
Table 1. Summary of crystallographic analysis.
Values in parentheses are for the outer resolution shell.
| Protein | FnIII-3 (14571548) | FnIII-4 (15721666) | ||
|---|---|---|---|---|
| Data set | Native | Native 1 | EMTS soak | Native 2 |
| Data collection | ||||
| Space group | I212121 | P41212 | P41212 | P41212 |
| Unit-cell parameters () | ||||
| a | 66.7 | 56.9 | 56.9 | 57.3 |
| b | 68.3 | 56.9 | 56.9 | 57.3 |
| c | 88.4 | 78.4 | 78.4 | 76.3 |
| Wavelength () | 1.5418 | 1.5418 | 1.5418 | 0.9793 |
| Resolution () | 1.60 (1.691.60) | 1.80 (1.901.80) | 1.95 (2.051.95) | 1.50 (1.541.50) |
| Unique reflections | 26721 | 11858 | 17812† | 19975 |
| Average multiplicity | 9.0 (8.1) | 19.7 (20.1) | 15.0 (14.6) | 24.3 (26.3) |
| Completeness (%) | 98.6 (91.2) | 95.0 (89.5) | 99.6 (99.9) | 95.3 (99.8) |
| R meas ‡ | 0.039 (0.313) | 0.052 (0.609) | 0.072 (0.420) | 0.062 (1.433) |
| CC1/2 | 1.00 (0.96) | 1.00 (0.98) | 1.00 (0.98) | 1.00 (0.89) |
| I/(I) | 41.7 (7.7) | 45.1 (7.8) | 32.4 (8.8) | 33.9 (3.3) |
| SIRAS phasing | ||||
| R iso § | 0.344 | |||
| Phasing power (iso, acentric/iso, centric/ano) | 2.38/2.06/2.36 | |||
| FOM (acentric/centric) | 0.59/0.51 | |||
| Model refinement | ||||
| No. of protein molecules in asymmetric unit | 2 | 1 | ||
| Resolution range () | 351.60 | 461.50 | ||
| Unique reflections (work/free) | 25369/1340 | 18911/992 | ||
| R work/R free ¶ | 0.151/0.180 | 0.195/0.218 | ||
| No. of non-H atoms | ||||
| Protein†† | 826/822 | 773 | ||
| Waters | 222 | 122 | ||
| Sulfates | 10 | |||
| PEGs | 20 | |||
| Average B values (2) | ||||
| Wilson plot | 14.2 | 20.0 | ||
| Protein†† | 19.2/20.9 | 28.5 | ||
| Waters/sulfates/PEGs | 30.2 | 34.8 | ||
| Sulfates | 31.3 | |||
| PEGs | 27.6 | |||
| R.m.s.d. from ideal geometry‡‡ | ||||
| Bond lengths () | 0.009 | 0.012 | ||
| Angles () | 1.2 | 1.3 | ||
| R.m.s.d. B factors§§ (2) | ||||
| Main chain | 1.5 | 3.9 | ||
| Side chain | 3.4 | 6.2 | ||
| Ramachandran plot¶¶ | ||||
| Residues in favoured regions | 234 [98.3] | 108 [98.2%] | ||
| Additionally allowed | 4 [1.7%] | 2 [1.8%] | ||
| Outliers | 0 [0.0%] | 0 [0.0%] | ||
| Side-chain rotamer outliers¶¶ | 1 [0.5%] | 1 [1.2%] | ||
| Clashscore¶¶ | 0.60 | 0.66 | ||
| PDB code | 4wtw | 4wtx | ||
Keeping Bijvoet pairs separate.
The redundancy-independent R factor R
meas = 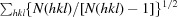
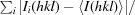
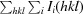 , where Ii(hkl) is the ith measurement and I(hkl) is the mean of all measurements of I(hkl) (Diederichs Karplus, 1997 ▶).
, where Ii(hkl) is the ith measurement and I(hkl) is the mean of all measurements of I(hkl) (Diederichs Karplus, 1997 ▶).
R
iso = 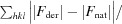
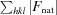 , where F
der is the heavy-atom-derivative structure factor and F
nat is the protein structure factor.
, where F
der is the heavy-atom-derivative structure factor and F
nat is the protein structure factor.
R
work = 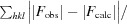
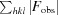 ; R
free was calculated using a randomly chosen 5% of reflections that were not included in the refinement and R
work was calculated for the remaining reflections.
; R
free was calculated using a randomly chosen 5% of reflections that were not included in the refinement and R
work was calculated for the remaining reflections.
Values for each protein chain.
Referred to the ideal geometry defined by Engh Huber (1991 ▶).
Calculated for bonded atom pairs with MOLEMAN2 (Kleywegt, 1997 ▶).
Values were obtained using MolProbity (Chen et al., 2010 ▶).