
FIG 2.
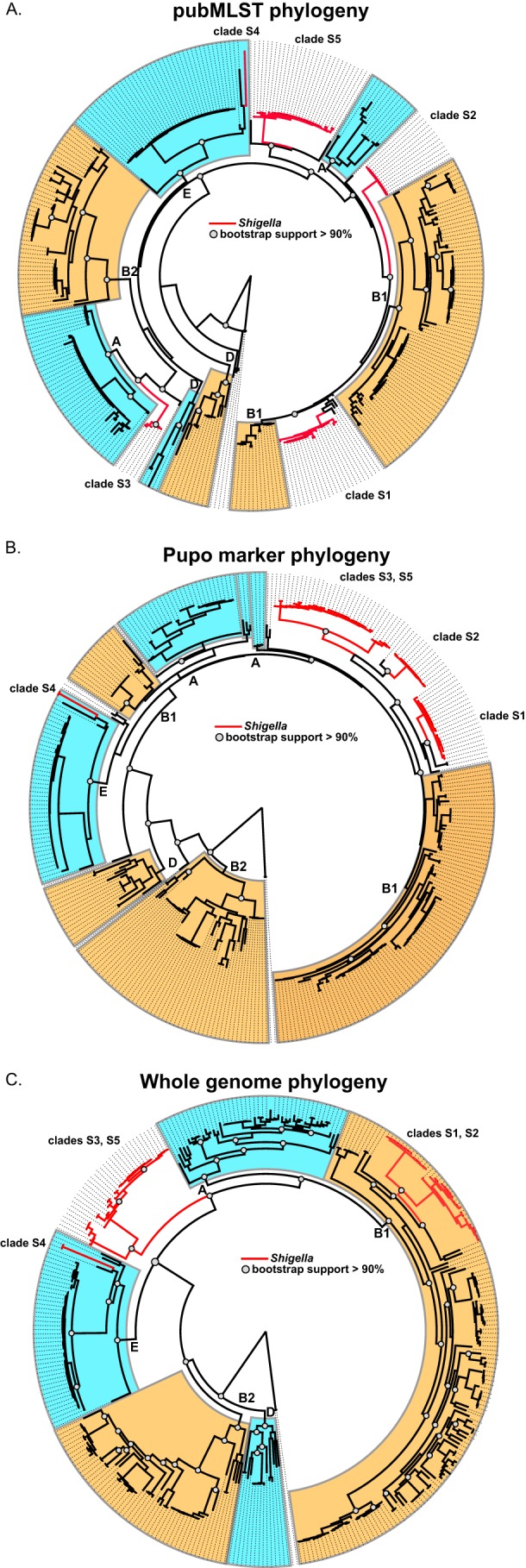
Phylogenies inferred from a diverse set of E. coli and Shigella genomes (n = 336). (A) A phylogeny inferred from a concatenation of sequences from multilocus sequence typing markers (see Table S3 in the supplemental material) from the E. coli pubMLST system (34). Conserved sequences were extracted from BLAST (31) alignments and were aligned with MUSCLE (32). The phylogenetic tree was inferred with FastTree2 (33), with 1,000 bootstrap replicates. (B) A phylogeny was inferred with FastTree2 from a concatenation of sequence markers (see Table S2) used in a previous study of Shigella evolution (15). (C) A phylogenetic tree of E. coli and Shigella isolates using whole-genome sequence data. Conserved genomic fragments were first identified in a core set of 40 E. coli genomes aligned with Mugsy (29). Conserved genomic regions were extracted by BLASTN, aligned with MUSCLE, and concatenated. A tree was then inferred on this alignment with FastTree2, with 1,000 bootstrap replicates.