulum stress. Am. J. Respir. Cell Mol. Biol. 2013;50(5):923–931. doi: 10.1165/rcmb.2013-0320OC. PMID: 24299608. [DOI] [PubMed] [Google Scholar]
- Malaiyandi LM, Honick AS, Rintoul GL, Wang QJ, Reynolds IJ. Zn2+ inhibits mitochondrial movement in neurons by phosphatidylinositol 3-kinase activation. J. Neurosci. 2005;25(41):9507–9514. doi: 10.1523/JNEUROSCI.0868-05.2005. PMID: 16221861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malli R, Frieden M, Osibow K, Zoratti C, Mayer M, Demaurex N, et al. Sustained Ca2+ transfer across mitochondria is essential for mitochondrial Ca2+ buffering, store-operated Ca2+ entry, and Ca2+ store refilling. J. Biol. Chem. 2003;278(45):44769–44779. doi: 10.1074/jbc.M302511200. PMID: 12941956. [DOI] [PubMed] [Google Scholar]
- Martino MB, Jones L, Brighton B, Ehre C, Abdulah L, Davis CW, et al. The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol. 2012;6:639–654. doi: 10.1038/mi.2012.105. PMID: 23168839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H. Effects of inflammatory cytokines on Ca2+ homeostasis in airway smooth muscle cells. In: Wang Y-X, editor. Calcium signaling in airway smooth muscle cells. Springer International Publishing; 2014. pp. 359–379. [Google Scholar]
- Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J. Physiol. 1999;519(3):829–840. doi: 10.1111/j.1469-7793.1999.0829n.x. PMID: 10457094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L, Zheng Y-M, Wang Y-X. Ryanodine and inositol trisphosphate receptors/Ca2+ release channels in airway smooth muscle cells. In: Wang Y-X, editor. Calcium signaling in airway smooth muscle cells. Springer International Publishing; 2014. pp. 1–20. [Google Scholar]
- Movassagh H, Shan L, Halayko AJ, Roth M, Tamm M, Chakir J, et al. Neuronal chemorepellent semaphorin 3E inhibits human airway smooth muscle cell proliferation and migration. J. Allergy Clin. Immunol. 2014;133(2):560–567. doi: 10.1016/j.jaci.2013.06.011. PMID: 23932461. [DOI] [PubMed] [Google Scholar]
- Munoz JP, Zorzano A. Endoplasmic reticulum stress enters a nogo zone. Sci. Transl. Med. 2011;3(88):88ps26. doi: 10.1126/scitranslmed.3002708. PMID: 21697529. [DOI] [PubMed] [Google Scholar]
- Munoz JP, Ivanova S, Sanchez-Wandelmer J, Martinez-Cristobal P, Noguera E, Sancho A, et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013;32(17):2348–2361. doi: 10.1038/emboj.2013.168. PMID: 23921556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RA, Rembold CM. The latch-bridge hypothesis of smooth muscle contraction. Can. J. Physiol. Pharmacol. 2005;83(10):857–864. doi: 10.1139/y05-090. PMID: 16333357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Stojanovski D, Ryan MT. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell. Signal. 2011;23(10):1534–1545. doi: 10.1016/j.cellsig.2011.05.021. PMID: 21683788. [DOI] [PubMed] [Google Scholar]
- Parris JR, Cobban HJ, Littlejohn AF, MacEwan DJ, Nixon GF. Tumour necrosis factor-alpha activates a calcium sensitization pathway in guinea-pig bronchial smooth muscle. J. Physiol. 1999;518(2):561–569. doi: 10.1111/j.1469-7793.1999.0561p.x. PMID: 10381600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul RJ. Smooth muscle energetics. Annu. Rev. Physiol. 1989;51:331–349. doi: 10.1146/annurev.ph.51.030189.001555. PMID: 2653187. [DOI] [PubMed] [Google Scholar]
- Perez-Vizcaino F, Cogolludo A, Moreno L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir. Physiol. Neurobiol. 2010;174(3):212–220. doi: 10.1016/j.resp.2010.08.009. PMID: 20797450. [DOI] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature. 2010;467(7313):291–296. doi: 10.1038/nature09358. PMID: 20693986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo P, Drago I, Filadi R, Pozzan T. Mitochondrial Ca2+ homeostasis: mechanism, role, and tissue specificities. Pflugers Arch. 2012;464(1):3–17. doi: 10.1007/s00424-012-1122-y. PMID: 22706634. [DOI] [PubMed] [Google Scholar]
- Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am. J. Physiol. Lung Cell. Mol. Physiol. 2013;305(12):L912–L933. doi: 10.1152/ajplung.00259.2013. PMID: 24142517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash YS, Kannan MS, Sieck GC. Regulation of intracellular calcium oscillations in porcine tracheal smooth muscle cells. Am. J. Physiol. 1997;272(3):C966–C975. doi: 10.1152/ajpcell.1997.272.3.C966. PMID: 9124533. [DOI] [PubMed] [Google Scholar]
- Prakash YS, Kannan MS, Walseth TF, Sieck GC. Role of cyclic ADP-ribose in the regulation of [Ca2+]i in porcine tracheal smooth muscle. Am. J. Physiol. 1998;274(6):C1653–C1660. doi: 10.1152/ajpcell.1998.274.6.C1653. PMID: 9611131. [DOI] [PubMed] [Google Scholar]
- Prakash YS, Pabelick CM, Kannan MS, Sieck GC. Spatial and temporal aspects of ACh-induced [Ca2+]i oscillations in porcine tracheal smooth muscle. Cell Calcium. 2000;27(3):153–162. doi: 10.1054/ceca.1999.0106. PMID: 11007128. [DOI] [PubMed] [Google Scholar]
- Prakash YS, Sathish V, Thompson MA, Pabelick CM, Sieck GC. Asthma and sarcoplasmic reticulum Ca2+ reuptake in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297(4):L794. doi: 10.1152/ajplung.00237.2009. PMID: 19783641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Rizzuto R. The mitochondrial Ca2+ uniporter. Cell Calcium. 2012;52(1):16–21. doi: 10.1016/j.ceca.2012.04.006. PMID: 22672876. [DOI] [PubMed] [Google Scholar]
- Rainbolt TK, Saunders JM, Wiseman RL. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014;25(10):528–537. doi: 10.1016/j.tem.2014.06.007. PMID: 25048297. [DOI] [PubMed] [Google Scholar]
- Ranieri M, Brajkovic S, Riboldi G, Ronchi D, Rizzo F, Bresolin N, et al. Mitochondrial fusion proteins and human diseases. Neurol. Res. Int. 2013;2013:293893. doi: 10.1155/2013/293893. PMID: 23781337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM) Biochim. Biophys. Acta. 2013;1833(1):213–224. doi: 10.1016/j.bbamcr.2012.04.013. PMID: 22575682. [DOI] [PubMed] [Google Scholar]
- Reynolds AM, Holmes MD, Scicchitano R. Cytokines enhance airway smooth muscle contractility in response to acetylcholine and neurokinin A. Respirology. 2000;5(2):153–160. doi: 10.1046/j.1440-1843.2000.00240.x. PMID: 10894105. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 2006;86(1):369–408. doi: 10.1152/physrev.00004.2005. PMID: 16371601. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262(5134):744–747. doi: 10.1126/science.8235595. PMID: 8235595. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci. STKE. 2004;2004(215):re1. doi: 10.1126/stke.2152004re1. PMID: 14722345. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, et al. Ca2+ transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta. 2009;1787(11):1342–1351. doi: 10.1016/j.bbabio.2009.03.015. PMID: 19341702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli A, Aguiari P, De Stefani D, Leo S, Marchi S, Rimessi A, et al. Endoplasmic reticulum/mitochondria calcium cross-talk. Novartis Found. Symp. 2007;287:122–131. discussion 131–129. PMID: 18074635. [PubMed] [Google Scholar]
- Sakai H, Otogoto S, Chiba Y, Abe K, Misawa M. TNF-alpha augments the expression of RhoA in the rat bronchus. J. Smooth Muscle Res. 2004;40(1):25–34. doi: 10.1540/jsmr.40.25. PMID: 15170075. [DOI] [PubMed] [Google Scholar]
- Samali A, Fitzgerald U, Deegan S, Gupta S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010;2010:830307. doi: 10.1155/2010/830307. PMID: 20169136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MJ, Delmotte P, Bai Y, Perez-Zogbhi JF. Regulation of airway smooth muscle cell contractility by Ca2+ signaling and sensitivity. Proc. Am. Thorac. Soc. 2008;5(1):23–31. doi: 10.1513/pats.200704-050VS. PMID: 18094081. [DOI] [PubMed] [Google Scholar]
- Sathish V, Leblebici F, Kip SN, Thompson MA, Pabelick CM, Prakash YS, et al. Regulation of sarcoplasmic reticulum Ca2+ reuptake in porcine airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;294(4):L787–L796. doi: 10.1152/ajplung.00461.2007. PMID: 18245264. [DOI] [PubMed] [Google Scholar]
- Sathish V, Thompson MA, Bailey JP, Pabelick CM, Prakash YS, Sieck GC. Effect of proinflammatory cytokines on regulation of sarcoplasmic reticulum Ca2+ reuptake in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297(1):L26–L34. doi: 10.1152/ajplung.00026.2009. PMID: 19395670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathish V, Delmotte PF, Thompson MA, Pabelick CM, Sieck GC, Prakash YS. Sodium-calcium exchange in intracellular calcium handling of human airway smooth muscle. PLoS One. 2011a;6(8):e23662. doi: 10.1371/journal.pone.0023662. PMID: 21858195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathish V, Yang B, Meuchel LW, VanOosten SK, Ryu AJ, Thompson MA, et al. Caveolin-1 and force regulation in porcine airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011b;300(6):L920–L929. doi: 10.1152/ajplung.00322.2010. PMID: 21421751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathish V, Abcejo AJ, Thompson MA, Sieck GC, Prakash YS, Pabelick CM. Caveolin-1 regulation of store-operated Ca2+ influx in human airway smooth muscle. Eur. Respir. J. 2012;40(2):470–478. doi: 10.1183/09031936.00090511. PMID: 22241747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible N, Delmotte P, Sieck G. Mitochondrial excitation–energy coupling in airway smooth muscle. In: Natarajan V, Parinandi NL, editors. Mitochondrial function in lung health and disease. Totowa, N.J.: Humana Press Inc.; 2014. pp. 96–116. [Google Scholar]
- Seow CY. Myosin filament assembly in an ever-changing myofilament lattice of smooth muscle. Am. J. Physiol. Cell Physiol. 2005;289(6):C1363–C1368. doi: 10.1152/ajpcell.00329.2005. PMID: 16275736. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion. 2010;10(6):640–648. doi: 10.1016/j.mito.2010.08.005. PMID: 20727425. [DOI] [PubMed] [Google Scholar]
- Sieck GC, Gransee HM. Colloquium series on integrated systems physiology, from molecule to function to disease #34. 3. Vol. 4. San Rafael, Calif.: Morgan & Claypool Life Sciences; 2012. Respiratory muscles structure, function & regulation; pp. 1–96. [Google Scholar]
- Sieck GC, Kannan MS, Prakash YS. Heterogeneity in dynamic regulation of intracellular calcium in airway smooth muscle cells. Can. J. Physiol. Pharmacol. 1997;75(7):878–888. PMID: 9315357. [PubMed] [Google Scholar]
- Sieck GC, Han YS, Prakash YS, Jones KA. Cross-bridge cycling kinetics, actomyosin ATPase activity and myosin heavy chain isoforms in skeletal and smooth respiratory muscles. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998;119(3):435–450. doi: 10.1016/s0305-0491(98)00005-4. PMID: 9734328. [DOI] [PubMed] [Google Scholar]
- Sieck GC, Han YS, Pabelick CM, Prakash YS. Temporal aspects of excitation–contraction coupling in airway smooth muscle. J. Appl. Physiol. 2001;91(5):2266–2274. doi: 10.1152/jappl.2001.91.5.2266. PMID: 11641370. [DOI] [PubMed] [Google Scholar]
- Sieck GC, White TA, Thompson MA, Pabelick CM, Wylam ME, Prakash YS. Regulation of store-operated Ca2+ entry by CD38 in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;294(2):L378–L385. doi: 10.1152/ajplung.00394.2007. PMID: 18178673. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynaminrelated protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 2001;12(8):2245–2256. doi: 10.1091/mbc.12.8.2245. PMID: 11514614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell. 2009;20(15):3525–3532. doi: 10.1091/mbc.E09-03-0252. PMID: 19477917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurway NC, Wray S. A phosphorus nuclear magnetic resonance study of metabolites and intracellular pH in rabbit vascular smooth muscle. J. Physiol. 1987;393:57–71. doi: 10.1113/jphysiol.1987.sp016810. PMID: 3446806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens NL, Li W, Jiang H, Unruh H, Ma X. The biophysics of asthmatic airway smooth muscle. Respir. Physiol. Neurobiol. 2003;137(2–3):125–140. doi: 10.1016/s1569-9048(03)00142-3. PMID: 14516721. [DOI] [PubMed] [Google Scholar]
- Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca2+ Cell Calcium. 2012;52(1):28–35. doi: 10.1016/j.ceca.2012.03.003. PMID: 22502861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumurugaan KG, Jude JA, Kang BN, Panettieri RA, Walseth TF, Kannan MS. TNF-alpha induced CD38 expression in human airway smooth muscle cells: role of MAP kinases and transcription factors NF-kappaB and AP-1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292(6):L1385–L1395. doi: 10.1152/ajplung.00472.2006. PMID: 17322278. [DOI] [PubMed] [Google Scholar]
- Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J. Exp. Med. 2007;204(13):3173–3181. doi: 10.1084/jem.20070956. PMID: 18056286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannuvel K, Renard P, Raes M, Arnould T. Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013;228(9):1802–1818. doi: 10.1002/jcp.24360. PMID: 23629871. [DOI] [PubMed] [Google Scholar]
- Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19(11):1880–1891. doi: 10.1038/cdd.2012.74. PMID: 22705852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012;197(7):857–867. doi: 10.1083/jcb.201110131. PMID: 22733998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welihinda AA, Tirasophon W, Kaufman RJ. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr. 1999;7(4–6):293–300. PMID: 10440230. [PMC free article] [PubMed] [Google Scholar]
- White TA, Xue A, Chini EN, Thompson M, Sieck GC, Wylam ME. Role of transient receptor potential C3 in TNF-alpha-enhanced calcium influx in human airway myocytes. Am. J. Respir. Cell Mol. Biol. 2006;35(2):243–251. doi: 10.1165/rcmb.2006-0003OC. PMID: 16574942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DB, Trian T, Siddiqui S, Pascoe CD, Johnson JR, Dekkers BG, et al. Phenotype modulation of airway smooth muscle in asthma. Pulm. Pharmacol. Ther. 2013a;26(1):42–49. doi: 10.1016/j.pupt.2012.08.005. PMID: 22939888. [DOI] [PubMed] [Google Scholar]
- Wright DB, Trian T, Siddiqui S, Pascoe CD, Ojo OO, Johnson JR, et al. Functional phenotype of airway myocytes from asthmatic airways. Pulm. Pharmacol. Ther. 2013b;26(1):95–104. doi: 10.1016/j.pupt.2012.08.003. PMID: 22921313. [DOI] [PubMed] [Google Scholar]
- Wright DB, Tripathi S, Sikarwar A, Santosh KT, Perez-Zoghbi J, Ojo OO, et al. Regulation of GPCR-mediated smooth muscle contraction: implications for asthma and pulmonary hypertension. Pulm. Pharmacol. Ther. 2013c;26(1):121–131. doi: 10.1016/j.pupt.2012.06.005. PMID: 22750270. [DOI] [PubMed] [Google Scholar]
- Wylam ME, Xue AL, Sieck GC. Mechanisms of intrinsic force in small human airways. Respir. Physiol. Neurobiol. 2012;181(1):99–108. doi: 10.1016/j.resp.2012.01.011. PMID: 22322114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JQ, Harder BA, Uman P, Craig R. Myosin filament structure in vertebrate smooth muscle. J. Cell Biol. 1996;134(1):53–66. doi: 10.1083/jcb.134.1.53. PMID: 8698822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 2004;167(4):661–672. doi: 10.1083/jcb.200406038. PMID: 15545319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. PMID: 22936770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZH, Wang YX, Song Y, Lu HZ, Hou LN, Cui YY, et al. Up-regulation of KCa3.1 promotes human airway smooth muscle cell phenotypic modulation. Pharmacol. Res. 2013;77:30–38. doi: 10.1016/j.phrs.2013.09.002. PMID: 24055799. [DOI] [PubMed] [Google Scholar]
- Zavaletta V, Delmotte PF, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. B18. Mitochondrial function in pulmonary health and disease. American Thoracic Society; 2011. Mitochondrial kinetics in human airway smooth muscle; pp. A2535–A2535. [Google Scholar]
- Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc. Am. Thorac. Soc. 2008;5(1):32–39. doi: 10.1513/pats.200704-048VS. PMID: 18094082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Otenbaker NP, Rose BA, Salisbury KS. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 2013;56(1–2):57–63. doi: 10.1016/j.molimm.2013.04.002. PMID: 23665383. [DOI] [PubMed] [Google Scholar]