

Abstract
Hybridization between diverged taxa tests the strength of reproductive isolation, and can therefore reveal mechanisms of reproductive isolation. However, it remains unclear how consistent reproductive isolation is across species’ ranges, and to what extent reproductive isolation might remain polymorphic as species diverge. To address these questions, we compared outcomes of hybridization across species pairs of Catostomus fishes in three rivers in the Upper Colorado River basin, where an introduced species, C. commersoni, hybridizes with at least two native species, C. discobolus and C. lattipinis. We observed substantial heterogeneity in outcomes of hybridization, both between species pairs and across geographically separate rivers within each species pair. We also observed hybridization of additional related species with our focal species, suggesting that reproductive isolation in this group involves interactions of multiple evolutionary and ecological factors. These findings suggest that a better understanding of the determinants of variation in reproductive isolation is needed, and that studies of reproductive isolation in hybrids should consider how the dynamics and mechanisms of reproductive isolation vary over ecological space and over evolutionary time. Our results also have implications for the conservation and management of native catostomids in the Colorado River basin. Heterogeneity in outcomes of hybridization suggests that the threat posed by hybridization and genetic introgression to the persistence of native species likely varies with extent of reproductive isolation, both across rivers and across species pairs.
Keywords: admixture, ancestry, Catostomus, hybridization, introduced species, reproductive isolation
Introduction
Reproductive isolation between differentiated populations can persist as a result of selection against phenotypes of hybrids or migrants. Because selection pressures and phenotypes typically vary among individuals, populations and environments (Wade & Kalisz 1990), reproductive isolation is likely to vary among recently diverged lineages (Cutter 2012). Nevertheless, many studies disregard this potential for variability by examining reproductive isolation through the lens of a single geographic instance of hybridization between a single pair of species. This approach assumes that a single instance of hybridization is representative of the mechanisms of reproductive isolation across the range of a species (McDermott & Noor 2011, Cutter 2012). However, each instance of hybridization represents a separate test of reproductive isolation (Buerkle & Rieseberg 2001, Teeter et al. 2010), and variability in outcomes of hybridization can reflect variability of reproductive isolation. If variability in mechanisms and genetic architectures of reproductive isolation is prevalent, our understanding of the origin of species should reflect the dynamics of genetic components of reproductive isolation that remain polymorphic within species over time. Furthermore, prevalent variability in isolation would call greater attention to potential environment-specific fitness and plasticity of phenotypes in contributing to reproductive isolation.
Research in plant and animal hybrid zones suggests that the frequency of hybrids and dynamics of introgression can vary substantially among geographic locations where species hybridize (Barton & Gale 1993, Buerkle & Rieseberg 2001, Morgan-Richards & Wallis 2003, Vines et al. 2003, Aldridge 2005, Borge et al. 2005, Božíková et al. 2005, Aldridge & Campbell 2009, Lepais et al. 2009, Nolte et al. 2009, Gompert et al. 2010, Lexer et al. 2010, Teeter et al. 2010, Haselhorst & Buerkle 2013), but also can be remarkably consistent (Larson et al. 2014). Additionally, studies of hybrid dysfunction in experimental crosses have revealed genetic variation for traits that are likely to contribute to reproductive isolation (Arabidopsis, Leppälä & Savolainen 2011; Caenorhabditis, Kozlowska et al. 2012; Chorthippus, Shuker et al. 2005; Drosophila, Reed & Markow 2004, Kopp & Frank 2005, McDermott & Noor 2011; Helianthus, Rieseberg 2000; Mimulus, Sweigart et al. 2007; Mus, Vyskočilová et al. 2005, Good et al. 2008; Tribolium, Wade et al. 1997; additional references in Cutter 2012). While research on hybrid zones and experimental crosses provides evidence of variability in isolating barriers in a variety of taxa, important gaps remain in our knowledge. For studies of wild populations, some of the observed variation could be due to differences in geographic sampling (Larson et al. 2014), ecological factors, or uncertain estimates of hybrid ancestry. Experimental crosses can reveal variation in potential mechanisms of reproductive isolation in a subset of amenable organisms. However, mechanisms identified in a laboratory setting might not be the same mechanisms acting in natural populations, and experimental crosses are impractical in most organisms. Further study of natural populations stands to improve our assessment of the prevalance and importance of variability in isolating barriers.
Few studies have compared hybridization in multiple species pairs within a genus to assess the rate at which incompabilities arise and become fixed in diverging species (Cutter 2012, Kozlowska et al. 2012). We have evidence that time since divergence is important in determining outcomes of secondary contact (e.g., Stelkens et al. 2010) and that pre- and postzygotic isolation in mating trials increase with evolutionary divergence (e.g., Presgraves 2002, Coyne & Orr 2004, Bolnick & Near 2005, Owens & Rieseberg 2014), consistent with an accumulation through time of trait differences and incompatibilities that contribute to reproductive isolation. However, we have relatively little information on the rate at which variability in isolating barriers is maintained or lost (Cutter 2012). By studying patterns of hybridization in multiple species pairs, and in multiple geographic locations, we can better assess variability in reproductive isolation and its persistence at different stages of evolutionary divergence and in different ecological contexts.
Understanding the evolutionary and ecological context of variability in reproductive isolation will be especially important for conservation of taxa that are threatened by hybridization, particularly in cases of hybridization that follow species introductions by humans (Allendorf et al. 2001). Introductions of related but previously allopatric species can result in introgressive hybridization, which is a major concern for species conservation (Allendorf et al. 2001, Fitzpatrick et al. 2010), and could potentially result in loss of evolutionary independence for the parental species (Rhymer & Simberloff 1996, Wolf et al. 2001, Buerkle et al. 2003, Seehausen et al. 2008). Interactions with non-native species, including hybridization, may be the most significant current threats to native North American freshwater fishes (Rael 2000). Understanding the consequences of hybridization for such species will require an understanding of the extent to which hybridization varies among geographic locations and different species pairs.
In this study, we consider multiple instances of geographic contact and hybridization between native and introduced fish species, both in the context of the evolution of reproductive isolation and the conservation of native species. We focus on three native and two introduced species (suckers, Catostomidae) in three rivers of the Upper Colorado River basin in Wyoming. Evolutionary relationships between Catostomus species have been inferred based on morphology and genetics, with some disagreement among phylogenies (Doosey et al. 2010). However, two recent studies concur on the relationships among our species of interest (McDonald et al. 2008, Hopken et al. 2013), and we present these evolutionary relationships in a schematic diagram (Fig. 1). Interspecific hybridization is known to be prevalent in the Catostomidae, and has previously been studied using morphological and molecular data, both among the species in our study region (Hubbs et al. 1943, Hubbs 1955, McDonald et al. 2008, Douglas & Douglas 2008, Quist et al. 2009), and among other sucker species (Hubbs et al. 1943, McPhee & Turner 2004, Mock et al. 2006). In one river in our study region, hybridization of two native Catostomus species with an introduced species has resulted in individual fish with ancestry from three different species, which McDonald et al. (2008) hypothesized was a result of introduced white suckers acting as a “genetic bridge”. Hybridization between the introduced and native species in this case led to admixture of genomes that remain isolated in the absence of the introduced species, but it is unknown how prevalent this outcome of hybridization is across natural populations. Despite longstanding interest in hybridization in the Catostomidae, previous studies have left considerable uncertainty about which hybrid combinations are produced, the genetic ancestry of individuals thought to be hybrids based on morphology, and the extent of hybridization in different species pairs and river drainages.
Figure 1.
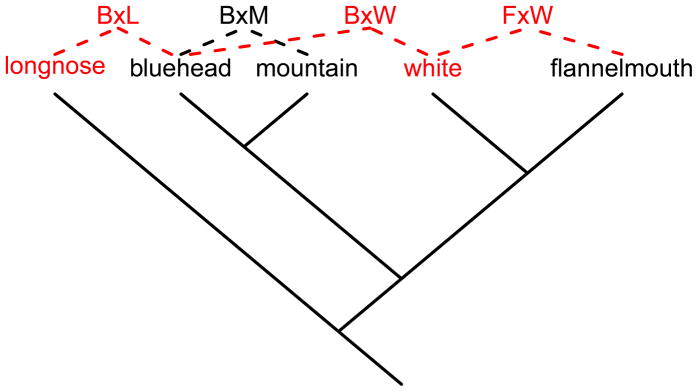
Schematic of hypothesized evolutionary relationships among Catostomus species in this study (based on McDonald et al. 2008, Hopken et al. 2013), with dashed lines showing hybridization documented in this study. Species and hybrids in black are native to the Upper Colorado River basin, while species and hybrids in red are introduced. Branch lengths are not to scale.
Here, we use high-throughput DNA sequencing and Bayesian modeling to estimate ancestry and compare outcomes of interspecific hybridization across multiple hybridizing species pairs in three rivers. Given that reproductive isolation is a consequence of potentially variable phenotypes and ecological context, and based on previous research, we expected isolation to vary among the three localities. If reproductive isolation evolves as a by-product of genetic divergence, we would expect stronger reproductive isolation and less hybridization in a more diverged species pair than in a more closely related pair (as in Presgraves 2002, Bolnick & Near 2005, Owens & Rieseberg 2014). Given the absence of formal theoretical predictions regarding the persistence of polymorphism for isolation (Cutter 2012), our comparison of hybridization across localities and species pairs can extend our understanding of evolutionary patterns. Finally, a robust genomic analysis of parental and hybrid ancestries in different ecological and evolutionary contexts is directly applicable to the conservation and management of these fish species.
Methods
Study species and sampling
Our analyses involve five Catostomus species in the Upper Colorado River basin (Fig. S1). Three of these species are native to our study area, and the other two are introduced. Flannel-mouth (Catostomus latipinnis) and bluehead (C. discobolus) suckers are native throughout the Upper Colorado River basin, according to 19th century surveys of fish diversity (Baxter et al. 1995). These species are geographically widespread, yet surveys suggest that population sizes are declining (Bezzerides & Bestgen 2002, Gill et al. 2007). Hubbs et al. (1943) documented low levels of hybridization between bluehead and flannelmouth suckers, but these two native suckers are not thought to hybridize extensively with one another (McDonald et al. 2008). Mountain suckers (C. platyrhynchus) are also native to rivers in the Upper Colorado River basin (Baxter et al. 1995), but typically occupy headwater areas upstream of the habitat utilized by flannelmouth and bluehead suckers (Weitzel 2002). Mountain suckers are morphologically similar to bluehead suckers, differing most obviously in smaller adult body size. Where these two species overlap there has been considerable taxonomic confusion and difficulty with phenotypic identification of small individuals (Baxter et al. 1995).
We also studied two Catostomus species that are not historically native to the Upper Colorado River basin (Gelwicks 2009). White suckers (C. commersoni) are native to river drainages east of the Continental divide in Wyoming and Colorado, where they are widespread and abundant. White suckers were introduced into the Upper Colorado River basin, west of the Continental Divide, probably in the first half of the 20th century (Baxter et al. 1995, Weitzel 2002, Gelwicks 2009). In their introduced range, white suckers hybridize with both native bluehead suckers and native flannelmouth suckers (McDonald et al. 2008, Douglas & Douglas 2008), and may pose a threat to the persistence of these native fish (Gelwicks 2009). Longnose suckers (C. catostomus) are also native to rivers east of the Continental Divide and have been introduced into the Upper Colorado River basin, but are not as widespread as white suckers in their introduced range. Prior to this study, they were not known to hybridize extensively with other Catostomidae (although infrequent hybridization between longnose and white suckers east of the Continental divide was previously inferred based on morphology; Hubbs et al. 1943).
We obtained fin tissue or existing DNA samples from 785 adult fish from the Upper Colorado River basin. Some tissue samples were collected by Wyoming Game and Fish personnel in summer 2011; we obtained other samples as archived tissue or extracted DNA, originally collected for McDonald et al. (2008). Sampling of fin tissue was sublethal, and fish were released following sampling. All of the summer 2011 fish were collected by electrofishing (either backpack or barge equipment) by Wyoming Game and Fish Department personnel conducting non-native fish removal. Fish were identified by field personnel based on qualititative assessment of external morphology as hybrids or pure parental species (Quist et al. 2009). Since the goal of this electrofishing was to remove all fish from a reach of river and was conducted by the same personnel in all three focal rivers, it is unlikely that our samples are biased towards particular Catostomus ancestries as a result of sampling practices. Electrofishing is inherently biased towards larger-bodied individuals, but likely is equally effective across similarly-sized species in a single genus. Sampling was non-random and does not reflect natural abundances of fish in the river. Instead, collection of fish samples for this study continued until at least 20–30 of any phenotypically identified species or hybrid present in a river had been collected.
We focus here on individuals found in three rivers in Wyoming (Fig. S1): Muddy Creek, the Big Sandy River, and Little Sandy Creek. We chose to focus on these three rivers because they have sympatric populations of bluehead, flannelmouth, and white suckers (Gill et al. 2007, Gelwicks 2009). Furthermore, all three are being targeted by the Wyoming Game and Fish Department for conservation of native flannelmouth and bluehead suckers (Senecal et al. 2010). In addition to fish from the three focal rivers, we included reference populations of each species from areas where they are unlikely to have hybridized with other species. These populations include flannelmouth suckers from Bitter Creek (WY), bluehead suckers from Ringdahl Reservoir (WY), white suckers from the Laramie River (WY), longnose suckers from Boulder Creek (CO) and North Crow Reservoir (WY), and mountain suckers from Littlefield Creek and Halfmoon Lake (WY).
DNA sequencing, assembly, and variant detection
We extracted DNA from fin clips using DNeasy Blood and Tissue Kits (Qiagen Inc.). We prepared two genomic libraries for genotyping by sequencing following protocols in Parchman et al. (2012). Briefly, we digested sample DNA with two different restriction enzymes, EcoRI and MseI, and ligated adaptors containing unique 8–10 base nucleotide barcodes to the resulting DNA fragments, thereby recognizably marking DNA fragments from each individual fish. We then pooled the barcoded restriction-ligation products and amplified barcoded fragments using PCR, with standard Illumina primers. We ran two replicate PCR reactions for each individual and then pooled the final PCR products for each of the two libraries.
Using gel electrophoresis, we size-separated DNA fragments in the pooled PCR product, and excised 350–500 base pair fragments from the gel. We used a QIAquick Gel Extraction kit (Qiagen Inc.) to purify gel punches, resulting in the final genomic libraries that were used for sequencing. Approximately 700 individual fish were included in one lane of Illumina sequencing. We chose this high level of multiplexing based on simulations that show that population genetic studies can optimize sequencing efficiency by increasing the number of individuals, at the cost of decreased sequence coverage, with little loss of ability to estimate key parameters (Li 2011, Buerkle & Gompert 2013, Skotte et al. 2013).
Genomic libraries were sequenced at the National Center for Genome Resources (Santa Fe, New Mexico) on the Illumina HiSeq 2000 platform, resulting in 114,936,817 short sequence reads (sequenced DNA fragments 84–86 nucleotides in length after removal of bar-codes and adaptor sequence). We completed a de novo assembly with a randomly chosen subset of 25 million reads, including reads from in all species, using the Seqman ngen software (DNASTAR, Inc.), and used this assembly to construct an artificial reference genome containing 592,018 contigs. We then completed independent, reference-based assemblies of reads from each individual fish to this artifical reference genome, using bwa (Burrows-Wheeler Aligner; Li & Durbin 2009). 77,467,467 total reads (67.4%) assembled to the artificial reference genome, corresponding to 39.7–76.8% of total sequence reads assembled for each individual fish. Similar numbers of reads assembled for all phenotypically identified species, suggesting that the artificial reference genome does not skew towards any one species.
We then identified 32,978 variable genetic sites (SNVs, single nucleotide variants) using samtools and bcftools (Li et al. 2009). For a variant site to be identified, we required that at least 50% of all individuals (>392 individuals) have at least 1 read at that locus. This filter ensured that sequence data for a locus would not be restricted to a single taxon, since no single Catostomus species comprised >50% of all individuals sampled. Rather than calling genotypes, most of our analyses integrated over genotype uncertainty by using genotype likelihoods as the data, which improves estimation of individual and population level parameters when working with low coverage sequence data (Li 2011, Skotte et al. 2013).
After identification of SNVs using samtools and bcftools, we converted the resulting genotype likelihoods (in Variant Call Format) to a simple composite genotype format for downstream analyses. We simultaneously removed variable sites with more than one alternate allele, so as to remove potential paralogs that may have been mistakenly assembled to the same reference locus. We stratified our variable sites by point estimates of allele frequency obtained from bcftools, and excluded rare variants (minor allele frequency less than or equal 5%). We sorted by allele frequency because common and rare variants may contain information about genetic structure at different temporal and spatial scales (Gompert et al. 2014b). To ensure independence of SNVs, we randomly selected one variable site per contig for contigs containing multiple SNVs. After filtering for allele frequency and independence, we retained genotype likelihood data for 4,095 common variants (hereafter referred to as SNPs), which we used in all further analyses. We calculated point estimates of locus-specific Hudson’s FST (Hudson et al. 1992) for each SNP in each pairwise comparison between species, which showed that many of these SNPs are diverged across species pairs, suggesting that these are informative loci.
Population genetic analyses
We estimated population genetic parameters using entropy (Gompert et al. 2014b), a program and model much like structure (Pritchard et al. 2000, Falush et al. 2003), which leads to estimates of allele frequencies in putative ancestral clusters and admixture proportions for individuals. entropy is a hierarchical Bayesian model that is closely related to the correlated allele frequency model in structure, and was previously described, tested and used in Gompert et al. 2014b. (Source code for entropy is available in the Dryad record associated with Gompert et al. 2014b, and is described in detail in the Supplemental Methods section of that paper.) Both entropy and structure only require a specification of the number of ancestral clusters to consider for modeling, and incorporate no a priori assumptions about assignment of individuals to clusters. Similar to the approach taken in Skotte et al. (2013), entropy makes use of more of the sequence data than would be possible if genotypes were called only for loci that surpass a sequence coverage threshold. Instead, the software incorporates uncertainty about the true genotype by taking genotype likelihoods from bcftools as input (Li 2011). Thus the model integrates over genotype uncertainty, and allows inference of updated genotype probabilities along with other model parameters, appropriately propagating uncertainty to higher levels of the model. Additionally, entropy implements calculations of DIC (deviance information criterion; a metric of model fit that corresponds to the likelihood of the model penalized by an estimate of the effective number of parameters) to support choice among models with different numbers of ancestral population clusters (k).
To compare statistical support for different numbers of genetic clusters (k), we ran entropy for k = 2 to k = 8. We chose this range of potential k values because we included five morphologically-identified species in our analysis. However, we also wanted to allow for the possibility that nominal species did not correspond to genetic clusters. We first used genotype likelihoods from bcftools to generate point estimates of genotype, and then calculate a genotype covariance matrix of all individuals. Following Jombart et al. (2010), we performed a principal components analysis of the genotype covariance matrix (prcomp in R; R Development Core Team 2012). We then applied k-means clustering (k means in R) and linear discriminant analysis (lda in R) to the PCA results to produce approximate starting values for q between 0 and 1 for each individual for each value of k. Starting values were informed by the data, but without reference to phenotypic species identification or collection location. We used this approach to speed convergence of our MCMC analysis, rather than starting at random values. These starting values do not constrain the posterior.
To obtain posterior probability distributions for population genetic parameters of interest (proportion of ancestry and genotype probability), we ran three independent 100,000 MCMC step chains of entropy, and discarded the first 50,000 values as burn-in. We retained every 10th value (thin = 10) and obtained 5,000 samples from the posterior distribution for each of three chains (15,000 posterior samples total). We estimated posterior means, medians, and 95% credible intervals for parameters of interest. MCMC chains were checked for mixing, stabilization, and convergence of parameter estimates by plotting a trace of the MCMC steps for a subset of parameter estimates.
Using the updated genotype estimates generated by entropy for each individual at each variant site, we calculated a new genotype covariance matrix for all individuals. We executed a principal components analysis of this genetic covariance matrix using the prcomp function in R (R Development Core Team 2012) and used this to summarize genotypic variation across all of the sampled individuals. Genotype estimates were robust to uncertainty about the number of ancestral population clusters, and were highly correlated across k values (mean correlation equals 0.923). We used the mean genotype probability across all k values in our principal components analysis (similar to Gompert et al. 2014b).
Genetic distance
Analyses with entropy produced informative estimates of genotype probability and proportion of ancestry, and allowed us to identify genetic clusters of individuals. However, entropy does not provide estimates of genetic distance between populations, so we also calculated Nei’s DA, a metric of genetic distance (Nei et al. 1983, Takezaki & Nei 1996). We used entropy q values to sort individuals by species (and to correct for a small number of phenotypically misidentified individuals), and then used genotype probabilities from entropy to calculate point estimates of allele frequency in each population of each species. We then calculated Nei’s DA (Nei et al. 1983, Takezaki & Nei 1996) from these allele frequencies, resulting in a matrix of genetic distance between all populations of all species (as in Gompert et al. 2014a). We did all calculations in R. To visualize the distance matrix, we constructed a neighbor-joining tree using the package ape in R (Paradis et al. 2004) and calculated mean Nei’s DA between and within species (Table 3).
Table 3.
Mean Nei’s D, a metric of genetic distance, both between populations of a single species, and between populations of different species. 21 populations spanning five species are represented here.
| Species | BHS (n=4) | FMS (n=5) | WHS (n=5) | LNS (n=3) | MTS (n=4) |
|---|---|---|---|---|---|
|
| |||||
| BHS | 0.0012 | 0.1045 | 0.1310 | 0.1168 | 0.0813 |
| FMS | 0.0025 | 0.1084 | 0.1000 | 0.0935 | |
| WHS | 0.0011 | 0.1105 | 0.1007 | ||
| LNS | 0.0041 | 0.0949 | |||
| MTS | 0.0037 | ||||
Genetic diversity
We estimated genetic diversity for each population of each species using samtools and bcftools (Li et al. 2009), and obtained estimates for both π, the expected heterogosity, and Watterson’s θ. We used the EM (expectation-maximization) algorithm, employing 20 iterations for each population to achieve convergence of estimates (Li 2011).
Results
Sequencing, assembly, and SNP identification
DNA sequencing resulted in 1.14× 108 short sequences (84–86 base pairs long) that matched to barcoded individuals. 7.75×107 reads (67.4%) assembled to the artificial reference genome of 592,018 contigs that resulted from the de novo assembly of a subset of 25 million reads. From the assembled contigs, we identified variable genetic sites using samtools and bcftools. After filtering by allele frequency and for independence of loci, we retained 4,095 SNPs for analysis. Sequence coverage for these retained SNPs was on average 2.65 reads per locus per individual (sd=2.32). Locus-specific FST estimates for retained SNPs confirm that these loci are informative for distinguishing parental and hybrid individuals (Fig. S2; mean locus-specific FST > 0.15).
Morphologically defined species correspond to genetic clusters
Despite known gene flow among taxa via interspecific hybridization, the five species form distinct genetic clusters across the first four principal components of genetic variation. Each species corresponds to a cluster in PC space, with members of a species across different rivers more similar to one another than to other species (Fig. 2). PC1 accounts for 72.3% of variation in the matrix of genotype covariances between individuals and separates bluehead suckers from other sucker species (Welch’s 2 sample t-test, t = 82.82, df = 765.14, p-value < 0.001). PC2 (19.6%) separates white suckers from the flannelmouth and bluehead suckers (Welch’s 2 sample t-test, t = −76.59, df = 774.92, p-value < 0.001). PC3 (6.79%) separates longnose suckers from the remaining species (Welch’s 2 sample t-test, t = 62.51, df = 128.13, p-value < 0.001). PC4 (1.08%) distinguishes mountain suckers from the other taxa (Welch’s 2 sample t-test, t = −28.20, df = 52.19, p-value < 0.001). This result that separated populations within species are genetically similar across rivers is supported by calculations of Nei’s DA (Table 3; summarized visually in Fig. 3). Values of Nei’s DA between populations of the same species are very low (all less than 0.005), suggesting that while populations of a species are not genetically identical, they are very similar to one another. In contrast, values of Nei’s DA between populations of different species are all greater than 0.08, one to two orders of magnitude higher than Nei’s DA between populations of the same species. Genetic diversity statistics for all populations of all species are shown in Fig. S3. Estimates of genetic diversity vary, but are relatively consistent across populations of a species.
Figure 2.

Principal components analysis of genotype probabilities for bluehead, flannel-mouth, white, longnose, and mountain suckers and their hybrids. Each point represents one individual fish, labeled according to phenotypic identification. Parental species are shown as solid points. Hybrid individuals are shown as crosses, and are abbreviated in the legend (e.g., “F × W” is a flannelmouth×white hybrid). Each principal component separates out a species based on individual-level genetic variation at 4,095 SNPs. A) PC1 separates bluehead suckers from flannelmouth and white suckers, while PC2 separates flannelmouth from white suckers. B) PC3 distinguishes longnose suckers from the other species. C) PC4 separates mountain suckers from the other species.
Figure 3.

An unrooted neighbor-joining tree of Neis genetic distances (D) between 210 pairs of populations shows that distances between populations within species are very small relative to differences between populations from different species. Distances are calculated based on point estimates of allele frequencies at 4,095 SNPs. The left tree has scaled branch lengths; the right hand tree is unscaled so labels are readable. Abbreviations on tips consist of the taxon abbreviation and sampling location (see Table 1, Table 2), separated by a period. Tips are color-coded by taxon, using the same colors as Fig. 2.
Individuals that were morphologically identified as hybrids occupy locations in PCA space intermediate between parental taxa. The results of our principal components analysis suggest that different pairs of hybridizing species produce very different distributions of hybrid ancestry. Bluehead×white sucker hybrids are tightly clustered in an intermediate space between the parental species on PC1, whereas flannelmouth×white hybrids are dispersed more widely along PC2, between the parental species. Flannelmouth×white hybrids exhibit variation in PC space consistent with backcrossing and introgression, while bluehead×white hybrids appear to have a more constrained, intermediate ancestry, consistent with F1 or later generation hybrids but no backcrossing. Bluehead×longnose hybrids also exhibit intermediate ancestry between parental species.
Individual admixture proportions indicate variation in hybridization by river and species pair
We estimated posterior distributions of admixture proportion (q) for individuals using entropy, for k = 2 to k = 8 genetic clusters (Fig. 4). We chose to interpret our results primarily using estimates of q from the k = 5 model. The k = 4 model was slightly more strongly supported by our model choice criterion (DIC; Fig. 4, S4), but the k = 5 model has similar statistical support and is more biologically sensible. The k = 5 model distinguishes longnose suckers from flannelmouth suckers, whereas the k = 4 model shows these two phenotypically and geographically distinct species as a single genetic cluster. Estimates of q are highly conserved across models for k. The correlation between maximum q for individuals for k = 4 and k = 5 is 0.9928, meaning that individuals are consistently identified as parental or hybrid individuals, with the same proportion of hybrid ancestry.
Figure 4.

Estimates of q from models with k = 2 to k = 8, with colors corresponding to the k = 5 model results (shown in greater detail in Fig. 5). We chose to use the k = 5 model because it is strongly supported by DIC, and is biologically sensible because each nominal species corresponds to a genetic cluster.
Under the k = 5 model, each of k genetic clusters corresponds to a morphologically identified species. Estimates of q for individual fish under the k = 5 model show variation in individual ancestry by river (Fig. 5), both in the range of hybrid ancestry and identity of hybrids produced. As shown in Fig. 6, hybrid ancestry varies by parental species pair as well as by river. Estimates of q for k = 5 have very little uncertainty. 95% credible intervals for q are not shown in Fig. 5 because for most estimates of q, the credible intervals are too narrow to be visible in a figure. Across individuals the median 95% credible interval for q was 0.0025 (the mean credible interval was 0.018, and 95% of credible intervals fell between 0.00032 and 0.13).
Figure 5.

Distribution of individual fish ancestries varies by species pair and across the three focal river drainages. Genetic clusters correspond to nominal species, and each is represented by a color. The proportion of ancestry from each cluster is shown by the height of each block of color; vertical bars composed of more than one color represent hybrid individuals. Estimates of q are shown sorted by focal river drainage, but the analysis was conducted for individuals from all rivers simultaneously.
Figure 6.

Distribution of proportion of ancestry differs in bluehead×white hybrids and flannelmouth×white hybrids. The top histograms are a comparison by species pair; the bottom histograms are a comparison of two rivers where flannelmouth×white hybrid have different ancestry distributions.
Estimates of q for each individual fish confirm that the distribution of hybrid ancestry varies by parental species pair, as previously suggested by the principal components analysis. Bluehead×white sucker hybrid ancestry in our samples was mostly intermediate between parental species, while flannelmouth×white sucker q estimates represented a much broader range of potential hybrid ancestries (Fig. 5; Fig. 6). Distributions of hybrid ancestry are statistically different in bluehead×white and flannelmouth×white hybrids (two-sample Kolmogorov-Smirnov test, D = 0.304, p-value < 0.05). Calculations of Nei’s DA confirm that bluehead and white suckers are more genetically distant from one another than flannelmouth and white suckers (Table 3). We constructed an unrooted neighbor-joining tree based on Nei’s DA to visualize genetic distance between all populations of parental species (Fig. 3). Although not a rigorously constructed phylogeny, this distance tree suggests that previously published phylogenies for this group of species based on one or two mitochondrial regions (e.g., McDonald et al. 2008, Hopken et al. 2013) might need to be revisited, since our distance tree suggests that longnose suckers and white suckers might be more closely related.
Individual estimates of q also revealed substantial variation in outcomes of hybridization across different rivers within hybridizing species pairs (Fig. 5, Fig. 6). For example, flannelmouth×white sucker hybrids occur in both Muddy Creek and the Little Sandy River, but the distribution of hybrid ancestry is different in each river (two-sample Kolmogorov-Smirnov test, D = 0.4588, p-value < 0.001). Among the rivers studied, hybridization of native fishes with introduced white suckers was most extensive in Muddy Creek, where both bluehead×white and flannelmouth×white sucker hybrids are common. Some individual suckers in this river are hybrids of three parental species (bluehead×flannelmouth×white suckers), as initially identified by McDonald et al. (2008). We did not observe three-way crosses or bluehead×flannelmouth hybrids in either of the other rivers studied. In the other two rivers, the Big Sandy River and Little Sandy Creek, bluehead×white sucker hybrids were rare or absent, despite sympatric distributions of the parental species (Sweet & Hubert 2010). Flannelmouth×white sucker hybrids also were infrequent in the Big Sandy River. Interestingly, in both of these rivers, native bluehead suckers hybridize with additional sympatric Catostomus species. In the Big Sandy River, bluehead×longnose sucker hybrids were present, while in Little Sandy Creek, bluehead×mountain sucker hybrids were present.
Discussion
A central challenge to understanding speciation is that species are to some extent evolutionarily cohesive, yet individual populations of a species also evolve somewhat independently from one another. Despite evidence that traits contributing to reproductive isolation can vary among genotypes and populations (e.g., Sweigart et al. 2007, Good et al. 2008), variation in reproductive isolation in nature is still poorly understood (Cutter 2012). If variation in realized reproductive isolation is common in nature, our conception of reproductive isolation as resulting from cohesive, species-level evolution might need to be reconsidered.
Our study complements and expands on existing work in two ways. First, by sampling at a genomic level (4,095 SNPs), we provide very precise estimates of ancestry in hybrids (median credible interval 0.0025) that are robust to model choice. Secondly, our study compares two species pairs across three instances of contact and hybridization, whereas many previous studies considered only a single species pair at each location. Our analyses revealed substantial heterogeneity in outcomes of contact between species pairs, especially in the case of hybridization between introduced white suckers and native Colorado River suckers. We focus here primarily on two dimensions of variability in outcomes of hybridization, evolutionary distance and geographic variability, and the implications for our understanding of reproductive isolation.
Isolation increases with evolutionary divergence
One view of speciation is that phenotypes contributing to reproductive isolation might evolve gradually, accumulating as a function of overall genomic divergence (Coyne & Orr 2004). If this is true, more diverged species pairs are expected to exhibit stronger reproductive isolation than less diverged species pairs. Reduction in viability of hybrid offspring with increased evolutionary distance between parental taxa has been demonstrated experimentally in diverse taxa, including sunfish (Bolnick & Near 2005), cichlids (Stelkens et al. 2010), butterflies (Presgraves 2002), and sunflowers (Owens & Rieseberg 2014). However, comparisons of hybridization across multiple species pairs have not typically been made in natural populations like those in this study.
Our comparison of hybridization of two sympatric species pairs provides some support for increased reproductive isolation at the species level, as a product of overall evolutionary divergence between two species. This inference is necessarily limited by number of rivers with hybrids that we were able to include (n=3), and we are currently working to expand our sampling. In our study rivers, the range of hybrids produced by the more diverged species pair (bluehead×white) was much more constrained than the range of hybrids produced by the less diverged species pair (flannelmouth×white; Fig. 2,5,6). For the observed range of flannelmouth×white hybrid ancestries to exist, F1 flannelmouth×white hybrids must back-cross to both parental species. In contrast, we observed few bluehead×white sucker hybrids with ancestry proportions characteristic of backcrossed hybrids (mean expectations of q = 0.25 or q = 0.75). Estimates of ancestry (q) and genotype had very little associated uncertainty, and were highly correlated across different models for k (correlations of 0.923 and 0.993, respectively), giving us high confidence in ancestry estimates.
The relative absence of bluehead×white sucker backcrosses suggests that some pre- or postzygotic mechanism of reproductive isolation, potentially operating at the species level, prevents F1 bluehead×white hybrids from successfully reproducing with parental species. If prezygotic mechanisms prevent backcrossing, several explanations are possible. Prezygotic isolation could result from hybrid sterility, spatial or temporal spawning isolation, or rejection of bluehead×white hybrids as potential mates by parental species. However, explanations related to temporal and spatial isolation appear unlikely for this system. Previous work in the Big Sandy River suggests that habitat use and timing of spawning overlap substantially for bluehead and white suckers (Sweet & Hubert 2010). Similarly, mate choice based on sexual signals is not known to be pronounced in the Catostomidae. Hybrid sterility is therefore the most plausible prezygotic mechanism, but there is currently no information on the fertility of bluehead×white sucker hybrids. Experimental crosses could provide a clear test of this hypothesis.
Postzygotic reproductive isolation could result in a narrow range of bluehead×white hybrid ancestry, if lower viability offspring are produced when an F1 bluehead×white hybrid backcrosses to bluehead or white suckers. A full range of hybrid ancestry might be produced, but selection might act to constrain the range of hybrid individuals surviving to adulthood, as in Populus hybrids (Lindtke et al. 2014). In Catostomus hybrids, it is plausible that selection against hybrids through ecological incompatibility could affect bluehead×white hybrids more strongly than flannelmouth×white hybrids. Bluehead and white suckers use different food resources (Baxter et al. 1995, Walsworth et al. 2013), and hybrids with intermediate phenotypes might be ecologically mismatched with available resources. In cichlids, it has been proposed that greater evolutionary distance between hybridizing species pairs might lead to more novel phenotypes in hybrids (Stelkens et al. 2009). Phenotypic novelty in bluehead×white hybrids could be detrimental if phenotypes do not match the environment and available resources. Finally, since fish from the three focal rivers were collected the same summer by the same personnel, it is extremely unlikely that the observed variation in hybridization across rivers and species pairs was a result of sampling bias.
Reproductive isolation is geographically variable
Some traits that contribute to reproductive isolation are likely to be consistent across the range of contact between a pair of species, but some components of reproductive isolation might also evolve at the population level. Results of this study provide support for geographic variation in realized reproductive isolation. Previous studies have identified variation in the range of hybrids produced by a species pair across geographic instances of hybridization (e.g., Aldridge 2005, Lepais et al. 2009, Haselhorst & Buerkle 2013, Pujolar et al. 2014), which corresponds to variation in realized reproductive isolation. Previous work has also described variable locus-specific introgression (e.g., Nolte et al. 2009, Teeter et al. 2010, Gompert et al. 2012), suggesting that genetic architecture of reproductive isolation might be variable in some species pairs.
In both Catostomus species pairs, replicate instances of hybridization between native and introduced species result in different ranges of hybrid ancestries. Flannelmouth×white hybrids vary from a continuum of ancestry in Muddy Creek to mostly intermediate ancestry in Little Sandy Creek and the Big Sandy River. Bluehead×white hybrids are present and are intermediate between parental species in Muddy Creek, but are infrequent or absent in the Big Sandy River and Little Sandy Creek, despite sympatry of bluehead and white suckers in these locations (Fig. 5). These results suggest that reproductive isolation and the resulting hybrid fitness vary across locations.
Potential explanations fall into two categories. First, the genetic mechanisms of reproductive isolation between species pairs could vary across rivers. Studies showing differential locus-specific introgression suggest that the genetic architecture and mechanisms of reproductive isolation may vary across replicate hybrid zones (Nolte et al. 2009, Teeter et al. 2010, Gompert & Buerkle 2010). Alternatively, differing ecological contexts across rivers could lead to different hybridization outcomes. In this case, the genetic architecture of reproductive isolation might be shared, but would have different effects in different environments. Environment-dependent fitness of interspecific hybrids in sticklebacks has been documented both in natural populations (Hatfield & Schluter 1999) and in an experimental setting (Arnegard et al. 2014), and is regarded as a major driver of divergence between pairs of benthic and limnetic species. Outcomes of hybridization might also be influenced by ecological factors such as relative abundance of parental species, as in oaks (Lepais et al. 2009). Although we do not currently have data to distinguish between these two possibilities, our results suggest that hybrid fitness and ecological compatibility of hybrids with their environments might be important for understanding why hybridization outcomes are consistent or variable.
Hybridization beyond a species pair
Typically, studies of hybridization focus on the interaction between two parental species. However, hybridizing species pairs often exist in sympatry with multiple closely related species (including some well-studied systems such as oaks and cichlid fish; Lepais et al. 2009, Keller et al. 2013). Two of our findings make the case for considering hybridization in the context of all species present. First, we confirmed a previous finding that hybrid individuals with ancestry from three parental species are present in Muddy Creek. Second, we observed hybridization of bluehead suckers with three different related species, not just with white suckers.
Hybrid suckers with ancestry from three Catostomus species were known to be present in Muddy Creek prior to this study (McDonald et al. 2008). McDonald et al. (2008) suggested that hybridization with introduced white suckers results in a “genetic bridge”, where hybridization with white suckers allows the previously isolated native species to exchange genetic material. Our analyses based on genome wide sampling confirm that three-way hybrids are present in Muddy Creek, using some of the same samples as McDonald et al. (2008), and some additional samples. With more precise estimates of ancestry in the hybrids, we are also now able to infer origins of three-way hybrids. Many of these individuals possess approximately 50% bluehead sucker genetic ancestry, with varying proportions of flannelmouth and white sucker ancestry. This suggests that in Muddy Creek, three-way hybrids are typically formed when a flannelmouth×white F1 hybrid mates with a bluehead sucker. We did not observe three-way hybrids in Little Sandy Creek or the Big Sandy River, indicating that the “genetic bridge” is not general to all rivers where these three species come into contact. This fits with our observation that reproductive isolation in this species complex is geographically variable, and indicates that both pairwise and more complex admixture might depend on ecological context. We are currently expanding our geographic sampling, and will soon be able to characterize the prevalence of three-way Catostomus hybrids in other drainages.
A second type of hybridization involving more than two species also occurs in the Catostomidae. Our genomic data indicate that native bluehead suckers hybridize with three different Catostomus species in the three rivers studied. In addition to bluehead×white sucker hybrids in Muddy Creek and the Big Sandy River, we also observed bluehead×mountain sucker hybrids in Little Sandy Creek and bluehead×longnose sucker hybrids in the Big Sandy River. It is possible that bluehead×mountain sucker hybrids are also present in the Big Sandy River, but since very few mountain suckers were sampled, it is likely that mountain sucker hybrids are very rare, if they are present. The Catostomidae are a hybridization-prone group (Hubbs et al. 1943, Hubbs 1955, Dowling & Secor 1997, McPhee & Turner 2004, Mock et al. 2006, McDonald et al. 2008), so it probable that additional hybrid combinations exist elsewhere. Unanticipated hybrid crosses involving bluehead suckers were associated with exceptionally low levels of hybridization between bluehead and white suckers in both the Big Sandy River and Little Sandy Creek. We do not currently have data from enough river drainages to comment on how strong this association is, or how common these hybrids are elsewhere in the Upper Colorado River basin. However, this result underscores the importance of studying evolutionary processes at multiple geographic locations and in more than a single pair of species.
Conservation implications
Hybridization between native and introduced species presents a challenge for conservation (Allendorf et al. 2001). Our analysis of hybridization between introduced and native suckers is directly relevant to ongoing conservation and management of native suckers (Gelwicks 2009, Senecal et al. 2010). The heterogeneous outcomes of hybridization discussed in the previous two sections suggest that successful conservation of native flannelmouth and bluehead suckers may require drainage-specific and species-specific strategies.
Additionally, our results suggest that the threat posed by introgressive hybridization to native species differs for flannelmouth suckers and bluehead suckers. In the rivers we have studied so far, hybridization between bluehead and white suckers represents a potential demographic sink. If F1 bluehead×white sucker hybrids are unable to reproduce, then hybridization with white suckers effectively removes individual bluehead suckers from the gene pool. Bluehead individuals that reproduce with white suckers are also likely to reproduce less with conspecifics, which is problematic for conservation of currently declining bluehead populations (Bezzerides & Bestgen 2002, Gill et al. 2007). These same demographic issues probably also affect flannelmouth suckers.
A second potential threat specific to flannelmouth suckers stems from the extensive back-crossing indicated by the wide range of flannelmouth×white hybrid ancestry. On a practical level, where hybridization and introgression produce a continuum of hybrid ancestry, there is also probably a continuum of hybrid phenotypes. This makes field identification of hybrids and parental species extremely difficult (Quist et al. 2009), complicating management. If backcrossing is extensive, as our results suggest, the genetic identity of the species has the potential to erode, leading to a local collapse of two species into one hybrid swarm. It is unclear how probable this outcome is, although several studies have addressed this problem (Rhymer & Simberloff 1996, Wolf et al. 2001, Buerkle et al. 2003). Species collapse by hybridization has occurred in sticklebacks (Taylor et al. 2006, Gow et al. 2006), cichlids (summarized in Seehausen et al. 2008), and whitefish (e.g., Vonlanthen et al. 2012), but many fish lineages experience extensive ongoing hybridization without loss of the parental species, including centrarchids (Bolnick & Near 2005) and salmonids (Hohenlohe et al. 2013). Since our results suggest variation in reproductive isolation in the Catostomidae, it is possible that if species collapse via hybridization occurs, it will be restricted to a subset of rivers.
Conclusions
This study makes several contributions to our understanding of hybridization, and the evolution and maintenance of reproductive isolation between species. Our overall conclusion is that realized reproductive isolation in the Catostomidae is extremely variable. From the observed variation in outcomes of hybridization, we can draw several additional conclusions. First, the results of this study are consistent with an accumulation of reproductive isolation as a consequence of species divergence. Our comparison of two species pairs across three rivers showed a more constrained range of hybrid ancestry in the more diverged species pair. Second, this study suggests that realized reproductive isolation can also be geographically variable, leading us to the contention that some components of reproductive isolation might be population-level rather than species-level phenomena. Broad geographic sampling is necessary to capture the range of variation in reproductive isolation that exists between a pair of species. Third, our results underscore the value of considering hybridization in the context of all related species present, to ensure detection of unanticipated hybrid pairings and complex hybridization involving more than two parental species. Considering the full complexity of hybridization in nature, including environmental dependence and variation in outcomes, is likely to substantially augment our understanding of the maintenance of reproductive isolation between species.
Supplementary Material
Table 1.
Five Catostomus species were sampled for this study. Species names, common names, abbreviations, and status in the Upper Colorado River basin are given in the following table.
| Species | Common name | Abbreviation | Status |
|---|---|---|---|
|
| |||
| Catostomus discobolus | bluehead sucker | BHS | native |
| Catostomus latipinnis | flannelmouth sucker | FMS | native |
| Catostomus commersoni | white sucker | WHS | introduced |
| Catostomus catostomus | longnose sucker | LNS | introduced |
| Catostomus platyrhynchus | mountain sucker | MTS | native |
Table 2.
Number of individual fish sampled from each focal river and reference population, and species sampled from each river.
| Location | Individuals | Parental Species | Hybrids |
|---|---|---|---|
|
| |||
| Big Sandy River (BS) | 224 | BHS, FMS, WHS, LNS, MTS | B×L, B×W, F×W |
| Little Sandy Creek (LS) | 164 | BHS, FMS, WHS, MTS | B×M, F×W |
| Muddy Creek (MC) | 243 | BHS, FMS, WHS | B×W, F×W, B×F×W |
| Ringdahl Reservoir (RI) | 24 | BHS | – |
| Bitter Creek (BC) | 14 | FMS | – |
| Laramie River (LR) | 8 | WHS | – |
| North Crow Reservoir (NC) | 34 | LNS | – |
| Boulder Creek (BO) | 16 | LNS | – |
| Littlefield Creek (LC) | 39 | MTS | B×W |
| Half Moon Lake (HM) | 5 | MTS | – |
| Green River, WY (GR) | 14 | FMS | F×W |
Acknowledgments
This research was funded through BLM Grant L10AC20067 to D.B. McDonald. E.G. Mandeville was supported by NIH INBRE funding to the University of Wyoming (NCRR P20RR016474/NIGMS P20GM103432), as well as scholarships from the Vern Bressler fund and the Haub School of Environment and Natural Resources. We would like to thank the Wyoming Game and Fish Department and Bureau of Land Management for assistance in obtaining samples, and for sharing their considerable experience in this study system. We specifically thank K. Gelwicks, R. Compton, D. Zafft, M. Smith, R. Keith, W. Cleary, P. Attwood, S. Albeke, and P. Lionberger. This manuscript was improved by comments from M. Haselhorst, four anonymous reviewers, and the editor.
Footnotes
Data Accessibility
Dryad Digital Repository (doi:10.5061/dryad.r31sg): Genotype likelihood data (VCF file format), artificial reference genome, reference-based assemblies, entropy input file, lists of barcodes for individuals, approximate locations of sampled populations, and input files and scripts for Nei’s D calculation and plotting of neighbor joining trees.
Raw sequence data (.fastq files) are deposited at the NCBI SRA: SRX877850.
References
- Aldridge G. Variation in frequency of hybrids and spatial structure among Ipomopsis (Polemoniaceae) contact sites. New Phytologist. 2005;167:279–288. doi: 10.1111/j.1469-8137.2005.01413.x. [DOI] [PubMed] [Google Scholar]
- Aldridge G, Campbell D. Genetic and morphological patterns show variation in frequency of hybrids between Ipomopsis (Polemoniaceae) zones of sympatry. Heredity. 2009;102:257–265. doi: 10.1038/hdy.2008.112. [DOI] [PubMed] [Google Scholar]
- Allendorf F, Leary R, Spruell P, Wenburg J. The problems with hybrids: setting conservation guidelines. Trends in Ecology & Evolution. 2001;16:613–622. [Google Scholar]
- Arnegard ME, McGee MD, Matthews B, et al. Genetics of ecological divergence during speciation. Nature. 2014 doi: 10.1038/nature13301. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH, Gale KS. Genetic analysis of hybrid zones. In: Harrison RG, editor. Hybrid Zones and the Evolutionary Process. Oxford University Press; New York: 1993. pp. 13–45. [Google Scholar]
- Baxter GT, Stone MD, Parker L. Fishes of Wyoming. Wyoming Game and Fish Department Cheyenne; 1995. [Google Scholar]
- Bezzerides N, Bestgen K. Tech rep. Larval Fish Laboratory, Colorado State University; Fort Collins: 2002. Status review of roundtail chub, flannelmouth sucker, and bluehead sucker in the colorado river basin. [Google Scholar]
- Bolnick DI, Near TJ. Tempo of hybrid inviability in centrarchid fishes (teleostei: Centrarchidae) Evolution. 2005;59:1754–1767. [PubMed] [Google Scholar]
- Borge T, Lindroos K, Nádvorník P, Syvänen AC, Sætre GP. Amount of introgression in flycatcher hybrid zones reflects regional differences in pre and post-zygotic barriers to gene exchange. Journal of Evolutionary Biology. 2005;18:1416–1424. doi: 10.1111/j.1420-9101.2005.00964.x. [DOI] [PubMed] [Google Scholar]
- Božiková E, Munclinger P, Teeter KC, Tucker PK, Machoĺan M, Píalek J. Mito-chondrial DNA in the hybrid zone between Mus musculus musculus and Mus musculus domesticus: a comparison of two transects. Biological Journal of the Linnean Society. 2005;84:363–378. [Google Scholar]
- Buerkle CA, Gompert Z. Population genomics based on low coverage sequencing: how low should we go? Molecular Ecology. 2013;22:3028–3035. doi: 10.1111/mec.12105. [DOI] [PubMed] [Google Scholar]
- Buerkle CA, Rieseberg LH. Low intraspecific variation for genomic isolation between hybridizing sunflower species. Evolution. 2001;55:684–691. doi: 10.1554/0014-3820(2001)055[0684:livfgi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Buerkle CA, Wolf DE, Rieseberg LH. The origin and extinction of species through hybridization. In: Brigham CA, Schwartz MW, editors. Population Viability in Plants: Conservation, Management, and Modeling of Rare Plants. Springer Verlag; New York: 2003. pp. 117–141. [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sinauer Associates; Sunderland, Massachusetts: 2004. [Google Scholar]
- Cutter AD. The polymorphic prelude to Bateson–Dobzhansky–Muller incompatibilities. Trends in Ecology and Evolution. 2012;27:209– 218. doi: 10.1016/j.tree.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Doosey MH, Bart HL, Saitoh K, Miya M. Phylogenetic relationships of catostomid fishes (actinopterygii: Cypriniformes) based on mitochondrial nd4/nd5 gene sequences. Molecular Phylogenetics and Evolution. 2010;54:1028–1034. doi: 10.1016/j.ympev.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Douglas M, Douglas M. Final Report to the Wyoming Game and Fish Department, WGFD Agreement 100/06. 2008. Molecular Genetic Assessment of Hybrid Suckers [Catostomidae] in the Upper Green River of Wyoming. [Google Scholar]
- Dowling TE, Secor CL. The role of hybridization and introgression in the diversification of animals. Annual Review of Ecology and Systematics. 1997;28:593–620. [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick BM, Johnson JR, Kump DK, Smith JJ, Voss SR, Shaffer HB. Rapid spread of invasive genes into a threatened native species. Proceedings of National Academy of Sciences. 2010;107:3606–3610. doi: 10.1073/pnas.0911802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelwicks K. Current Status of Roundtail Chub, Flannelmouth Sucker and Bluehead Sucker in the Green River Drainage of Wyoming. Wyoming Game and Fish Department, Fish Division; 2009. [Google Scholar]
- Gill CJ, Gelwicks KR, Keith RM. American Fisheries Society Symposium. Vol. 53. American Fisheries Society; 2007. Current distribution of bluehead sucker, flannel-mouth sucker, and roundtail chub in seven subdrainages of the green river, wyoming; p. 121. [Google Scholar]
- Gompert Z, Buerkle CA. introgress: a software package for mapping components of isolation in hybrids. Molecular Ecology Resources. 2010;10:378–384. doi: 10.1111/j.1755-0998.2009.02733.x. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Comeault AA, Farkas TE, et al. Experimental evidence for ecological selection on genome variation in the wild. Ecology Letters. 2014a;17:369–379. doi: 10.1111/ele.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert Z, Lucas LK, Buerkle CA, Forister ML, Fordyce JA, Nice CC. Admixture and the organization of genetic diversity in a butterfly species complex revealed through common and rare genetic variants. Molecular Ecology. 2014b:n/a–n/a. doi: 10.1111/mec.12811. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Lucas LK, Fordyce JA, Forister ML, Nice CC. Secondary contact between Lycaeides idas and L. melissa in the Rocky Mountains: extensive introgression and a patchy hybrid zone. Molecular Ecology. 2010;19:3171–3192. doi: 10.1111/j.1365-294X.2010.04727.x. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Lucas LK, Nice CC, Fordyce JA, Forister ML, Buerkle CA. Genomic regions with a history of divergent selection affect fitness of hybrids between two butterfly species. Evolution. 2012;66:2167–2181. doi: 10.1111/j.1558-5646.2012.01587.x. [DOI] [PubMed] [Google Scholar]
- Good JM, Handel MA, Nachman MW. Asymmetry and polymorphism of hybrid male sterility during the early stages of speciation in house mice. Evolution. 2008;62:50–65. doi: 10.1111/j.1558-5646.2007.00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow JL, Peichel CL, Taylor EB. Contrasting hybridization rates between sympatric three-spined sticklebacks highlight the fragility of reproductive barriers between evolutionarily young species. Molecular Ecology. 2006;15:739–752. doi: 10.1111/j.1365-294X.2006.02825.x. [DOI] [PubMed] [Google Scholar]
- Haselhorst MSH, Buerkle CA. Population genetic structure of Picea engelmannii, P. glauca and their previously unrecognized hybrids in the central Rocky Mountains. Tree Genetics & Genomes. 2013;9:669–681. [Google Scholar]
- Hatfield T, Schluter D. Ecological speciation in sticklebacks: environment-dependent hybrid fitness. Evolution. 1999:866–873. doi: 10.1111/j.1558-5646.1999.tb05380.x. [DOI] [PubMed] [Google Scholar]
- Hohenlohe PA, Day MD, Amish SJ, et al. Genomic patterns of introgression in rainbow and westslope cutthroat trout illuminated by overlapping paired-end rad sequencing. Molecular ecology. 2013;22:3002–3013. doi: 10.1111/mec.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopken MW, Douglas MR, Douglas ME. Stream hierarchy defines riverscape genetics of a north american desert fish. Molecular ecology. 2013;22:956–971. doi: 10.1111/mec.12156. [DOI] [PubMed] [Google Scholar]
- Hubbs C. Hybridization between fish species in nature. Systematic zoology. 1955:1–20. [Google Scholar]
- Hubbs C, Hubbs L, Johnson R. Hybridization in Nature Between Species of Catostomid Fishes. University of Michigan Press; 1943. [Google Scholar]
- Hudson RR, Slatkin M, Maddison W. Estimation of levels of gene flow from dna sequence data. Genetics. 1992;132:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller I, Wagner CE, Greuter L, et al. Population genomic signatures of divergent adaptation, gene flow and hybrid speciation in the rapid radiation of lake victoria cichlid fishes. Molecular Ecology. 2013;22:2848–2863. doi: 10.1111/mec.12083. [DOI] [PubMed] [Google Scholar]
- Kopp A, Frank A. Speciation in progress? a continuum of reproductive isolation in Drosophila bipectinata. Genetica. 2005;125:55–68. doi: 10.1007/s10709-005-4787-8. [DOI] [PubMed] [Google Scholar]
- Kozlowska JL, Ahmad AR, Jahesh E, Cutter AD. Genetic variation for postzygotic reproductive isolation between Caenorhabditis briggsae and C aenorhabditis Sp. 9. Evolution. 2012;66:1180–1195. doi: 10.1111/j.1558-5646.2011.01514.x. [DOI] [PubMed] [Google Scholar]
- Larson EL, White TA, Ross CL, Harrison RG. Gene flow and the maintenance of species boundaries. Molecular Ecology. 2014:n/a–n/a. doi: 10.1111/mec.12601. [DOI] [PubMed] [Google Scholar]
- Lepais O, Petit R, Guichoux E, et al. Species relative abundance and direction of introgression in oaks. Molecular Ecology. 2009;18:2228–2242. doi: 10.1111/j.1365-294X.2009.04137.x. [DOI] [PubMed] [Google Scholar]
- Leppälä J, Savolainen O. Nuclear-cytoplasmic interactions reduce male fertility in hybrids of Arabidopsis lyrata subspecies. Evolution. 2011;65:2959–2972. doi: 10.1111/j.1558-5646.2011.01361.x. [DOI] [PubMed] [Google Scholar]
- Lexer C, Joseph JA, van Loo M, et al. Genomic admixture analysis in European Populus spp. reveals unexpected patterns of reproductive isolation and mating. Genetics. 2010;186:699–712. doi: 10.1534/genetics.110.118828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindtke D, Gompert Z, Lexer C, Buerkle CA. Unexpected ancestry of populus seedlings from a hybrid zone implies a large role for postzygotic selection in the maintenance of species. Molecular ecology. 2014 doi: 10.1111/mec.12759. [DOI] [PubMed] [Google Scholar]
- McDermott SR, Noor MAF. Genetics of hybrid male sterility among strains and species in the Drosophila pseudoobscura species group. Evolution. 2011;65:1969–1978. doi: 10.1111/j.1558-5646.2011.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DB, Parchman TL, Bower MR, Hubert WA, Rahel FJ. An introduced and a native vertebrate hybridize to form a genetic bridge to a second native species. Proceedings of National Academy of Sciences. 2008;105:10837–10842. doi: 10.1073/pnas.0712002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee MV, Turner TF. No genetic evidence for hybridization between rio grande sucker, catostomus plebeius, and the introduced white sucker, catostomus commersoni, in the rio grande. Environmental biology of fishes. 2004;71:85–93. [Google Scholar]
- Mock KE, Evans R, Crawford M, Cardall B, Janecke SU, Miller M. Rangewide molecular structuring in the utah sucker (catostomus ardens) Molecular Ecology. 2006;15:2223–2238. doi: 10.1111/j.1365-294X.2006.02932.x. [DOI] [PubMed] [Google Scholar]
- Morgan-Richards M, Wallis GP. A comparison of five hybrid zones of the weta Hemideina thoracica (Orthoptera: Anostostomatidae): degree of cytogenetic differentiation fails to predict zone width. Evolution. 2003;57:849–861. doi: 10.1111/j.0014-3820.2003.tb00296.x. [DOI] [PubMed] [Google Scholar]
- Nei M, Maruyama T, Wu C. Models of evolution of reproductive isolation. Genetics. 1983;103:557–579. doi: 10.1093/genetics/103.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte AW, Gompert Z, Buerkle CA. Variable patterns of introgression in two sculpin hybrid zones suggest that genomic isolation differs among p opulations. Molecular Ecology. 2009;18:2615–2627. doi: 10.1111/j.1365-294X.2009.04208.x. [DOI] [PubMed] [Google Scholar]
- Owens GL, Rieseberg LH. Hybrid incompatibility is acquired faster in annual than in perennial species of sunflower and tarweed. Evolution. 2014;68:893–900. doi: 10.1111/evo.12297. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Parchman TL, Gompert Z, Mudge J, Schilkey F, Benkman CW, Buerkle CA. Genome-wide association genetics of an adaptive trait in lodgepole pine. Molecular Ecology. 2012;21:2991–3005. doi: 10.1111/j.1365-294X.2012.05513.x. [DOI] [PubMed] [Google Scholar]
- Presgraves DC. Patterns of postzygotic isolation in Lepidoptera. Evolution. 2002;56:1168–1183. doi: 10.1111/j.0014-3820.2002.tb01430.x. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujolar JM, Jacobsen M, Als TD, et al. Assessing patterns of hybridization between north atlantic eels using diagnostic single-nucleotide polymorphisms. Heredity. 2014;112:627–637. doi: 10.1038/hdy.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quist MC, Bower MR, Hubert WA, Parchman TL, McDonald DB. Morphometric and Meristic Differences among Bluehead Suckers, Flannelmouth Suckers, White Suckers, and Their Hybrids: Tools for the Management of Native Species in the Upper Colorado River Basin. North American Journal of Fisheries Management. 2009;29:460–467. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2012. [Google Scholar]
- Rahel FJ. Homogenization of fish faunas across the united states. Science. 2000;288:854–856. doi: 10.1126/science.288.5467.854. [DOI] [PubMed] [Google Scholar]
- Reed LK, Markow TA. Early events in speciation: Polymorphism for hybrid male sterility in Drosophila. Proceedings of the National Academy of Sciences. 2004;101:9009–9012. doi: 10.1073/pnas.0403106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhymer JM, Simberloff D. Extinction by hybridization and introgression. Annual Review of Ecology and Systematics. 1996;27:83–109. [Google Scholar]
- Rieseberg LH. Crossing relationships among ancient and experimental sunflower hybrid lineages. Evolution. 2000;54:859–865. doi: 10.1111/j.0014-3820.2000.tb00086.x. [DOI] [PubMed] [Google Scholar]
- Seehausen O, Takimoto G, Roy D, Jokela J. Speciation reversal and biodiversity dynamics with hybridization in changing environments. Molecular Ecology. 2008;17:30–44. doi: 10.1111/j.1365-294X.2007.03529.x. [DOI] [PubMed] [Google Scholar]
- Senecal AC, Gelwicks KR, Cavalli PA, Keith RM. WGFD Short-term Plan for the Three Species in the Green River Drainage of Wyoming; 2009–2014. Wyoming Game and Fish Department, Fish Division; 2010. [Google Scholar]
- Shuker DM, Underwood K, King TM, Butlin RK. Patterns of male sterility in a grasshopper hybrid zone imply accumulation of hybrid incompatibilities without selection. Proceedings of the Royal Society B: Biological Sciences. 2005;272:2491–2497. doi: 10.1098/rspb.2005.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotte L, Korneliussen TS, Albrechtsen A. Estimating individual admixture proportions from next generation sequencing data. Genetics. 2013;195:693–702. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelkens R, Schmid C, Selz O, Seehausen O. Phenotypic novelty in experimental hybrids is predicted by the genetic distance between species of cichlid fish. BMC Evolutionary Biology. 2009;9:283. doi: 10.1186/1471-2148-9-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelkens RB, Young KA, Seehausen O. The accumulation of reproductive incompatibilities in african cichlid fish. Evolution. 2010;64:617–633. doi: 10.1111/j.1558-5646.2009.00849.x. [DOI] [PubMed] [Google Scholar]
- Sweet DE, Hubert WA. Seasonal movements of native and introduced catostomids in the big sandy river, wyoming. The Southwestern Naturalist. 2010;55:382–389. [Google Scholar]
- Sweigart AL, Mason AR, Willis JH. Natural variation for a hybrid incompatibility between two species of Mimulus. Evolution. 2007;61:141–151. doi: 10.1111/j.1558-5646.2007.00011.x. [DOI] [PubMed] [Google Scholar]
- Takezaki N, Nei M. Genetic distances and reconstruction of phylogenetic trees from microsatellite dna. Genetics. 1996;144:389–399. doi: 10.1093/genetics/144.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB, Boughman JW, Groenenboom M, Sniatynski M, Schluter D, Gow JL. Speciation in reverse: morphological and genetic evidence of the collapse of a three-spined stickleback (Gasterosteus aculeatus) species pair. Molecular Ecology. 2006;15:343–355. doi: 10.1111/j.1365-294X.2005.02794.x. [DOI] [PubMed] [Google Scholar]
- Teeter KC, Thibodeau LM, Gompert Z, Buerkle CA, Nachman MW, Tucker PK. The variable genomic architecture of isolation between hybridizing species of house mouse. Evolution. 2010;64:472–485. doi: 10.1111/j.1558-5646.2009.00846.x. [DOI] [PubMed] [Google Scholar]
- Vines TH, Kohler SC, Thiel A, et al. The maintenance of reproductive isolation in a mosaic hybrid zone between the fire-bellied toads Bombina bombina and B. variegata. Evolution. 2003;57:1876–1888. doi: 10.1111/j.0014-3820.2003.tb00595.x. [DOI] [PubMed] [Google Scholar]
- Vonlanthen P, Bittner D, Hudson AG, et al. Eutrophication causes speciation reversal in whitefish adaptive radiations. Nature. 2012;482:357–362. doi: 10.1038/nature10824. [DOI] [PubMed] [Google Scholar]
- Vyskočilová M, Trachtulec Z, Forejt J, Piálek J. Does geography matter in hybrid sterility in house mice? Biological Journal of the Linnean Society. 2005;84:663–674. [Google Scholar]
- Wade MJ, Johnson NA, Jones R, Siguel V, McNaughton M. Genetic variation segregating in natural populations of Tribolium castaneum affecting traits observed in hybrids with T. freemani. Genetics. 1997;147:1235–1247. doi: 10.1093/genetics/147.3.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade MJ, Kalisz S. The causes of natural selection. Evolution. 1990;40:1947–1955. doi: 10.1111/j.1558-5646.1990.tb04301.x. [DOI] [PubMed] [Google Scholar]
- Walsworth TE, Budy P, Thiede GP. Longer food chains and crowded niche space: effects of multiple invaders on desert stream food web structure. Ecology of Freshwater Fish 2013 [Google Scholar]
- Weitzel DL. Conservation and Status Assessment of Wyoming’s Common Suckers: White Sucker (Catostomus commersoni), Mountain Sucker (Catostomus platyrhynchus), Longnose Sucker (Catostomus catostomus), Utah Sucker (Catostomus ardens), Quill-back (Carpoides cyprinus), River Carpsucker (Carpoides carpio), and Shorthead Redhorse (Maxostoma macrolepidotum) Wyoming Fish and Game [sic] Department, Fish Division; 2002. [Google Scholar]
- Wolf DE, Takebayashi N, Rieseberg LH. Predicting the risk of extinction through hybridization. Conservation Biology. 2001;15:1039–1053. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.