
Figure 2.
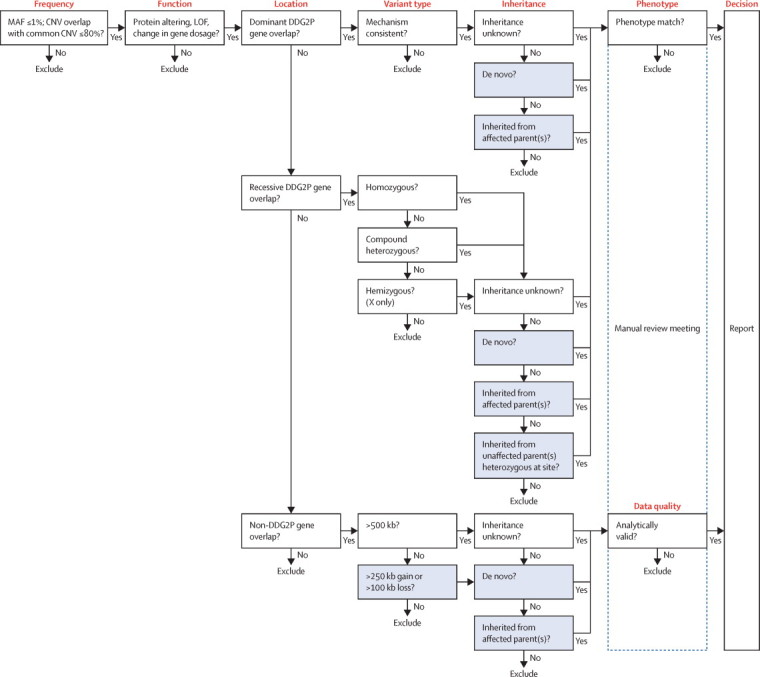
Variant filtering logic for clinical reporting within the study
Genomic variants were filtered on the basis of six factors, of which the first five were automated and the final one was done manually: (1) frequency, prevalence of the variant in the general population (MAF ≤1%); (2) function, most severe predicted functional consequence, such as LOF, defined by specific sequence ontology terms (transcript ablation, splice donor variant, splice acceptor variant, stop-gained, frameshift variant, stop-lost, initiator codon variant, in-frame insertion, in-frame deletion, missense variant, transcript amplification, and coding sequence variant); (3) location, genomic location compared with DDG2P of published genes; (4) variant type, genotype (eg, heterozygous or homozygous) and loss or gain for small CNVs (which were only considered when they contained entire genes in which LOF or dominant negative mutations had been previously reported, and gains were only considered when they overlapped genes in which increased gene dosage mutations had been previously reported); (5) inheritance, aspects of the pipeline that are dependent on inheritance information derived from parental data are shaded; and (6) phenotype, patient phenotype was manually compared against published phenotypes for a particular gene. MAF=minor allele frequency. CNV=copy number variant. LOF=loss of function. DDG2P=Developmental Disorders Genotype-to-Phenotype database.