

Abstract
Others and we have reported that prior methamphetamine (METH) exposure attenuates the persistent striatal dopaminergic deficits caused by a subsequent high-dose “binge” METH exposure. The current study investigated intermediate neurochemical changes that may contribute to, or serve to predict, this resistance. Rats self-administered METH or saline for 7 d. On the following day (specifically, 16 h after the conclusion of the final METH self-administration session), rats received a binge exposure of METH or saline (so as to assess the impact of prior METH self-administration), or were sacrificed without a subsequent METH exposure (i.e., to assess the status of the rats at what would have been the initiation of the binge METH treatment). Results revealed that METH self-administration per se decreased striatal dopamine (DA) transporter (DAT) function and DA content, as assessed 16 h after the last self-administration session. Exposure to a binge METH treatment beginning at this 16-h time point decreased DAT function and DA content as assessed 1 h after the binge METH exposure: this effect on DA content (but not DAT function) was attenuated if rats previously self-administered METH. In contrast, 24 h after the binge METH treatment prior METH self-administration: 1) attenuated deficits in DA content, DAT function and vesicular monoamine transporter-2 function; and 2) prevented increases in glial fibrillary acidic protein and DAT complex immunoreactivity. These data suggest that changes 24 h, but not 1 h, after binge METH exposure are predictive of tolerance against the persistence of neurotoxic changes following binge METH exposures.
Keywords: methamphetamine, self-administration, dopamine, striatum, tolerance
1. Introduction
Methamphetamine (METH) is a widely abused psychostimulant that can cause persistent alterations in monoaminergic neuronal function. Preclinical studies demonstrate that exposure to high-dose METH, administered to mimic some aspects of human “binge” usage, causes persistent dopaminergic deficits. METH-induced alterations in dopamine (DA) content, DA transporter (DAT) function, vesicular monoamine transporter-2 (VMAT2) function/distribution, and tyrosine hydroxylase (TH) activity 1 – 24 h post-treatment may contribute to these long-term deficits (Brennan et al., 2010; Chu et al., 2010; Eyerman and Yamamoto, 2007; Hadlock et al., 2009, 2010; Hotchkiss and Gibb, 1980; McFadden et al., 2012A). Increases in extracellular glutamate, reactivity species and oxidative stress formation, and glial activation are also thought to contribute to the dopaminergic deficits following binge METH exposure (for reviews see Krasnova and Cadet, 2009; Tata and Yamamoto, 2007).
Researchers have noted that administration of escalating doses or intake of METH models human METH patterns of use (Fischman and Schuster, 1974; Schmidt et al., 1985B). Further, preclinical studies indicate that pretreatment with escalating doses of METH attenuates the deficits induced by a large neurotoxic binge exposure to METH (Schmidt et al., 1985B), including persistent losses of DA and serotonin (5HT) content (Cadet et al., 2009; Hodges et al., 2011; Johnson-Davis et al., 2003; McFadden et al., 2012A; McFadden et al., 2012B; Segal et al., 2003), DAT function/immunoreactivity/binding (Belcher et al., 2008; Krasnova et al., 2011; McFadden et al., 2012A; McFadden et al., 2012B; Segal et al., 2003), VMAT2 function (McFadden et al., 2011), and glial activation (McFadden et al., 2012A; Thomas et al., 2005). These studies are important since most individuals who abuse METH receive multiple exposures to the drug. However, relatively little is known of the mechanisms underlying the “neuroprotection” afforded by prior METH exposure.
Similar to the escalating-dose studies described above, our laboratory reported that prior METH self-administration reduced the persistent neurotoxic decreases in striatal DA content and DAT function caused by a subsequent binge METH exposure (McFadden et al., 2012A). These findings are of potential clinical relevance in that the resistance to binge-induced dopaminergic deficits caused by the repeated METH exposures may provide a model to explain why human METH abusers generally do not display dopaminergic deficits as great in magnitude as those resulting pre-clinically from a binge METH treatment, even after using large quantities of METH (McFadden et al., 2012A, McFadden et al., 2012B). Further, preclinical studies have suggested that contingent drug administration may lead to distinct neurochemical changes compared to non-contingent administration (Frankel et al., 2012; Hanson et al., 2012; Hanson et al., 2013; Lominac et al., 2012; Stefanski et al., 2004).
Given the importance noted above of alterations 1 – 24 h post-METH treatment, the current study focused on such changes that may contribute to and/or predict this resistance to METH-induced deficits. Results revealed that METH self-administration per se decreased striatal DAT function and DA content, as assessed 16 h after the last self-administration session. Exposure to a binge METH treatment beginning at this 16-h time point decreased DAT function and DA content as assessed 1 h after the binge METH exposure: this effect on DA content, but not DAT function, was attenuated if rats were exposed previously to METH self-administration. In contrast, prior METH self-administration attenuated deficits in DA content, as well as DAT function and VMAT2 function as assessed 24 h after the binge treatment. Further, prior METH self-administration prevented increases in glial fibrillary acidic protein (GFAP) and DAT complex immunoreactivity compared to saline self-administrating/binge METH exposed rats at this 24 h time point. These findings suggest that striatal changes occurring 24 h after the binge exposure to METH are predictive of persistent deficits induced by a binge exposure to METH as previously reported (McFadden et al., 2012A).
2. Methods
2.1 Animals
Male Sprague-Dawley rats (275–300 g; Charles River Laboratories, Portage, MI) were housed four rats/cage (35×30×16 cm). Following surgery, each rat was individually housed in a transparent plastic cage (45×23×21 cm). Water was available in their home cage ad libitum. During food training, rats were food restricted such that no rat dropped below 90% of their starting body weight. Rats were maintained under the same 14:10 h light/dark cycle in the animal facility and in the operant chambers. All experiments were approved by the University of Utah’s Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2 Drugs
Racemic-METH hydrochloride (Research Triangle Institute; Research Triangle Park, NC) was dissolved in 0.9% sterile saline, with the dose described as the free-base form. Ketamine (90 mg/kg; Hospira Inc., Lake Forest, IL, USA) and xylazine (7 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) were used to anesthetize animals. The antibiotic cefazolin (10 mg/ml; Schein Pharmaceutical, Florham Park, NJ, USA) was dissolved in heparinized saline (63.33 U/ml; Sigma, St. Louis, MO, USA). Flunixin meglumine (1.1 mg/kg; MWI Veterinary Supply, Meridian, ID, USA) was used for post-surgery analgesia.
2.3 Food Training and Surgery
Food training and self-administration occurred in an operant chamber (30.5 cm x 25.5 cm x 30.5 cm; Coulbourn Instruments, Whitehall, PA USA) as described in McFadden et al., 2012A. Prior to surgery, each rat was trained to press for a 45-mg food pellet during four overnight 14-h sessions. Following food training, rats were anesthetized and an indwelling catheter was implanted. The catheter was constructed as described previously (Frankel et al., 2010). Each rat received flunixin meglumine on the day of and following the surgery. Immediately following surgery and daily thereafter, each rat was infused with 0.1 ml of cefazolin followed by 0.05 ml of heparinized saline and heparinized glycerol. Catheter patency was confirmed by infusing 0.03 ml (20 mg/ml) of xylazine. The animals’ food was reduced to 25 g the day before self-administration began. On all following days, animals were allowed to free feed.
2.4 Self-Administration and METH Challenge
Figure 1 illustrates the time course of experiments. All rats underwent 7 d of self-administration (8 h/session; FR1; 0.12 mg/infusion METH or saline; with associated lever pressing presented as Supplemental Figure 1) during the light cycle in a room maintained at 29±1°C to promote lever pressing (Cornish et al., 2008). For each active lever press, an infusion pump connected to a liquid swivel (Coulbourn Instruments) delivered 10 μl of METH or saline per infusion over a 5-s duration through a polyethylene tube located within a spring leash (Coulbourn Instruments) tethered to the rat. During this period, both levers were retracted. Following the infusion, the levers remained retracted for an additional 20 s. The active lever was counterbalanced within each group. Pressing the inactive lever resulted in no programmed consequences although it was recorded. METH self-administering rats were only included in the analysis if they: 1) pressed an average of more than 10 active lever presses per d; and 2) the ratio of active/inactive lever presses was ≥ 2:1. These criteria were largely based upon those of Brennan et al., 2010. METH intake and body weight changes were similar between experiments and therefore representative pressing behavior and weights are illustrated in Figures 2A and 2B. Animals were sacrificed 16 h after the end of the last self-administration session or received a binge of METH or saline as described below (Figure 1).
Figure 1.
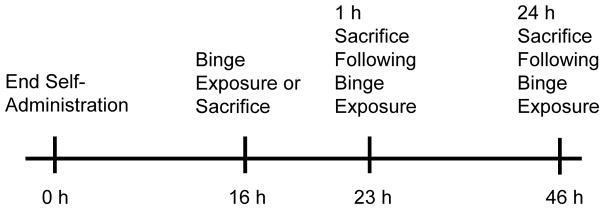
Sacrifice times of rats in the manuscript. Animals were allowed to self-administer METH or Saline for 7 d. Sixteen h after the last self-administration session rats were sacrificed or received a binge exposure to METH or Saline. Rats were sacrificed 1 h or 24 h after the binge exposure.
Figure 2.

Representative METH intake and body weight during self-administration, and hyperthermia during the binge exposure to METH. Rats were allowed to self-administer METH (0.12 mg/infusion; normalized to mg/kg) for 7 d (8 h/d; Panel A). Body weight was recorded daily (Panel B). Sixteen h after the last self-administration session rats received a binge exposure to METH (7.5 mg/kg) or saline (1 mL/kg) and hyperthermia was recorded 30 min and 90 minutes following each injection (as indicated by the arrows; Panel C). *p<0.05 METH/METH compared to Saline/METH
Sixteen h after the end of the last self-administration session, rats were challenged with 4 injections of METH (7.5 mg/kg/injection; 2-h interval) or saline (1 ml/kg/injection). This time point was chosen based on research utilizing escalating doses of METH followed by a binge exposure (see Danaceau et al., 2007; O’Neil et al., 2006) and to replicate previous methods (McFadden et al., 2012A; McFadden et al., 2012B). Previous studies (McFadden et al., 2012A; McFadden et al., 2012B) have demonstrated that hyperthermia is attenuated during the binge exposure to METH in animals that had previously self-administered METH. To promote hyperthermia, METH self-administering rats challenged with METH (METH/METH) were maintained in a warm environment (25°C). Rats that self-administered saline prior to the binge of METH (Saline/METH) were maintained in 22.5°C environment to promote similar hyperthermia between METH/METH and Saline/METH groups. Previous research in our laboratory has shown that this higher ambient temperature permits binge METH-induced neurotoxicity that would have otherwise been attenuated by prior exposure to METH during development (see McFadden et al., 2011). Representative temperature data during the binge exposure to METH are provided in Figure 2C. Animals were sacrificed 1 h or 24 h after the binge exposure (Figure 1).
2.5 Tissue Preparation
Tissue preparation was conducted as previously described (Hanson et al., 2009). Following decapitation, the striata were quickly dissected out and the right striatum was homogenized in ice-cold sucrose buffer (0.32 M sucrose, 3.8 mM NaH2PO4, and 12.7 mM Na2HPO4). The left striatum was quickly frozen on dry ice.
2.6 Plasmalemmal & Vesicular [3H]DA Uptake Assays
[3H]DA uptake assays were conducted according to Johnson-Davis et al. (2004). For plasmalemmal uptake of [3H]DA, striatal synaptosomes were prepared accordingly and resuspended in ice-cold Krebs’ buffer (126 nM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 16 mM sodium phosphate, 1.4 mM MgSO4, 11 mM dextrose, 1 mM ascorbic acid, pH 7.4). Assay tubes containing 1.5 mg striatal tissue and 1 μM pargyline were incubated (3 min, 37°C) with [3H]DA (0.5 nM final concentration, Perkin Elmer, Boston, MA). Nonspecific values were ascertained in the presence of 10 μM cocaine. Samples were filtered using a filtering manifold (Brandel, Inc Gaithersburg, MD) through Whatman GF/B filters (Whatman International LTD, Maidstone, England) soaked previously in 0.05% polyethylenimine and washed three times with 3 ml of ice-cold 0.32 M sucrose.
For vesicular [3H]DA uptake, synaptic vesicles were isolated according to Johnson-Davis et al. (2004) and resuspended in assay buffer (25 mM HEPES, 100 mM potassium tartrate, 1.7 mM ascorbic acid, 0.05 mM EGTA, 0.1 mM EDTA and 1.8 mM ATP-Mg2+; pH 7.5). Vesicles were incubated (3 min, 30°C) in the presence of [3H]DA (30 nM final concentration, Perkin Elmer, Boston, MA). Nonspecific values were found by measuring vesicular [3H]DA uptake at 4°C in the absence of ATP. Samples were filtered through GF/F filters (Whatman International LTD, Maidstone, England) previously soaked in 0.5% polyethylenimine and washed 3 times with cold wash buffer. The radioactivity trapped in filters was counted using a liquid scintillation counter. Protein concentrations were determined using the Bradford Protein Assay.
2.7 Western Blotting
Western blotting was conducted according to Hadlock et al. (2010). Equal quantities of protein (8 μg) were loaded into each well of a 4 to 12% NuPAGE Novex Bis-Tris Midi gradient gel (Invitrogen, Carlsbad, CA) and electrophoresed using an XCell4 Surelock Midi-cell (Invitrogen). Membranes were blocked for 30 min with Starting Block Blocking Buffer (Pierce Chemical, Rockford, IL) and incubated for 1 h at room temperature with a D2 antibody (SC-7522; Santa Cruz Biotechnology, Santa Cruz, CA), DAT antibody (a generous gift from Dr. Roxanne Vaughan, University of North Dakota; Freed et al., 1995), or GFAP (556329; BD Biosciences, San Jose, CA). The polyvinylidene difluoride membrane was then washed five times in Tris-buffered saline with Tween (250 mM NaCl, 50 mM Tris, and 0.05% Tween 20, pH 7.4). The membranes were then incubated for 1 h with a horseradish peroxidase-conjugated secondary antibody (BioSource International, Camarillo, CA). After five washes in Tris-buffered saline with Tween, the bands were visualized using Western Lightning Chemiluminescence Reagents Plus (PerkinElmer Life and Analytical Sciences) and were quantified by densitometry using a FluorChem SP Imaging System (Alpha Innotech, San Leandro, CA). Protein concentrations were determined using the Bradford Protein Assay.
2.8 DA and 3,4-Dihydroxyphenylacetic Acid (DOPAC) Content
The anterior portion of the left striatum was sonicated for 3–5 s in 1 ml tissue buffer (0.1 M phosphate/citrate buffer, pH=2.5, containing 10% methanol) and were prepared according to Haughey et al., (2000). Fifty μL was injected onto a partisphere C-18 reverse-phase analytical column (5-μm spheres; 250 X 4.6 mm; Whatman, Clifton, NJ, USA). For vesicular DA content, vesicles were prepared as described in Chu et al., (2010), resuspended in tissue buffer and 100 μL was injected. Mobile phase consisting of 0.05 M sodium phosphate, 0.03 M citrate buffer, 0.1 M EDTA, 0.035% sodium octylsulfate, and 15% methanol (pH= 2.8, flow rate=0.7 ml/min) was used. DA and DOPAC was detected using an ampherometric electrochemical detector with the working electrode potential set at +0.70 V relative to an Ag+/AgCl reference electrode. Protein concentrations were determined using the Bradford Protein Assay for tissue content. Vesicular content was normalized to wet weights.
2.9 Statistical Analysis
Statistical analysis was conducted in GraphPad Prism (5.1; La Jolla, CA, USA). Statistical analyses among groups were conducted using a t-test or one-way analysis of variance (ANOVA) followed by Newman-Keuls posthoc analyses. The data represent means ± standard error of the mean (S.E.M.) of 5–11 rats/group.
3. Results
METH self-administration decreased DAT function and striatal DA content compared to saline self-administration when assessed 16 h after the last session (Figure 3). METH self-administration decreased DAT (t(13)=3.63, p<0.05; Figure 3A) function, but VMAT2 function was not altered in either the cytoplasmic (t(10)=1.43, ns; Saline: 110.20±8.03 fmol DA/μg protein; METH: 93.33±7.73 fmol DA/μg protein) or membrane-associated fractions (t(10)=0.68, ns; Saline: 13.50±1.15 fmol DA/μg protein; METH: 12.50±0.88 fmol DA/μg protein). A trend towards a decrease in dopamine D2 receptor immunoreactivity was observed in the METH self-administering group compared to the saline controls (t(12)=1.84, p=0.09; Figure 3B). A slight but significant decrease in DA (t(13)=2.41, p<0.05; Figure 3C), as well as an increase in DOPAC (t(13)=2.56, p<0.05; Figure 3D) content was observed in METH self-administering compared to saline self-administering rats.
Figure 3.

Dopaminergic changes in the striatum 16 h following the last self-administration session. Rats were allowed to self-administer METH (0.12 mg/infusion) or saline (10 μl/infusion) for 7 d (8 h/d) and were sacrificed 16 h after the last session. METH self-administration resulted in a reduction in DAT function (Panel A) and a trend toward a reduction in D2 receptor immunoreactivity (Panel B). Striatal DA content was reduced in METH self-administering rats and DOPAC content was increased compared to saline self-administering rats (Panels C & D). Insert: Representative D2 immunoreactivity of Saline (left) and METH (right) self-administering rats depicted in Panel B. *p<0.05 compared to saline self-administrating rats. †p<0.10 compared to saline self-administering rats.
Prior METH self-administration attenuated the decrease in DA content, but not striatal DAT function, as assessed 1 h following the binge of METH (Figure 4). Specifically, the binge METH exposure similarly decreased DAT function in both binge METH groups (F(2,24)=52.75, p<0.05; Figure 4A). Prior METH self-administration attenuated the decrease in striatal DA content (F(2,24)=18.00, p<0.05; Figure 4B). Of note, prior METH self-administration did not alter binge METH-induced decreases in striatal DOPAC concentrations compared to the Saline/METH group (F(2,24)=47.09, p<0.05; Figure 4C).
Figure 4.

Prior METH self-administration attenuated decreases in DA content but not DAT function 1 h after binge METH exposure. Rats were allowed to self-administer METH (0.12 mg/infusion) or saline (10 μl/infusion) for 7 d (8 h/d), received a binge exposure to METH (7.5 mg/kg/injection; 4 injections) 16 h after the last session, and were sacrificed 1 h following the last injection. Binge exposure to METH similarly reduced DAT function in the METH/METH and Saline/METH groups (Panel A). Prior METH exposure attenuated reductions in striatal DA content (Panel B) following the binge exposure, but did not attenuate decreases in DOPAC content (Panel C). *p<0.05 compared to Saline/Saline. #p<0.05 compared to all other groups.
To further investigate the attenuation in DA content, vesicular content was determined in animals sacrificed 1 h following the binge exposure to METH. Prior METH self-administration did not alter the binge METH-induced decreases in cytosolic (F(2,20)=21.41, p<0.05; Figure 5A) or membrane-associated (F(2,20)=8.99, p<0.05; Figure 5B) vesicular DA content. Similarly, prior METH self-administration did not attenuate the binge METH-induced decreases in cytosolic VMAT2 immunoreactivity (F(2,19)=7.06, p<0.05; Figure 5C). As in other experiments described in this manuscript and of interest because of its role in METH-induced toxicity, binge METH-induced hyperthermia was attenuated in the METH/METH group 1 h following the first injection of METH as assessed in the METH/METH group compared to the Saline/METH group in these animals (F(14,140)=7.13, p<0.05; Figure 2B).
Figure 5.

Binge METH exposure resulted in similar decreases in vesicular DA content and cytosolic VMAT2 immunoreactivity 1 h following binge exposure. Rats were treated as described in Figure 4. Prior METH self-administration did not alter reductions in cytosolic vesicular DA content (Panel A) or membrane-associated vesicular DA content (Panel B) following the binge exposure to METH. Similar reductions in cytosolic VMAT2 immunoreactivity were observed between the METH/METH and Saline/METH group (Panel C). Insert: Representative VMAT2 immunoreactivity of Saline/Saline, METH/METH and Saline/METH (left to right) shown in Panel C. *p<0.05 compared to Saline/Saline. #p<0.05 compared to all other groups.
In contrast to effects at 1-h, prior METH self-administration attenuated all of the binge METH-induced alterations investigated as assessed 24 h after treatment. In particular, prior METH self-administration attenuated decreases in DAT function (F(2,16)=17.49, p<0.05; Figure 6A), DA content (F(2,17)=77.77, p<0.05; Figure 6B) and DOPAC content (F(2,18)=32.92, p<0.05; Figure 6C) compared to the Saline/METH group. Further, prior METH self-administration attenuated binge METH-induced decreases in cytosolic VMAT2 function (F(2,16)=28.96, p<0.05; Figure 7A), membrane-associated VMAT2 function (F(2,18)=30.50, p<0.05; Figure 7B) and membrane-associated VMAT2 DA content (F(2,21)=71.09, p<0.05; Saline/Saline: 104.5±2.62 pg DA/mg wet weight; METH/METH: 55.98±2.51 pg DA/mg wet weight; Saline/METH 44.53±4.39 pg DA/mg wet weight). Prior METH self-administration also attenuated binge METH-induced increases in GFAP (F(2,18)=3.55, p=0.05; Figure 8A) and DAT complex immunoreactivity (F(2,18)=6.92, p<0.05; Figure 8B) at this 24-h timepoint. Consistent with DA uptake, prior METH self-administration also attenuated decreases in DAT monomer immunoreactivity (F(2,18)=28.28, p<0.05; Saline/Saline: 197.20±54.41 arbitrary units; METH/METH: 117.70±38.71 arbitrary units; Saline/METH: 44.56±12.40 arbitrary units).
Figure 6.

Prior METH self-administration attenuated dopaminergic deficits 24 h following binge METH exposure. Rats were allowed to self-administer METH or saline for 7 d, received a binge exposure to METH or saline, and were sacrificed 24 h after the last injection. Prior METH self-administration attenuated binge METH-induced deficits in DAT function (Panel A), DA content (Panel B), and DOPAC content (Panel C). *p<0.05 compared to Saline/Saline. #p<0.05 compared to all other groups.
Figure 7.
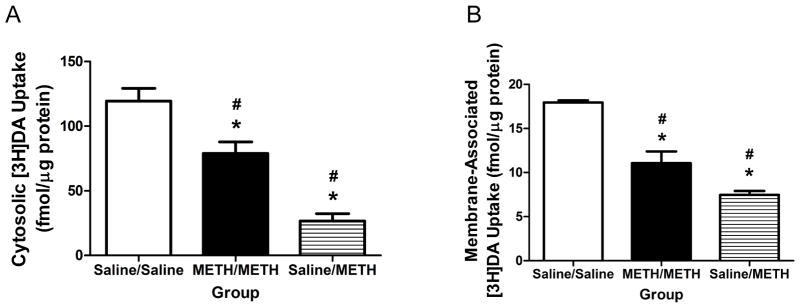
Prior METH self-administration attenuated VMAT2 functional deficits induced by a binge exposure to METH as assessed 24 h later. Rats were treated as described in Figure 6. Striatal VMAT2 function was assessed in the cytosolic fraction (Panel A) and membrane-associated fraction (Panel B).
Figure 8.
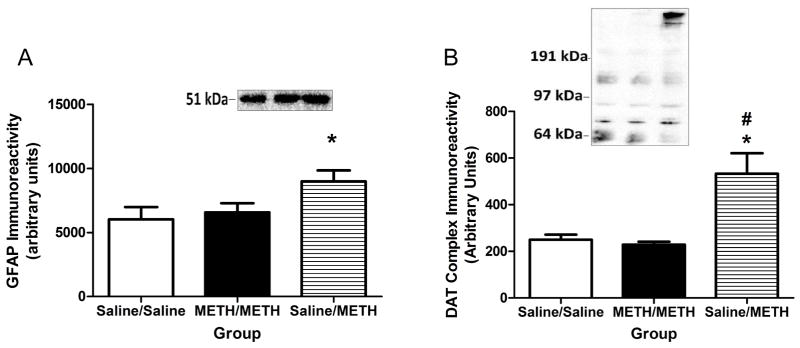
Other markers of METH-induced toxicity were not elevated in rats that self-administered METH prior to the binge exposure as assessed 24 h later. Rats were treated as described in Figure 6. Increases in striatal GFAP (Panel A; Insert: Representative GFAP immunoreactivity of Saline/Saline, METH/METH, and Saline/METH [left to right] exposed rats) and DAT complexes immunoreactivity (Panel B; Insert: Representative DAT complex immunoreactivity spanning from above 97 kDa to the top of the gel for Saline/Saline-, METH/METH-, and Saline/METH-exposed rats [left to right]).
4. Discussion
Previous studies have demonstrated that prior METH self-administration attenuates the persistent dopaminergic deficits caused by a subsequent binge METH exposure (McFadden et al., 2012A). We have reported that this protective effect was not likely due to alterations in the pharmacokinetics of METH caused by prior METH self-administration (McFadden et al., 2012A). Further, the attenuation of binge METH-induced hyperthermia afforded by prior METH self-administration likely contributes to protection. Still, additional mechanisms underlying this phenomenon have not been elucidated. The present study focused attention on dopaminergic deficits occurring prior to and 1 – 24 h after a binge METH treatment, as these have been suggested to be important contributors to and/or predictors of METH-induced persistent dopaminergic deficits (McFadden et al., 2012A).
Striatal dopaminergic changes induced by METH self-administration were assessed 16 h after the last self-administration session to determine if preexisting dopaminergic changes may have contributed to differences following the binge exposure (Figure 3). Consistent with previous reports using escalating METH intake and measuring short-term effects, the current study found no changes in VMAT2 function (Lacan et al., 2013). Slight decreases in DA content also occurred in METH self-administering rats that were associated with increases in DOPAC content. In contrast to these previously mentioned effects, DAT function was decreased in METH self-administering rats when animals were sacrificed at this time point. Inhibiting the DAT with selective drugs such as amfonelic acid attenuates persistent DA depletions and inhibition of TH activity caused by binge METH treatments (Marek et al., 1990; Schmidt et al., 1985A). Thus, the decreased DAT function caused by METH self-administration present at the time of the initiation of the binge METH exposure may have acted similarly to amfonelic acid and thereby contribute to tolerance against METH-induced dopaminergic deficits. Alternatively, altering DAT densities or function has been shown to affect DA release after administration of the METH-related stimulant, amphetamine (Calipari et al., 2013; Siciliano et al., 2014). Reduction of DAT function in the current study may have reduced the impact of METH during the binge exposure and thus contributed to reductions in its neurotoxic effects.
The observed changes in dopaminergic markers at 1 h following the binge METH exposure suggest that the acute pharmacological effects of METH on DAT and VMAT2 are unaltered by prior METH exposure (Figures 4 & 5). Similar decreases in DAT function and decreases in vesicular content 1 h following the binge exposure to METH suggest that the ability of METH to inhibit DAT function and redistribute DA from the vesicles to the cytosol are unaffected by prior METH self-administration. Previously our laboratory has reported similar brain METH content in the METH/METH and Saline/METH groups, further suggesting that many of the acute effects of the binge METH exposure are unaltered (McFadden et al., 2012A).
Of interest, DA content was approximately 25% higher in the METH/METH group compared to the Saline/METH group 1 h following the binge exposure (Figure 4B). Prior exposure to METH may have attenuated METH’s ability to decrease TH activity (Schmidt et al., 1985B). A one week escalating-dose pretreatment paradigm attenuated decreases in TH activity 18 h following the binge exposure and was associated with increases in DA content (Schmidt et al., 1985B). A similar phenomenon could also occur when METH is self-administered prior to the binge exposure, such that TH activity is not inhibited as greatly in the METH/METH group compared to the Saline/METH group.
No differences in vesicular DA content were found in the METH/METH group in comparison to the Saline/METH group 1 h following the binge exposure to METH (Figure 5A & 5B), which suggests that the 25% increase in total DA content may not reflect DA sequestered in vesicles, but rather in the cytosol of the dopaminergic neurons. Previous work suggests decreasing DA sequestered by vesicles increases METH-induced toxicity (Guillot et al., 2008; Thomas et al., 2008). Inhibited VMAT2 function either by prior treatment with reserpine or through genetic manipulation leads to increased markers of striatal METH-induced toxicity including greater loss in DA content, and DAT and TH immunoreactivity, and increases in GFAP immunoreactivity and oxidative stress (Guillot et al., 2008; Thomas et al., 2008). However, these effects lasted for at least 2 d following the METH exposure (Guillot et al., 2006; Thomas et al., 2008). In the current study, many of these markers showed signs of recovery at 24 h following the binge METH exposure in the METH/METH group which may have minimized the potentially toxic effects of the unsequestered DA.
As previously mentioned, VMAT2 is thought to play a pivotal role in METH toxicity (Fumagalli et al., 1999; Guillot et al., 2008; Thomas et al., 2008). An important finding of the current study is that prior exposure to METH through self-administration is associated with attenuated deficits in VMAT2 function at 24 h following the binge exposure (Figure 7). The attenuation in deficits of VMAT2 function may have decreased reactive species formation and METH-induced toxicity. Previous research suggests protein-bound DA quinones are elevated 3 d and 14 d following binge exposure but not at 1 d following METH in mice (Miyazaki et al., 2006), suggesting that DA quinone formation may increase with time. In the current study, the attenuation of the reductions in VMAT2 function observed in the METH/METH group compared to the Saline/METH group 24 h after the binge exposure may have mitigated the formation of DA quionones by sequestering DA into the vesicles.
METH self-administration prior to binge-METH exposure blocked the formation of DAT complexes as assessed at 24 h and 7 d after binge exposure (Figure 8B; McFadden et al., 2012A). This mitigation in the formation of DAT complexes was also associated with an attenuation in binge METH-induced decreases in DAT function (Figures 6A). Reactive species have been implicated in the formation of DAT complexes, further implicating their role in the toxicity to METH (Baucum et al., 2004; Hadlock et al., 2009). Pretreatment with D2 receptor antagonists also attenuated DAT complex formation (Hadlock et al., 2009; Hadlock et al., 2010), as well as the long-term deficits caused by METH. METH self-administration decreases D2 receptor function (Lacan et al., 2013). This decrease in D2 receptors caused by METH self-administration may cause pharmacological effects similar to that of D2 receptor antagonist and result in a decrease in the formation of DAT complexes. Thus, the reduction in D2 receptors may contribute to the protection afforded by METH self-administration by reducing the formation of DAT complexes and increasing DAT function.
Astrocytes are important regulators glutamate (Cisneros and Ghorpade, 2012; Lau et al., 2000). Of relevance, METH also induces reactive astrogliosis (McFadden et al., 2012A; Hadlock et al., 2010; Thomas et al., 2004; Zhu et al., 2005). Current (Figure 8A) and previous studies suggest that reactive astrogliosis is significantly increased in the Saline/METH group (McFadden et al., 2012A), and is attenuated by prior METH self-administration. It can thus be speculated that the lack of reactive astrogliosis in the METH/METH group may be an indicator of a reduction in glutamate release caused by the binge exposure of METH.
In conclusion, the current study provides novel insight into the changes associated with tolerance to the neurotoxic effects of METH within the striatum. Effects occurring 24 h after the binge METH exposure appear particularly linked to persistent neurotoxicity, although it is unclear if these actually contribute to the persistent dopaminergic deficits or are a result of other changes that may contribute to this toxicity. It is also unclear if the neuroprotection afforded by prior contingent METH administration differs in magnitude from the effects of prior non-contingent METH exposure. This is an important issue, as that the learned associations between the physiological effects of the drug and the behavioral output may contribute to differences among the various paradigms. Of note, there is precedence for differences between contingent and noncontingent METH exposure. For example, prior METH self-administration increases levels of the neuropeptide, neurotensin, compared to yoked-METH rats (Frankel et al., 2011; Hanson et al., 2012). Given the role of neurotensin as a neuromodulator of DA, differential neuroprotection as a consequence of contingency is indeed possible and an important area for future study. Future studies are needed to elucidate how these dopaminergic changes at 24 h following binge METH exposure are associated with tolerance to the neurotoxic events following binge METH exposure and if these changes are associated with contingency.
Supplementary Material
Supplemental Figure 1. Rats were allow to press for METH (0.12 mg/infusion) for 7 d (8 h/d). Panel A: Pressing behavior corresponding to Figure 3. Average total METH intake over the course of the 7 d was 22.33 mg ± 1.82 mg. Panel B: Pressing behavior for corresponding to animals used to assess VMAT2 uptake at 16 h after the last self-administration session. Average total METH intake over the course of the 7 d was 19.16 mg ± 2.11 mg. Panel C: Pressing behavior corresponding to Figure 4. Average total METH intake over the course of the 7 d was 19.96 mg ± 1.24 mg. Panel D: Pressing behavior corresponding to Figures 2A, 5A, and 5B. Average total METH intake over the course of the 7 d was 20.29 mg ± 1.01 mg. Panel E: Pressing behavior corresponding to Figure 5C. Average total METH intake over the course of the 7 d was 18.94 mg ± 1.63 mg. Panel F: Pressing behavior corresponding to Figure 6A. Average total METH intake over the course of the 7 d was 21.92 mg ± 2.48 mg. Panel G: Pressing behavior corresponding to Figures 6B, 6C, and 7. Average total METH intake over the course of the 7 d was 16.26 mg ± 1.33 mg. Panel H: Pressing behavior corresponding to Figure 7. Average total METH intake over the course of the 7 d was 21.24 mg ± 1.40 mg.
Highlights.
Contingent METH exposure decreased dopamine transporter (DAT) function 16 h later.
Prior contingent METH did not attenuate acute binge METH-induced DAT deficits.
24 h after the binge treatment, prior contingent METH attenuated striatal deficits.
Acknowledgments
Role of Funding Source
Funding for this study was provided by National Institute of Health, National Institute on Drug Abuse grants: DA033097, 036012, 011389, 019447, 000378, 013367, 031883. The National Institute of Health had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Abbreviations
- DOPAC
3,4-dihydroxyphenylacetic acid
- DA
dopamine
- DAT
dopamine transporter
- GFAP
glial fibrillary acidic protein
- METH
methamphetamine
- 5HT
serotonin
- TH
tyrosine hydroxylase
- VMAT2
vesicular monoamine transporter-2
Footnotes
Authorship Contributions
Participated in research design: McFadden, Vieira-Brock, Hanson, and Fleckenstein.
Conducted experiments: McFadden and Vieira-Brock.
Performed data analysis: McFadden.
Wrote or contributed to the writing of the manuscript: McFadden, Vieira-Brock, Hanson, and Fleckenstein.
All authors contributed to and have approved the final manuscript.
Conflict of Interests
All authors declare that they have no conflicts of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baucum AJ, 2nd, Rau KS, Riddle EL, Hanson GR, Fleckenstein AE. Methamphetamine increases dopamine transporter higher molecular weight complex formation via a dopamine- and hyperthermia-associated mechanism. J Neurosci. 2004;24:3436–3443. doi: 10.1523/JNEUROSCI.0387-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher AM, Feinstein EM, O’Dell SJ, Marshall JF. Methamphetamine influences on recognition memory: comparison of escalating and single-day dosing regimens. Neuropsychopharmacology. 2008;33:1453–1463. doi: 10.1038/sj.npp.1301510. [DOI] [PubMed] [Google Scholar]
- Brennan KA, Colussi-Mas J, Carati C, Lea RA, Fitzmaurice PS, Schenk S. Methamphetamine self-administration and the effect of contingency on monoamine and metabolite tissue levels in the rat. Brain Res. 2010;1317:137–146. doi: 10.1016/j.brainres.2009.11.069. [DOI] [PubMed] [Google Scholar]
- Cadet JL, McCoy MT, Cai NS, Krasnova IN, Ladenheim B, Beauvais G, Wilson N, Wood W, Becker KG, Hodges AB. Methamphetamine preconditioning alters midbrain transcriptional responses to methamphetamine-induced injury in the rat striatum. PLoS One. 2009;4:e7812. doi: 10.1371/journal.pone.0007812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Ferris MJ, Salahpour A, Caron MG, Jones SR. Methylphenidate amplifies the potency and reinforcing effects of amphetamine by increasing dopamine transporter expression. Nat Commun. 2013;4:2720. doi: 10.1038/ncomms3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu PW, Hadlock GC, Vieira-Brock P, Stout K, Hanson GR, Fleckenstein AE. Methamphetamine alters vesicular monoamine transporter-2 function and potassium-stimulated dopamine release. J Neurochem. 2010;115:325–332. doi: 10.1111/j.1471-4159.2010.06922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisneros IE, Ghorpade A. HIV-1, methamphetamine and astrocyte glutamate regulation: combined excitotoxic implications for neuro-AIDS. Curr HIV Res. 2012;10:392–406. doi: 10.2174/157016212802138832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish JL, Clemens KJ, Thompson MR, Callaghan PD, Dawson B, McGregor IS. High ambient temperature increases intravenous methamphetamine self-administration on fixed and progressive ratio schedules in rats. J Psychopharmacol. 2008;22:100–110. doi: 10.1177/0269881107082286. [DOI] [PubMed] [Google Scholar]
- Danaceau JP, Deering CE, Day JE, Smeal SJ, Johnson-Davis KL, Fleckenstein AE, Wilkins DG. Persistence of tolerance to methamphetamine-induced monoamine deficits. Eur J Pharmacol. 2007;559:46–54. doi: 10.1016/j.ejphar.2006.11.045. [DOI] [PubMed] [Google Scholar]
- Eyerman DJ, Yamamoto BK. A rapid oxidation and persistent decrease in the vesicular monoamine transporter 2 after methamphetamine. J Neurochem. 2007;103:1219–1227. doi: 10.1111/j.1471-4159.2007.04837.x. [DOI] [PubMed] [Google Scholar]
- Fischman MW, Schuster CR. Tolerance development to chronic methamphetamine intoxication in the rhesus monkey. Pharmacol Biochem Behav. 1974;2:503–508. doi: 10.1016/0091-3057(74)90010-0. [DOI] [PubMed] [Google Scholar]
- Frankel PS, Hoonakker AJ, Alburges ME, McDougall JW, McFadden LM, Fleckenstein AE, Hanson GR. Effect of methamphetamine self-administration on neurotensin systems of the basal ganglia. J Pharmacol Exp Ther. 2011;336:809–815. doi: 10.1124/jpet.110.176610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed C, Revay R, Vaughan RA, Kriek E, Grant S, Uhl GR, Kuhar MJ. Dopamine transporter immunoreactivity in rat brain. J Comp Neurol. 1995;359:340–349. doi: 10.1002/cne.903590211. [DOI] [PubMed] [Google Scholar]
- Frankel PS, Hoonakker AJ, Alburges ME, McDougall JW, McFadden LM, Fleckenstein AE, Hanson GR. Effect of methamphetamine self-administration on neurotensin systems of the basal ganglia. J Pharmacol Exp Ther. 2010;336:809–815. doi: 10.1124/jpet.110.176610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knockout mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li Y, Emson PC, Miller GW. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlock GC, Chu PW, Walters ET, Hanson GR, Fleckenstein AE. Methamphetamine-induced dopamine transporter complex formation and dopaminergic deficits: the role of D2 receptor activation. J Pharmacol Exp Ther. 2010;335:207–212. doi: 10.1124/jpet.110.166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlock GC, Baucum AJ, 2nd, King JL, Horner KA, Cook GA, Gibb JW, Wilkins DG, Hanson GR, Fleckenstein AE. Mechanisms underlying methamphetamine-induced dopamine transporter complex formation. J Pharmacol Exp Ther. 2009;329:169–174. doi: 10.1124/jpet.108.145631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Birdsall E, Seferian KS, Crosby MA, Keefe KA, Gibb JW, Hanson GR, Fleckenstein AE. Methamphetamine-induced dopaminergic deficits and refractoriness to subsequent treatment. Eur J Pharmacol. 2009;607:68–73. doi: 10.1016/j.ejphar.2009.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GR, Hoonakker AJ, Alburges ME, McFadden LM, Robson CM, Frankel PS. Response of limbic neurotensin system to methamphetamine self-administration. Neuroscience. 2012;203:99–107. doi: 10.1016/j.neuroscience.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GR, Hoonakker AJ, Robson CM, McFadden LM, Frankel PS, Alburges ME. Response of neurotensin basal ganglia systems during extinction of methamphetamine self-administration in rats. J Pharmacol Exp Ther. 2013;346:173–181. doi: 10.1124/jpet.113.205310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey HM, Fleckenstein AE, Metzger RR, Hanson GR. The effects of methamphetamine on serotonin transporter activity: role of dopamine and hyperthermia. J Neurochem. 2000;75:1608–1617. doi: 10.1046/j.1471-4159.2000.0751608.x. [DOI] [PubMed] [Google Scholar]
- Hodges AB, Ladenheim B, McCoy MT, Beauvais G, Cai N, Krasnova IN, Cadet JL. Long-term protective effects of methamphetamine preconditioning against single-day methamphetamine toxic challenges. Curr Neuropharmacol. 2011;9:35–39. doi: 10.2174/157015911795017344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- Johnson-Davis KL, Fleckenstein AE, Wilkins DG. The role of hyperthermia and metabolism as mechanisms of tolerance to methamphetamine neurotoxicity. Eur J Pharmacol. 2003;482:151–154. doi: 10.1016/j.ejphar.2003.09.063. [DOI] [PubMed] [Google Scholar]
- Johnson-Davis KL, Truong JG, Fleckenstein AE, Wilkins DG. Alterations in vesicular dopamine uptake contribute to tolerance to the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 2004;309:578–586. doi: 10.1124/jpet.103.062695. [DOI] [PubMed] [Google Scholar]
- Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009;60:379–407. doi: 10.1016/j.brainresrev.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnova IN, Ladenheim B, Hodges AB, Volkow ND, Cadet JL. Chronic methamphetamine administration causes differential regulation of transcription factors in the rat midbrain. PLoS One. 2011;6:e19179. doi: 10.1371/journal.pone.0019179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laćan G, Hadamitzky M, Kuczenski R, Melega WP. Alterations in the striatal dopamine system during intravenous methamphetamine exposure: Effects of contingent and noncontingent administration. Synapse. 2013;67:476–488. doi: 10.1002/syn.21654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau JW, Senok S, Stadlin A. Methamphetamine-induced oxidative stress in cultured mouse astrocytes. Ann N Y Acad Sci. 2000;914:146–156. doi: 10.1111/j.1749-6632.2000.tb05192.x. [DOI] [PubMed] [Google Scholar]
- Lominac KD, Sacramento AD, Szumlinski KK, Kippin TE. Distinct neurochemical adaptations within the nucleus accumbens produced by a history of self-administered vs non-contingently administered intravenous methamphetamine. Neuropyschopharmacology. 2012;37:707–722. doi: 10.1038/npp.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, Vosmer G, Seiden LS. Dopamine uptake inhibitors block long-term neurotoxic effects of methamphetamine upon dopaminergic neurons. Brain Res. 1990;513:274–279. doi: 10.1016/0006-8993(90)90467-p. [DOI] [PubMed] [Google Scholar]
- McFadden LM, Hadlock GC, Allen SC, Vieira-Brock PL, Stout KA, Ellis JD, Hoonakker AJ, Andrenyak DM, Nielsen SM, Wilkins DG, Hanson GR, Fleckenstein AE. Methamphetamine self-administration causes persistent striatal dopaminergic alterations and mitigates the deficits caused by a subsequent methamphetamine exposure. J Pharmacol Exp Ther. 2012A;340:295–303. doi: 10.1124/jpet.111.188433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden LM, Hoonakker AJ, Vieira-Brock PL, Stout KA, Sawada KA, Ellis JD, Allen SC, Walters ET, Nielsen SM, Gibb JW, Alburges ME, Wilkins DG, Hanson GR, Fleckenstein AE. Methamphetamine treatment during development attenuates the dopaminergic deficits caused by subsequent high-dose methamphetamine administration. Synapse. 2011;65:771–777. doi: 10.1002/syn.20902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden LM, Hunt MM, Vieira-Brock PL, Muehle J, Nielsen SM, Allen SC, Hanson GR, Fleckenstein AE. Prior methamphetamine self-administration attenuates serotonergic deficits induced by subsequent high-dose methamphetamine administrations. Drug Alcohol Depend. 2012B;126:87–94. doi: 10.1016/j.drugalcdep.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Diaz-Corrales FJ, Fukuda M, Kitaichi K, Miyoshi K, Ogawa N. Methamphetamine-induced dopaminergic neurotoxicity is regulated by quinone-formation-related molecules. FASEB J. 2006;20:571–573. doi: 10.1096/fj.05-4996fje. [DOI] [PubMed] [Google Scholar]
- O’Neil ML, Kuczenski R, Segal DS, Cho AK, Lacan G, Melega WP. Escalating dose pretreatment induces pharmacodynamic and not pharmacokinetic tolerance to a subsequent high-dose methamphetamine binge. Synapse. 2006;60:465–473. doi: 10.1002/syn.20320. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Gibb JW. Role of the dopamine uptake carrier in the neurochemical response to methamphetamine: Effects of amfonelic acid. Eur J Pharmacol. 1985A;109(1):73–80. doi: 10.1016/0014-2999(85)90541-2. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Sonsalla PK, Hanson GR, Peat MA, Gibb JW. Methamphetamine-induced depression of monoamine synthesis in the rat: development of tolerance. J Neurochem. 1985B;44:852–855. doi: 10.1111/j.1471-4159.1985.tb12893.x. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R, O’Neil ML, Melega WP, Cho AK. Escalating dose methamphetamine pretreatment alters the behavioral and neurochemical profiles associated with exposure to a high-dose methamphetamine binge. Neuropsychopharmacology. 2003;28:1730–1740. doi: 10.1038/sj.npp.1300247. [DOI] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Jones SR. Amphetamine potency varies with dopamine uptake rate across striatal subregions. J Neurochem. 2014;131:348–355. doi: 10.1111/jnc.12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanski R, Justinova Z, Hayashi T, Takebayashi M, Goldberg SR, Su TP. Sigma1 receptor upregulation after chronic methamphetamine self-administration in rats: a study with yoked controls. Psychopharmacology. 2004;175:68–75. doi: 10.1007/s00213-004-1779-9. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett. 2004;367:349–354. doi: 10.1016/j.neulet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2007.05155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata DA, Yamamoto BK. Interactions between methamphetamine and environmental stress: role of oxidative stress, glutamate and mitochondrial dysfunction. Addiction. 2007;102(Suppl 1):49–60. doi: 10.1111/j.1360-0443.2007.01770.x. [DOI] [PubMed] [Google Scholar]
- Zhu JP, Xu W, Angulo JA. Disparity in the temporal appearance of methamphetamine-induced apoptosis and depletion of dopamine terminal markers in the striatum of mice. Brain Res. 2005;1049:171–181. doi: 10.1016/j.brainres.2005.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Rats were allow to press for METH (0.12 mg/infusion) for 7 d (8 h/d). Panel A: Pressing behavior corresponding to Figure 3. Average total METH intake over the course of the 7 d was 22.33 mg ± 1.82 mg. Panel B: Pressing behavior for corresponding to animals used to assess VMAT2 uptake at 16 h after the last self-administration session. Average total METH intake over the course of the 7 d was 19.16 mg ± 2.11 mg. Panel C: Pressing behavior corresponding to Figure 4. Average total METH intake over the course of the 7 d was 19.96 mg ± 1.24 mg. Panel D: Pressing behavior corresponding to Figures 2A, 5A, and 5B. Average total METH intake over the course of the 7 d was 20.29 mg ± 1.01 mg. Panel E: Pressing behavior corresponding to Figure 5C. Average total METH intake over the course of the 7 d was 18.94 mg ± 1.63 mg. Panel F: Pressing behavior corresponding to Figure 6A. Average total METH intake over the course of the 7 d was 21.92 mg ± 2.48 mg. Panel G: Pressing behavior corresponding to Figures 6B, 6C, and 7. Average total METH intake over the course of the 7 d was 16.26 mg ± 1.33 mg. Panel H: Pressing behavior corresponding to Figure 7. Average total METH intake over the course of the 7 d was 21.24 mg ± 1.40 mg.