

Abstract
Background
Phosphohistidine phosphatase 1 (PHPT1), also named protein histidine phosphatase (PHP), is a eukaryotic enzyme dephosphorylating proteins and peptides that are phosphorylated on a histidine residue. A preliminary finding that histone H1, which lacks histidine, was phosphorylated by phosphoramidate and dephosphorylated by PHPT1 prompted the present investigation.
Methods
Histone H1 and polylysine were phosphorylated at a low concentration (3.9 mM) of phosphoramidate. Their dephosphorylation by recombinant human PHPT1 was investigated by using a DEAE-Sepharose spin column technique earlier developed by us for studies on basic phosphoproteins and phosphopeptides. Determination of protein-bound, acid-labile phosphate was performed by a malachite green method. Mass spectrometry (MS) was used to investigate the occurrence of N-ε-phospholysine residues in a phosphorylated histone H1.2 preparation, and to measure the activity of PHPT1 against free N-ω-phosphoarginine.
Results
Histone H1.2, which lacks histidine, was phosphorylated by phosphoramidate on several lysine residues, as shown by MS. PHPT1 was shown to dephosphorylate phosphohistone H1 at a rate similar to that previously described for the dephosphorylation of phosphohistidine-containing peptides. In addition, phosphopolylysine was an equally good substrate for PHPT1. However, no dephosphorylation of free phosphoarginine by PHPT1 could be detected.
Conclusion
The finding that PHPT1 can dephosphorylate phospholysine in chemically phosphorylated histone H1 and polylysine demonstrates a broader specificity for this enzyme than known so far.
Keywords: Histone H1, phosphohistidine phosphatase, phospholysine, phospholysine phosphatase, PHP, PHPT1, protein histidine phosphatase
Introduction
The discovery of protein phosphorylation on histidine (1) was made 52 years ago (2). Most of the work related to phosphohistidine has so far been performed on the bacterial phosphoenolpyruvate-glucose phosphotransferase system (3) and two-component systems (4). In eukaryotic cells, protein-bound phosphohistidine constitutes a significant amount of the total phospho-amino acid (5). Still, only a very low number of phosphohistidine-containing proteins have been identified in contrast to the great number of proteins phosphorylated on serine, threonine and tyrosine (2,6,7). The proteins phosphorylated on histidine are to a large extent represented by catalytic intermediates of metabolic enzymes, such as the extensively studied nucleoside diphosphate kinase (NDPK) that transiently forms 1-phosphohistidine (8,9) and ATP-citrate lyase and succinyl-CoA synthetase (succinate thiokinase) that similarly form 3-phosphohistidine in their active sites (10-15). The slow progress in eukaryotic phosphohistidine research can be explained by the lability of the N–P bonds of phosphohistidines at the acid conditions that are routinely used in most phosphoprotein research (5,16-18). However, recent developments of specific antibodies in combination with mass-spectrometric methods adapted to the detection of phosphohistidine (19) give hope for a change in the near future.
In 2002, a new tool for studies of eukaryotic histidine phosphorylation was obtained in the form of a 14 kDa phosphohistidine phosphatase (PHPT1) that was independently discovered and characterized by Ek et al. (20) and Klumpp et al. (21). The latter group named the enzyme protein histidine phosphatase (PHP). No activity of this enzyme could be detected toward a set of O-phosphorylated peptides containing phosphoserine, phosphothreonine, or phosphotyrosine (20). The activity was independent of divalent cations, and okadaic acid did not inhibit the enzyme (20,21). By mutational studies of the recombinant human enzyme, His-53 was shown to be essential for the phosphatase activity toward phosphohistidine (22). The 3D structure of the enzyme was determined both by X-ray crystallography (23) and NMR (24). ATP-citrate lyase and the β-subunit of G-protein were soon identified as potential physiological substrates (25,26). Convincing evidence of a physiological role for PHPT1 has been obtained from studies of the potassium channel KCa3.1 that is phosphorylated by nucleoside diphosphate kinase B (NDPK-B) on histidine residue 358 (27,28). In these experiments, ion transportation was activated by the phosphorylation and deactivated by a PHPT1-dependent dephosphorylation. Similar results have been obtained for the calcium channel TRPV5, although a direct proof of the formation of phosphohistidine in this case is still lacking (29). The interesting possibility of NDPK acting as a protein kinase with PHPT1 as the balancing phosphoprotein phosphatase has been discussed in a review by Wieland et al. (30). Furthermore, evidence for a possible role of PHPT1 in cytoskeletal reorganization (31,32) and in hepatocellular carcinoma cell proliferation (33) has been presented. In several of the experiments above, the possibilities to modulate the expression of the PHPT1 gene have been crucial.
Another approach to study the substrate specificity of PHPT1 was described by Attwood et al. in their study of chemically phosphorylated histone H4 peptides containing His-18 and His-75 (34). These phosphopeptides were good substrates of PHPT1. In parallel, our group designed a new method for the determination of phosphohistidine phosphatase activity based on highly basic substrates, such as chemically phosphorylated histone H4 and Ac-VRLKHRKLR-pNA, a peptide representing the amino acid sequence around His-358 of KCa3.1 (35). During preliminaries to the latter work several histone preparations were tested beside histone H4. Commercial preparations of histone H1 from calf thymus, in spite of their lack of histidine, were phosphorylated to the highest degree of all histones tested and were dephosphorylated by PHPT1 at high rates. These unexpected results have been investigated in the present study.
Materials and methods
Materials
Human recombinant phosphohistidine phosphatase (PHPT1) was expressed and purified as described by Ma et al. (22). Calf thymus histone H1 (IIIS) and polylysine were from Sigma-Aldrich (Stockhom, Sweden). Calf thymus histone H1 was also from Calbiochem(Stockhom, Sweden), Abcam (Cambridge, UK), Santa Cruz (Heidelberg, Germany), and SignalChem (Stockhom, Sweden). Trypsin was from Promega (Stockhom, Sweden). N-ω-phospho-L-arginine was from Sigma-Aldrich. Non-radioactive phosphoramidate was synthesized by applying the method described for [32P]phosphoramidate by Buckler and Stock (36). Malachite green reagent was Biomolgreen from AH Diagnostics (Stockholm, Sweden). DEAE-Sephacel was from GE Health Care (Uppsala, Sweden). Micro Bio-Spin columns were obtained from BioRad (Stockholm, Sweden).
Methods
Protein phosphorylation and dephosphorylation. Histone H1 and polylysine were chemically phosphorylated by 3.9 mM phosphoramidate as described for histone H4 (35). Thus, 50 μg of histone H1, 100 μg of 30 kDa polylysine, or 100 μg of 90 kDa polylysine, each dissolved in 25 μL 10 mM HCl, were separately mixed with 25 μL 25 mM Tris/HCl (pH 8.5) and 1 μL 0.2 M phosphoramidate to give pH 7.0 and incubated for at least 24 h. Tween 20 was then added to give the final concentration 0.04% (w/v), and the incubation was interrupted, either by freezing at 80°C or by centrifugation for 2 min at 900 g on a 200-μL DEAE-Sephacel spin column equilibrated in 25 mM Tris/HCl (pH 8.5) as described (35). The eluted volume was immediately centrifuged on a second DEAE-Sephacel column, and the eluted volume from the latter column was used for dephosphorylation experiments and analysis of the acid-labile phosphate bound to the basic protein, as described (35). Biomolgreen was used as the malachite green reagent.
Dephosphorylation was performed at pH 7.5 and 30°C by incubating a 50-μL aliquot of the eluted volume from the second spin column with 5 pmol of PHPT1 in 1 μl 25 mM Hepes (pH 7.5) for indicated times. The incubation was interrupted by an immediate centrifugation at 900 g on a DEAE-Sephacel spin column to bind PHPT1 and any released orthophosphate. The eluted volume from this column was analyzed for remaining acid-labile, protein-bound phosphate as described above. Histone H1 and polylysine concentrations were estimated by UV spectrometry at 280 nm and 215 nm, respectively.
Investigation of PHPT1 activity against N-ω-phosphoarginine. N-ω-phosphoarginine (250 µM) was incubated for 40 min with recombinant PHPT1 in the same buffers and volumes as described above for phosphohistone H1 and phosphopolylysine. N-ω-phosphoarginine and arginine were analyzed by MS, as described below.
Mass-spectrometric analysis. Chromatographically purified histone H1 from SignalChem was chemically phosphorylated as described above for the H1 preparation from Sigma-Aldrich, and then diluted with nine volumes of 0.1 M ammonium hydrogen carbonate and digested with 1 µg trypsin/100 µg histone for 3 h. The digestion was interrupted by freezing the sample at –20°C. A 1-µL thawed sample was run on a Thermo Orbitrap Velos (Termo Scientific, Stockholm, Sweden) in 0.1% (w/v) formic acid, using a gradient from 4% to 30% acetonitrile in 120 min, followed by a steep gradient to 48% in 10 min, and finally by a washing step at 80% acetonitrile. MS1 resolution was held at 60,000 (full width at half maximum), and fragments were measured between 300 and 1800 Da. Fragmentation was generated with collision-induced dissociation (CID). The LC-column was a Thermo Easy column C18 (100 mm × 75 µm and 3 µm beads) from Termo Scientific. Buffer A was 0.1% formic acid in H2O, and buffer B was 0.1% formic acid in acetonitrile. The instrument was using lock-mass for improved mass accuracy. The resulting raw-format files were analyzed by XTandem over the web. Beside the default settings, phosphorylated serine, threonine, and lysine were chosen as possible modifications.
For MS analysis of phosphoarginine and arginine, a Thermo Velos equipment was used in infusion mode. A syringe pump delivered 3 µL/min of 50% methanol (no acid) into which the sample was diluted with four parts of solvent. The instrument’s standard capillary inlet was used, and when the analysis seemed stable several manual recordings of 1 min were made. The spectra were averaged, and the 255.08 Da peak was used for comparison of the plus with the minus dephosphorylation samples. To strengthen further the identification of phosphoarginine, a brief fragmentation was also recorded showing the characteristic loss of HPO3 (80 Da).
Results
Phosphorylation of histone H1
In the pre-study of commercial histones mentioned in the Introduction, the chemical phosphorylation by phosphoramidate was found to be highest for histone H1 from calf thymus. Under the conditions used, all histone H1 preparations could be chemically phosphorylated to a level of 1–1.5 mol phosphate/mol protein. By amino acid analysis, performed by the Amino Acid Analysis Center at Uppsala University, Sweden, the histidine content of the histone H1 preparation from Sigma-Aldrich (IIIS) was found to be less than 0.2 mol/mol protein and could thus not account for the observed level of acid-labile phosphorylation. This histone is obtained from the supernatant after precipitation of nuclear fraction with 5% trichloroacetic acid (37). The amino acid analysis data were compatible with reported primary structures of histones H1.2–5. Therefore, this histone H1 preparation was judged to be suitable for the present phosphorylation/dephosphorylation experiments. Although the content of arginine was found to be 4.4 mol/mol protein, the possibility of arginine phosphorylation by phosphoramidate can probably be excluded (38). Since, however, a phosphorylation of peptide-bound lysine residues by phosphoramidate has been demonstrated (38), a phosphorylation of some of the numerous lysine residues (e.g. 59 lysine residues/mol protein in histone H1.2) was considered possible. Compared to this high number of lysine residues, the phosphorylation of histone H1 is quite low. This can be explained by the initial concentrations used for phosphoramidate (3.9 mM) and histone H1 (0.05 mM), which are much lower than used for histidine-containing peptides (20,34) and lysine-containing peptides (38). The low initial concentration of phosphoramidate was, however, a deliberate choice in order not to exceed the binding capacity of the DEAE-Sephacel spin columns used as described under Methods.
Dephosphorylation by PHPT1 of chemically phosphorylated histone H1
All preparations of phosphorylated histone H1 could be dephosphorylated by PHPT1. One set of experiments is described in Figure 1. The initial rate of the PHPT1-catalyzed dephosphorylation was 1.0 ± 0.1 mol/s per mol of enzyme (mean and SD of seven separate experiments). This rate is in the same order of magnitude as previously described for the dephosphorylation of chemically phosphorylated histone H4 (35) and a chemically phosphorylated histidine-containing peptide (20). In the absence of PHPT1, phosphorylated histone H1 was stable under the conditions used, i.e. the mean remaining phosphohistone H1 for seven separate experiments at 0, 5, and 10 min was 101% ± 2%. Other commercial histone H1 preparations, phosphorylated to about the same degree, were dephosphorylated by PHPT1 at similar initial rates (data not shown).
Figure 1.
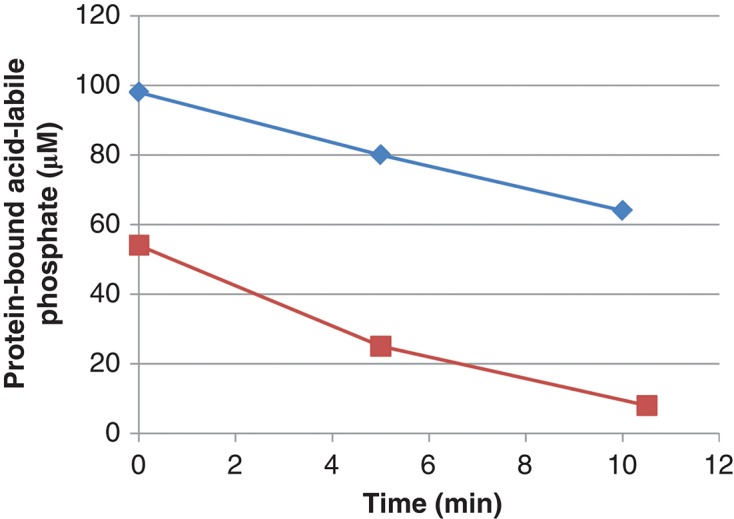
Decrease in phosphate in phosphoramidate phosphorylated histone H1 (▪) and 30 kDa polylysine (⧫) during incubation with PHPT1. The concentration was 1 mg/mL of phosphohistone and 2 mg/mL of phosphopolylysine. An amount of 5 pmol PHPT1 was added per 51 µL incubation volume. The reaction was performed at pH 7.5 and 30°C during indicated times and was interrupted by centrifugation of 50 µL of the reaction mixture through a spin column containing 200 µL of DEAE-Sepharose equilibrated in 25 mM Tris/HCl pH 8.5. The protein-bound, acid-labile phosphate in the final eluate was analyzed as described under Methods. Each time point was analyzed in duplicate.
Phosphorylated polylysine and its dephosphorylation by PHPT1
To confirm that protein-bound lysine can be N-ε-phosphorylated by phosphoramidate and then dephosphorylated by PHPT1, polylysine (30 kDa) was investigated. When phosphorylated under conditions identical to those used for histone H1, a phosphorylation of 1.5–2 mol/mol polylysine was usually obtained. Phosphopolylysine was dephosphorylated by PHPT1 at a rate that was 70% ± 8% (n = 3) of that of phosphorylated histone H1 (Figure 1). In the absence of PHPT1, phosphopolylysine was stable under the conditions used, i.e. the mean remaining phosphopolylysine for three separate experiments at 0, 5, and 10 min was 100% ± 1%.
To exclude the possibility that N-terminal N-α-phospholysine in the 30 kDa polylysine was the only substrate of PHPT1, polylysine with a greater mean molecular size (90 kDa) was investigated together with the 30 kDa polylysine. Under the conditions of this experiment, performed three times, the phosphorylation was 6.0 ± 2.6 and 1.6 ± 0.5 mol/mol protein for the 90 kDa and 30 kDa polylysine, respectively. Upon extended incubation of both types of phosphorylated polylysines with PHPT1, all phosphate was released (data not shown). This shows that beside any N-terminal phospholysine, several lysine residues were N-ε-phosphorylated and could be dephosphorylated by PHPT1.
PHPT1 and phosphoarginine
No dephosphorylation of N-ω-phosphoarginine by PHPT1 could be detected, although the incubation time was 4-fold that under which phosphohistidine H1 and phosphopolylysine were significantly dephosphorylated. The intensity reported for the phosphoarginine peak was 1.33 × 106 for the control without dephosphorylation and 1.52 × 106 after attempting dephosphorylation, while the reported intensity for arginine was negligible.
MS-sequencing of chemically phosphorylated histone H1: identification of phosphorylated lysine residues
In order to control that the phosphorylation of histone H1 really occurred on lysine residues, the calf thymus histone H1 obtained from SignalChem was phosphorylated to 1.5 mol phosphate/mol protein by phosphoramidate as described above and subjected to MS-sequencing after trypsination. This chromatographically purified histone H1 preparation was chosen in order to minimize the risk of interference with the LC-MS/MS. From the MS-data, this histone H1 was interpreted by the XTandem program to be histone H1c, which corresponds to histone H1.2 according to an alternative nomenclature (39). The high reliability of this interpretation is apparent from the log(e) for this assignment, which was –683 and defined as ‘the base-10 log of the expectation that any particular protein assignment was made at random (E-value)’. Sequence data are given in Figure 2. Seven out of the 59 lysine residues were identified as targets for the chemical phosphorylation and are highlighted. Of the 305 tryptic peptides that were used to identify the protein, 14 were reported to contain phospholysine, and, of these, two contained phospholysine as the C-terminal residue (both representing Lys-46). This seems to be incompatible with the substrate specificity of trypsin, and the interpretation of Lys-46 as a target for phosphorylation may therefore be questioned. However, a phosphorylation of at least six different lysine residues seems to have been verified. Since only a fraction of each target lysine would be phosphorylated at the low phosphoramidate concentration used, this number is compatible with the total phosphorylation of 1.5 mol/mol protein.
Figure 2.

Amino acid sequence and phosphorylated sites of bovine histone H1.2 as determined by mass spectrometry. Lysine residues are marked red, and those identified as targets for the phosphorylation by phosphoramidate (i.e. seven residues) are highlighted. Out of the 305 peptides that were used to identify the protein, 14 peptides were reported to contain phospholysine.
MS spectra
Figure 3 shows the fragmentation pattern of a phospholysine-containing peptide representing residues 82–90 of histone H1.2. The peptide was selected as doubly charged, and if one follows the y-ion series an almost complete ladder is found (only the C-terminal K is missing). The bound phosphate seems to be quite stable, and the expected phospho-fragments with higher mass are all present both as doubly and singly charged, implying that the phospholysine is capable of carrying a plus charge even if phosphorylated. The lysine-bound phosphate is removed between peaks 741.3 and 533.3 in the y-series, showing that the phosphorylated residue is Lys-85. On top of the figure the small insert depicts the y- and b-ions series in a mini-format, where the size of each staple is proportional to the abundance of the corresponding fragment in the main spectrum. The main spectrum also shows peaks corresponding to 18 Da-losses of both y- and b-ions.
Figure 3.

MS/MS spectrum showing the fragmentation pattern of one of the peptides obtained after trypsin treatment of histone H1 from SignalChem. The y-ion series is shown in red, and the b-ions series is in blue. Also shown is y- and b-ions fitting with the loss of 18 Da in violet and turquoise, respectively. Other observed but unspecified mached ions are grey. The small inset at the top shows only the y- and b-ions, with the length of the staples representing the intensity of the ions.
Discussion
The present study shows that phosphohistidine phosphatase (PHPT1) can dephosphorylate a chemically phosphorylated histone H1 that does not contain histidine residues. This is explained by the finding that some of the lysine residues of histone H1 were phosphorylated during the incubation with phosphoramidate and became good substrates of PHPT1.
It is known that peptide-bound lysine residues can be N-ε-phosphorylated by phosphoramidate (38). As seen from the sequencing of the phosphorylated histone H1.2 by MS (Figure 2), several lysine residues were identified as targets for the phosphoramidate. Kowalewska et al. found that fragmentation of phospholysine peptides by CID gave a too low number of fragments to establish the phosphorylation site and suggested that this was caused by extensive neutral losses of HPO3 (80 Da) and water (18 Da) (38). However, interpretation of the spectrum in Figure 3 suggests that not all phospholysine-containing peptides show this extensive loss of HPO3 and water since it showed a sufficient number of phosphorylated fragments to allow the identification of the phosphorylated site.
It is worth noting that most of the lysine residues of histone H1, that were identified as targets for phosphoramidate, are also subject to natural post-translational modifications (39,40). Therefore, it is reasonable to suppose that some of these sites may be accessible also to enzyme-catalyzed phosphorylations, e.g. by histone H1 kinases, such as those described by Smith and co-workers (41-44) and Sikorska et al. (45). The same sites, when phosphorylated, may then be accessed also by PHPT1. One prerequisite for this to happen would be that lysine-phosphorylated histone H1, usually located in the nuclei, and PHPT1, usually located in the cytosol, can meet at least temporarily in the same cell compartment. Interestingly, it has been shown by immunohistochemical methods that part of the PHPT1 can be located also in the nuclei (www.proteinatlas.org/search/PHPT1) (46).
PHPT1 could also dephosphorylate phosphopolylysine at a rate similar to that of the phosphorylated histone H1. Whether this means that a phospholysine residue itself, rather than the amino acid sequence around the phospholysine, defines the substrate specificity of PHPT1 may be a subject of future investigations. Of interest in this context is the finding by Attwood et al. that the kinetics of the dephosphorylation of short phosphohistidine-containing peptides depends on the amino acid sequence around the phosphohistidine (34).
A free α-amino group of an N-terminal amino acid may be phosphorylated by phosphoramidate (47), although to a lesser extent than the ε-amino group of lysine residues (38). We therefore cannot entirely exclude the possibility of an N-terminal phosphorylation of histone H1 or polylysine. However, N-ε-phosphorylation actually occurred of histone H1, as judged from the MS-spectra. A similar phosphorylation occurred of polylysine, as judged from the finding that the phosphorylation increased almost four times when a 90 kDa polylysine was compared to a 30 kDa polylysine as phosphate acceptor.
The finding that PHPT1 shows activity toward phospholysine in addition to phosphohistidine adds to the fact that PHPT1 can also dephosphorylate free phosphoramidate (20,34). This activity towards phosphoramidate actually made Attwood and Wieland suggest, in a recent review, that PHPT1 (PHP) ‘may not be quite so specific for phosphohistidine’ (48).
The enzyme is, however, not active against all N–P bonds, since it did not cleave N-ω-phosphoarginine under conditions used in the present study. This result raises the question of whether PHPT1 is in fact similar to, or even identical with, the partially characterized 13 kDa phosphoamidase described in 1999 by Hiraishi et al. (49). According to these authors, the latter enzyme dephosphorylated NDPK that was autophosphorylated on a histidine residue. This is, however, in contrast to a result by Klumpp et al. who, in their studies on the dephosphorylation of ATP citrate lyase, did not obtain any dephosphorylation of autophosphorylated NDPK by PHPT1 (25). Therefore, the current evidence suggests that the two enzymes are not identical.
Other phosphatases, with activity against phospholysine, have also been reported. Comprehensive reviews on this matter have been published (50,51). In addition to the 13 kDa phosphatase, Hiraishi et al. have studied a bovine liver 56 kDa phosphatase that is active toward free phosphohistidine and phospholysine (52). The corresponding human enzyme has been cloned and further characterized, and the chromosomal location of its gene was determined to be 10q26.13 (53). According to a BLAST/Align search at www.ncbi.nlm.nih.gov its amino acid sequence shows no similarity to that of PHPT1. In addition, the PHPT1 gene has a different chromosomal location, i.e. 9q34.3 (20).
Wong et al. (54,55) investigated the presence of phospholysine phosphatase activity in soluble rat tissue extracts and in a partially purified form by using phosphorylated polylysine as the probe. Interestingly, the activity against this substrate in extracts from rat liver corresponds to the activity of PHPT1 in pig liver cytosol, as measured with a phosphohistidine peptide as the substrate (20). To our knowledge, further purification and characterization of the enzyme described by Wong et al. have not been reported, and its identity with PHPT1 is thus unknown.
In conclusion, the results of the present study suggest that PHPT1 would be active against several phospholysine-containing proteins in vivo. With the exception of histone H1, which is already shown to be enzymatically phosphorylated in vivo and in vitro (41-44), such proteins remain to be identified. We assume that the well-characterized phosphohistidine phosphatase PHPT1 will become a valuable tool in the investigation of the role also of phospholysine in eukaryotic regulatory protein phosphorylation. The value of this tool may add to future characterizations of putative lysine kinases and to new phosphoproteomic approaches of the type recently outlined for phosphorylated lysine peptides (56).
Acknowledgements
Gunilla Pettersson and Elvy Netzel are gratefully acknowledged for performing some of the experiments in this work.
Funding
The work was supported by the Medical Faculty of Uppsala University.
Footnotes
Declaration of interest The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Boyer PD, DeLuca M, Ebner KE, Hultquist DE, Peter JB. Identification of phosphohistidine in digests from a probable intermediate of oxidative phosphorylation. J Biol Chem. 1962;237:PC3306–8. [PubMed] [Google Scholar]
- 2.Kee JM, Muir TW. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family . ACS Chem Biol. 2012;7:44–51. doi: 10.1021/cb200445w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Västermark Å, Saier MH., Jr The involvement of transport proteins in transcriptional and metabolic regulations . Curr Opin Microbiol. 2014;18:8–15. doi: 10.1016/j.mib.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Podgornaia AI, Laub MT. Determinants of specificity in two-component signal transduction . Curr Opin Microbiol. 2013;16:156–62. doi: 10.1016/j.mib.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Matthews HR. Protein kinases and phosphatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: a possible regulator of the mitogen-activated protein kinase cascade . Pharmac Ther. 1995;67:323–50. doi: 10.1016/0163-7258(95)00020-8. [DOI] [PubMed] [Google Scholar]
- 6.Klumpp S, Krieglstein J. Reversible phosphorylation of histidine residues in proteins from vertebrates. Sci Signal. 2009;2:pe13. doi: 10.1126/scisignal.261pe13. [DOI] [PubMed] [Google Scholar]
- 7.Attwood PV. Histidine kinases from bacteria to humans . Biochem Soc Trans. 2013;41:1023–8. doi: 10.1042/BST20130019. [DOI] [PubMed] [Google Scholar]
- 8.Edlund B, Rask L, Olsson P, Wålinder O, Zetterqvist Ö, Engström L. Preparation of crystalline nucleoside diphosphate kinase from baker’s yeast and identification of 1 [32P]phosphohistidine as the main phosphorylated product of an alkaline hydrolysate of enzyme incubated with adenosine [32P]triphosphate . Eur J Biochem. 1969;9:451–5. doi: 10.1111/j.1432-1033.1969.tb00630.x. [DOI] [PubMed] [Google Scholar]
- 9.Moréra S, Chiadmi M, LeBras G, Lascu I, Janin J. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium . Biochemistry. 1995;34:11062–70. [PubMed] [Google Scholar]
- 10.Mårdh S, Ljungström O, Högstedt S, Zetterqvist Ö. Studies on a rat-liver cell-sap protein yielding 3 [32P]phosphohistidine after incubation with [32P]ATP and alkaline hydrolysis. Identification of the protein as ATP citrate lyase . Biochim Biophys Acta. 1971;251:419–26. doi: 10.1016/0005-2795(71)90131-0. [DOI] [PubMed] [Google Scholar]
- 11.Williams SP, Sykes BD, Bridger WA. Phosphorus-31 nuclear magnetic resonance study of the active site phosphohistidine and regulatory phosphoserine residues of rat liver ATP citrate lyase . Biochemistry. 1985;24:5527–31. doi: 10.1021/bi00341a037. [DOI] [PubMed] [Google Scholar]
- 12.Fan F, Williams HJ, Boyer JG, Graham TL, Zhao H, Lehr R, et al. On the catalytic mechanism of human ATP citrate lyase . Biochemistry. 2012;51:5198–211. doi: 10.1021/bi300611s. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell RA, Butler LG, Boyer PD. The association of readily-soluble bound phosphohistidine from mitochondria with succinate thiokinase . Biochem Biophys Res Commun. 1964;16:545–50. doi: 10.1016/0006-291x(64)90190-1. [DOI] [PubMed] [Google Scholar]
- 14.Hultquist DE, Moyer RW, Boyer PD. The preparation and characterization of 1 phosphohistidine and 3 phosphohistidine . Biochemistry. 1966;5:322–31. doi: 10.1021/bi00865a041. [DOI] [PubMed] [Google Scholar]
- 15.Fraser ME, James MNG, Bridger WA, Wolodko WT. Phosphorylated and dephosphorylated structures of pig heart, GTP specific succinyl CoA synthetase . J Mol Biol. 2000;299:1325–39. doi: 10.1006/jmbi.2000.3807. [DOI] [PubMed] [Google Scholar]
- 16.Attwood PV, Piggott MJ, Zu XL, Besant PG. Focus on phosphohistidine . Amino Acids. 2007;32:145–56. doi: 10.1007/s00726-006-0443-6. [DOI] [PubMed] [Google Scholar]
- 17.Cieśla J, Frączyk T, Rode W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim Pol. 2011;58:137–47. [PubMed] [Google Scholar]
- 18.Gonzalez Sanchez MB, Lanucara F, Helm M, Eyers CE. Attempting to rewrite history: challenges with the analysis of histidine phosphorylated peptides . Biochem Soc Trans. 2013;41:1089–95. doi: 10.1042/BST20130072. [DOI] [PubMed] [Google Scholar]
- 19.Oslund RC, Kee JM, Couvillon AD, Bhatia VN, Perlman DH, Muir TW. A phosphohistidine proteomics strategy based on elucidation of a unique gas-phase phosphopeptide fragmentation mechanism . J Am Chem Soc. 2014;136:12899–911. doi: 10.1021/ja507614f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ek P, Pettersson G, Ek B, Gong F, Li J-P, Zetterqvist Ö. Identification and characterization of a mammalian 14-kDa phosphohistidine phosphatase . Eur J Biochem. 2002;269:5016–23. doi: 10.1046/j.1432-1033.2002.03206.x. [DOI] [PubMed] [Google Scholar]
- 21.Klumpp S, Hermesmeier J, Selke D, Bechmann G, van den Brulle J, Weidner G, et al. Protein histidine phosphatase: a novel enzyme with potency for neuronal signaling . J Cereb Blood Flow Metab. 2002;22:1420–4. doi: 10.1097/01.wcb.0000045041.03034.99. [DOI] [PubMed] [Google Scholar]
- 22.Ma R, Kanders E, Beckman Sundh U, Geng M, Ek P, Zetterqvist Ö, et al. Mutational study of human phosphohistidine phosphatase: effect on enzyme activity . Biochem Biophys Res Commun. 2005;337:887–91. doi: 10.1016/j.bbrc.2005.09.134. [DOI] [PubMed] [Google Scholar]
- 23.Busam RD, Thorsell AG, Flores A, Hammarström M, Persson C, Hallberg BM. First structure of a eukaryotic phosphohistidine phosphatase . J Biol Chem. 2006;281:33830–4. doi: 10.1074/jbc.C600231200. [DOI] [PubMed] [Google Scholar]
- 24.Gong W, Li Y, Cui G, Hu J, Fang H, Jin C, et al. Solution structure and catalytic mechanism of human protein histidine phosphatase 1 . Biochem J. 2009;418:337–44. doi: 10.1042/BJ20081571. [DOI] [PubMed] [Google Scholar]
- 25.Klumpp S, Bechmann G, Mäurer A, Selke D, Krieglstein J. ATP-citrate lyase as a substrate of protein histidine phosphatase in vertebrates . Biochem Biophys Res Commun. 2003;306:110–15. doi: 10.1016/s0006-291x(03)00920-3. [DOI] [PubMed] [Google Scholar]
- 26.Mäurer A, Wieland T, Meissl F, Niroomand F, Mehringer R, Krieglstein J, et al. The β-subunit of G proteins is a substrate of protein histidine phosphatase . Biochem Biophys Res Commun. 2005;334:1115–20. doi: 10.1016/j.bbrc.2005.06.200. [DOI] [PubMed] [Google Scholar]
- 27.Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AK, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells . Mol Cell. 2006;24:665–75. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Srivastava S, Zhdanova O, Di L, Li Z, Albaqumi M, Wulff H, et al. Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1 . Proc Natl Acad Sci USA. 2008;105:14442–6. doi: 10.1073/pnas.0803678105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai X, Srivastava S, Surindran S, Li Z, Skolnik EY. Regulation of the epthelial Ca2+ channel TRPV5 by reversible histidine phosphorylation mediated by NDPK B and PHPT1 . Mol Biol Cell. 2014;25:1244–50. doi: 10.1091/mbc.E13-04-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wieland T, Hippe HJ, Ludwig K, Zhou XB, Korth M, Klumpp S. Reversible histidine phosphorylation in mammalian cells: a teeter totter formed by nucleoside diphosphate kinase and protein histidine phosphatase 1 . Methods Enzymol. 2010;471:379–402. doi: 10.1016/S0076-6879(10)71020-X. [DOI] [PubMed] [Google Scholar]
- 31.Xu A, Hao J, Zhang Z, Tian T, Jiang S, Hao J, et al. 14 kDa phosphohistidine phosphatase and its role in human lung cancer cell migration and invasion . Lung Cancer. 2010;67:48–56. doi: 10.1016/j.lungcan.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Gaji RY, Huynh MH, Carruthers VB. A novel high throughput invasion screen identifies host actin regulators for efficient cell entry by Toxoplasma gondii . PLoS One. 2013;8:e64693. doi: 10.1371/journal.pone.0064693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han SX, Wang LJ, Zhao J, Zhang Y, Li M, Zhou X, et al. 14-kDa phosphohistidine phosphatase plays an important role in hepatocellular carcinoma cell proliferation . Oncol Lett. 2012;4:658–64. doi: 10.3892/ol.2012.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Attwood PV, Ludwig K, Bergander K, Besant PG, Adina-Zada A, Krieglstein J, et al. Chemical phosphorylation of histidine containing peptides based on the sequence of histone H4 and their dephosphorylation by protein histidine phosphatase . Biochim Biophys Acta. 2010;1804:199–205. doi: 10.1016/j.bbapap.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Beckman Sundh U, Ek B, Zetterqvist Ö, Ek P. A screening method for phosphohistidine phosphatase 1 activity . Ups J Med Sci. 2011;116:161–8. doi: 10.3109/03009734.2011.585253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buckler DR, Stock AM. Synthesis of [32P]phosphoramidate for use as a low molecular weight phosphodonor reagent . Anal Biochem. 2000;283:222–7. doi: 10.1006/abio.2000.4639. [DOI] [PubMed] [Google Scholar]
- 37.de Nooij EH, Westenbrink HGK. Isolation of a homogenous lysine-rich histone from calf thymus. Biochim Biophys Acta. 1962;62:608–9. doi: 10.1016/0006-3002(62)90254-8. [DOI] [PubMed] [Google Scholar]
- 38.Kowalewska K, Stefanowicz P, Ruman T, Frączyk T, Rode W, Szewczuk Z. Electron capture dissociation mass spectrometric analysis of lysine phosphorylated peptides . Biosci Rep. 2010;30:433–43. doi: 10.1042/BSR20090167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Happel N, Doenecke D. Histone H1 and its isoforms: contribution to chromatin structure and function . Gene. 2009;431:1–12. doi: 10.1016/j.gene.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Wiśniewski JR, Zougman A, Krüger S, Mann M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics. 2007;6:72–87. doi: 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Smith DL, Bruegger BB, Halpern RM, Smith RA. New histone kinases in nuclei of rat tissues. Nature. 1973;246:103–4. doi: 10.1038/246103a0. [DOI] [PubMed] [Google Scholar]
- 42.Smith DL, Chen CC, Bruegger BB, Holtz SL, Halpern RM, Smith RA. Characterization of protein kinases forming acid labile histone phosphates in Walker 256 carcinosarcoma cell nuclei . Biochemistry. 1974;13:3780–5. doi: 10.1021/bi00715a025. [DOI] [PubMed] [Google Scholar]
- 43.Chen CC, Smith DL, Bruegger BB, Halpern RM, Smith RA. Occurrence and distribution of acid labile histone phosphates in regenerating rat liver . Biochemistry. 1974;13:3785–9. doi: 10.1021/bi00715a026. [DOI] [PubMed] [Google Scholar]
- 44.Chen CC, Bruegger BB, Kern CW, Lin YC, Halpern RM, Smith RA. Phosphorylation of nuclear proteins in rat regenerating liver . Biochemistry. 1977;16:4852–5. doi: 10.1021/bi00641a016. [DOI] [PubMed] [Google Scholar]
- 45.Sikorska M, Whitfield JF. Isolation and purification of a new 105 kDa protein kinase from rat liver nuclei . Biochim Biophys Acta. 1982;703:171–9. doi: 10.1016/0167-4838(82)90045-0. [DOI] [PubMed] [Google Scholar]
- 46.Zhang XQ, Beckman Sundh U, Jansson L, Zetterqvist Ö, Ek P. Immunohistochemical localization of phosphohistidine phosphatase PHPT1 in mouse and human tissues . Ups J Med Sci. 2009;114:65–72. doi: 10.1080/03009730802642337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalbitzer HR, Rösch P. The effect of phosphorylation of the histidyl residue in the tetrapeptide Gly Gly His Ala. Changes of chemical shift and pK values in 1H and 31P NMR spectra. Org Magn Resonance. 1981;17:88–91. [Google Scholar]
- 48.Attwood PV, Wieland T. Nucleoside diphosphate kinase as protein histidine kinase . Naunyn Schmiedebergs Arch Pharmacol. 2014 doi: 10.1007/s00210-014-1003-3. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 49.Hiraishi H, Yokoi F, Kumon A. Bovine liver phosphoamidase as a protein histidine/lysine phosphatase . J Biochem. 1999;126:368–74. doi: 10.1093/oxfordjournals.jbchem.a022459. [DOI] [PubMed] [Google Scholar]
- 50.Besant PG, Attwood PV, Piggott MJ. Focus on phosphoarginine and phospholysine . Curr Protein Pept Sci. 2009;10:536–50. doi: 10.2174/138920309789630598. [DOI] [PubMed] [Google Scholar]
- 51.Attwood PV. P-N bond protein phosphatases. Biochim Biophys Acta. 2013;1834:470–8. doi: 10.1016/j.bbapap.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 52.Hiraishi H, Yokoi F, Kumon A. 3-Phosphohistidine and 6-phospholysine are substrates of a 56 kDa inorganic pyrophosphatase from bovine liver . Arch Biochem Biophys. 1998;349:381–7. doi: 10.1006/abbi.1997.0480. [DOI] [PubMed] [Google Scholar]
- 53.Yokoi F, Hiraishi H, Izuhara K. Molecular cloning of a cDNA for the human phospholysine phosphohistidine inorganic pyrophosphate phosphatase . J Biochem. 2003;133:607–14. doi: 10.1093/jb/mvg078. [DOI] [PubMed] [Google Scholar]
- 54.Wong C, Faiola B, Wu W, Kennely J. Phosphohistidine and phospholysine phosphatase activities in the rat: potential protein lysine and protein histidine phosphatases? . Biochem J. 1993;296:293–6. doi: 10.1042/bj2960293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong C, Wu W, Obenshain SA, Diah SK, Faiola B, Kennely PJ. High molecular weight polypeptide substrates for phospholysine phosphatases . Anal Biochem. 1994;222:14–18. doi: 10.1006/abio.1994.1447. [DOI] [PubMed] [Google Scholar]
- 56.Bertran-Vicente J, Serwa RA, Schümann M, Schmieder P, Krause E, Hackenberger CPR. Site specifically phosphorylated lysine peptides . J Am Chem Soc. 2014;136:13622–8. doi: 10.1021/ja507886s. [DOI] [PubMed] [Google Scholar]