

Abstract
Angiogenesis, the formation of new vessels from pre-existing ones, is essential during ontogenetic development and is related to many important physio-pathological processes in the adult. In fact, a persistent and deregulated angiogenesis is a required event for many diseases and pathological situations, including cancer progression and metastasis. Some rare diseases are also angiogenesis-related pathologies. However, there is a lack of an exhaustive review on the topic. The main purpose of this work is to carry out a systematic review of literature to determine what (and how much) scientific information concerning angiogenesis-related rare diseases can be extracted from available sources. After exhaustive searches in bibliographic databases, preselected data were filtered by selecting only those articles on rare diseases with an Orpha number hosted in the Orphanet web. The selected bibliographic references were further curated manually. With the 187 selected references, a critical reading and analysis was carried out allowing for an identification and classification of angiogenesis-related rare diseases, the involved genes and the drugs available for their treatment, all on the basis of the information available in Orphanet database.
Keywords: angiogenesis, rare diseases, orphanet
Introduction
A disease is considered rare when its low incidence becomes a problem to be added, making more difficult its accurate diagnosis and decreasing the interest in its research and the development of drugs for its treatment. According to the definition provided by the European Union, rare diseases are those with prevalence values lesser than 5/10,000 and leading patients to higher risk of death or chronic disability. Although it is difficult to estimate the exact number of rare diseases, most probably this number is within the range 6000–8000. More than 2000 rare diseases have already one or more genes assigned; see (http://www.rdplatform.org) and 1. The greatest database on the subject, Orphanet (http://www.orpha.net/consor/cgi-bin/index.php) contains information on almost 6000 rare diseases. Therefore, although each rare disease is infrequent, altogether rare diseases affect to 6–8% of total population in developed countries. Currently, more than 30 millions of European citizens suffer from some kind of rare disease and are exposed to discriminatory medical care benefits. Different projects have been financed under the Programme for Community Action on Rare Diseases in 1999–2003; the EU Public Health Programme 2003–2007 and the second EU Health Programme 2008–2013 (http://ec.europa.eu/health/rare_diseases/projects/cooperation/index_en.htm).
A fundamental question arises: how to get access to this highly specialized and very reduced knowledge area where each described rare disease becomes an isolated particularity? A reasonable option could be the comparative study of analogies among different rare diseases (http://www.orpha.net/consor/cgi-bin/Education_Home.php?lng=EN). However, the low prevalence (the fact of being rare) is the only descriptor that—by definition—can be applied in any methodology for the systemic study of all or a group of rare diseases. This description based on low prevalence is crucial for the integration of medical policies in the framework of rare diseases, but on the other hand it contributes nothing to the molecular description of rare diseases and the physio-pathological relationships they could share. This is the current situation, in spite of the fact that next-generation sequencing technologies and exome analysis are making possible to identify molecular markers for some rare diseases 2.
The specification of classification systems able to order the collected information on a disease according to coherent categories and criteria plays an essential role in biomedical sciences. In particular, this is of paramount importance for the description and diagnosis of rare diseases and for the adoption of decisions concerning health care, clinical and therapeutical indications and derivation of patients to specialists. In this context, Orphanet and WHO (World Health Organization) are currently making important efforts for the future specification of the international classification of diseases ICD11 (http://www.who.int/classifications/icd/revisionnews/en/), which will include rare diseases for the first time 3. Unfortunately, there is still a lack of a clinical identification accredited for this kind of diseases. In consequence, the search for information on rare diseases remains complex and diffuse.
Specific problems also arise from the usual way in which the scientific literature is written. Unfortunately, scientific literature does not use to identify rare diseases as such and the identifiers for them proposed (i.e. ORPHA or OMIM IDs) are scarcely mentioned. In consequence, the search of information on rare diseases through the primary sources of scientific documentation is not an easy task. In fact, the finding of common relationships among diseases depends mainly on the reduced and specialized area of knowledge that dominates each rare disease, a fact that severely limits the visibility of these relationships.
Far beyond the epidemiological, clinical and molecular studies and decision making in public health, the adoption of wider and more general and holistic approaches could become a new way to access to new knowledge in this area. The implementation of informatics resources to extract data from scientific literature and to analyze and enrich this information is currently contributing to the advancement of knowledge in biomedicine. This could be a powerful strategy to establish emerging sources of knowledge also in the specific field of rare diseases.
A systematic review and collection of relevant information contained in diverse sources of scientific information and documentation could provide new valuable, emergent information concerning rare diseases. In the present work, we apply this systemic approach to get a deeper insight on the current knowledge of angiogenesis-related rare diseases (A-RDs).
Angiogenesis, the formation of new vessels from the pre-existing vasculature, is one main mechanism of vascularisation during normal and specific physiological processes, such as embryonic development, growth, regeneration, wound healing and formation of corpus luteum and endometrium. Angiogenesis attracted wide interest in the scientific community when the pioneering hypothesis of Judah Folkman in 1971 that tumor progression and metastasis are dependent on angiogenesis (and, as such, cancer could be therapeutically attacked by inhibiting angiogenesis) began to be confirmed by experimental studies since the eighties 4–6. In fact, inhibition of this process has become a major challenge in the development of new anticancer agents, with more than 40,000 scientific papers published on this subject, and about a hundred anti-angiogenic compounds entered in clinical trials, and numerous others in preclinical development 7–9. Currently, it is well established that a deregulated and persistent angiogenesis is one of the hallmarks of cancer 10,11. Furthermore, there is overwhelming evidence on the involvement of deregulated angiogenesis in many other pathological situations, which are currently described as angiogenesis-dependent diseases 12. The interest and impact of angiogenesis as a new therapeutical target from the treatment of non-oncological angiogenesis-dependent diseases is well represented by the recent concession of the Lasker-Debakey Clinical Medical Research Award 2010 to Dr. Napoleone Ferrara for the discovery of VEGF as a major mediator of angiogenesis and the development of an effective anti-VEGF therapy for wet macular degeneration, a leading cause of blindness in the elderly 13. This is a type of age-related macular degeneration, included as a rare disease in the Orphanet website with the Orpha number ORPHA279. Other two examples of A-RDs are POEMS syndrome (ORPHA2905) 14,15 and Amyotrophic lateral sclerosis (ORPHA803) 16.
In fact, a number of the so far described rare diseases are infrequent types of neoplasia, most probably related to angiogenesis. Furthermore, many other rare diseases could be related to angiogenesis. However, there is a lack of an exhaustive and systematic review on the topic “angiogenesis related rare diseases”. To contribute to fill this gap is the main aim of the present review. To reach this goal, our group can make use or our previous experience in the management of databases and the implementation of bioinformatics tools 17–19. As a research group integrated in the Spanish Network of Rare Disease Research (http://www.ciberer.es/index.php?lang=english), we are actively involved in the search of new sources of knowledge in this research area. In the present work, we aim to evaluate how much and what kind of information we are able to uncover in the context of A-RDs. Furthermore, we also evaluate what kind of information contained in Orphanet can be extracted within the frame of our systematic search.
Methods
State of the art
The aim of this study was to perform a systematic study on the following question: what scientific information can be drawn from those rare diseases that are related to the topic called angiogenesis? To this end, we adapted the methodology stated in PRISMA statement (http://www.prisma-statement.org/statement.htm) for the systematic review of the whole set of documents that could be extracted from the databases used in this study 20,21. Table S1 in Supplementary material is the completed PRISMA checklist.
Strategy for the literature search
Eligibility criteria
Types of studies: This is a bibliometric study for the capture and identification of rare diseases appearing in different sources of scientific documentation and that are somehow related to angiogenesis.
Report eligibility: The scientific information specified in the previous section was exhaustively collected from scientific publications, namely, reviews, articles, case reports, proceedings and patents. The publication time ranged from 1991 to 2010 and there was no restriction concerning language or kind of publication. This search was dated in June of 2010 and updated in December of 2010. The whole set of collected rare diseases related to angiogenesis was submitted to a filter and selection procedure, according to their presence or not within a specialized database for rare diseases (Orphanet).
Information sources
For the selection of scientific publications concerning rare diseases somehow related to angiogenesis, an online search of literature was carried out in both PubMed and ISI Web of Knowledge databases.
Search terms used were “rare disease” as a generic term and “angiogen*”. The use of the two terms “rare” and “disease” juxtaposed allowed to include any pathology with its name before the search term “disease” (i.e. Castleman disease, Von Willebrand disease, Wilson disease, Menkes disease, Crohn disease, among others) and made possible to associate the search term “rare” with other related semantic terms, such as in the cases of rare tumor, rare pathology or rare disorder, among others. Finally, the term “angiogen*” was used as a root belonging to the generic term of angiogenesis and/or all possible variants.
As mentioned above, to validate the rare diseases found in this initial literature search, Orphanet database was used. Only those rare diseases contained and indexed within this database were confirmed and used for the rest of the study.
Search
In PubMed database, search terms were introduced in Advanced search without time limit or any other limit and listed as “(rare disease) AND angiogen*”, according to the following structured search: “rare diseases”[MeSH Terms] OR (“rare”[All Fields] AND “diseases”[All Fields]) OR “rare diseases”[All Fields] OR (“rare”[All Fields] AND “disease”[All Fields]) OR “rare disease”[All Fields], along with more specific terms that appear by default in Medical Subject Heading (MeSH) hierarchical structure in Medline. MeSH categories were: Orphan Drug Production / All MeSH Categories / Diseases Category / Pathological Conditions, Signs and Symptoms / Pathologic Processes / Disease Attributes / Rare Diseases. The MeSH terms were: Disease, Rare / Diseases, Rare / Rare Disease / Orphan Diseases / Disease, Orphan / Diseases, Orphan / Orphan Disease.
Search terms were introduced in ISI Web of Knowledge database as topics in both cases. The annotated search structure was as follows: Topic=(rare disease) AND Topic=(angiogen*). Timespan=All Years.
Study selection
Bibliography selection
The two lists of publications mentioning one or several rare diseases somehow related to angiogenesis obtained by searching PubMed and ISI Web of Knowledge as described above were merged into a unique list containing all the entrances without repetition. In order to reduce the bias among the selected data and the data that really fit the list of search terms, all the information extracted was reviewed and verified. To reach this goal, all the publications contained in the merged list were revised one by one by making a strategic reading of them 22. An analysis of matches was performed looking for the occurrence of the following terms “rare disease”, and/or “rare”, and/or “disease” and “angiogenic *” or “VEGF” in all titles, abstracts, author keywords and keywords plus found and MeSH terms. All the references that did not support this exclusion analysis were removed.
Rare diseases selection
Only those references containing explicit reference to one or more rare diseases (and therefore listed in Orphanet database with an Orpha identification number) were used to select those rare diseases included in the set to be further analyzed.
Data collection process and synthesis of indexed information
Data bibliographic systematic review
Once all the bibliographic information related to rare disease and angiogenesis was confirmed and validated, we proceeded to the analysis and compilation of all data of interest that Orphanet website could bring to the state of the art. All the collected data were allocated in sheets for the management of data in Excel format. This election was based on: (i) the easy access and operability of this format, (ii) the easy identification of data through the search option, and (iii) the capability to recover the original source of information through online links.
In a first column, the titles of all the selected bibliographic references in the study were included. The order or entrance was assigned according to publication dates, from the most recent to the oldest one.
All found and selected bibliographic references were categorized for each Orpha identification number validated with the applied selection criteria.
Initial classification of angiogenesis-related rare diseases
A first general classification criterion was established according to which each disease was assigned to one of the three following disjoined subsets: A (A-RDs with cancerous phenotype in all their features), B (A-RDs with cancerous phenotype only in some of their features) and C (A-RDs without cancerous phenotype). To determine the three subsets, a systematic search of the semantic terms “tumor” and “cancer” and the sufix “-ome” was carried out within the classification that orpahanet exposes for rare diseases. The terms glaucome, angiokeratome, lysosome, pseudoxanthome and hamartome were excluded due to obvious reasons. After this, all corresponding genes and orphan drugs were seized from Orphanet database, categorized for each Orpha identification number and associated to A-RDs in different sheets.
Bioinformatics analysis of bibliographic systematic review
To validate our manual systematic review of literature, we carried out an additional bioinformatics text mining study. The list of PubMed IDs (PMIDs) was obtained from the initial systematic review (all references of supplementary material, with the exception of references 5, 41, 44, 63 and 71) and uploaded into the text mining web application SciMiner 23. A biomedical literature mining analysis and the subsequent enrichment analysis were carried out concerning to those Medical Subject Headings (MeSH) associated with the retrieved PMIDs from the systematic review.
Results of the literature search and orphanet analysis
Bibliographic systematic review
As shown in the scheme depicted in Figure1, the search in ISI Web of Knowledge yielded 284 references, whereas that carried out in PubMed yielded 263 references. From all these references, only 187 fulfilled the selection criteria for their inclusion in the set to be further analyzed. The complete set of the 187 selected bibliographic references conforms the bibliography included in the Supplementary material and they validate the entrance of 180 A-RDs indexed in the database Orphanet (see Methods). An evaluation of this bibliographic set using PMIDs was carried out as described (see Methods). To avoid the occurrence of trivial MeSH terms, we have considered statistically significant only those terms with a P-value lower than 1.0E-5 and an enrichment (t-ratio/b-ratio) greater than 40. The results are shown in Table1. Figure S1 (Supplementary material) shows the PRISMA flow diagram corresponding to this systematic review.
Fig 1.
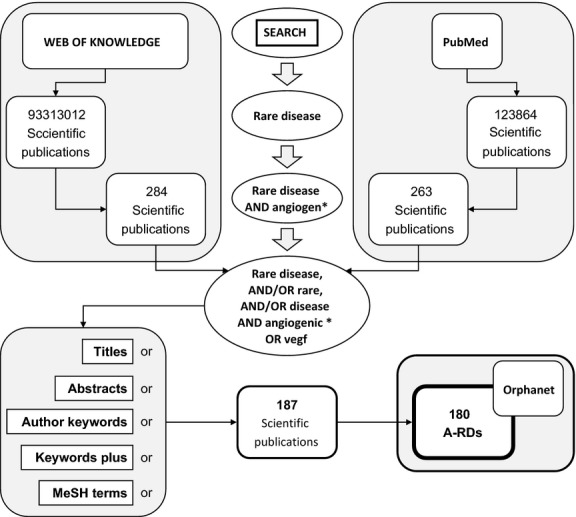
Flow of information through the different phases of bibliographic systematic review.
Table 1.
Bioinformatic analysis of bibliographic systematic review
| MeSH | t+ | t− | b+ | b− | WholeSize | t-Ratio | b-Ratio | t-ratio/b-ratio | P-value |
|---|---|---|---|---|---|---|---|---|---|
| Angiogenesis inhibitors | 33 | 149 | 8120 | 19,435,023 | 19,443,143 | 0.18 | 4.18E-04 | 434.2 | 6.18E-76 |
| Neovascularization, pathologic | 31 | 151 | 22,709 | 19,420,434 | 19,443,143 | 0.17 | 1.17E-03 | 145.8 | 9.89E-57 |
| Vascular endothelial growth factor A | 18 | 164 | 20,847 | 19,422,296 | 19,443,143 | 0.1 | 1.07E-03 | 92.2 | 9.41E-30 |
| Thalidomide | 12 | 170 | 4628 | 19,438,515 | 19,443,143 | 0.07 | 2.38E-04 | 277 | 6.17E-26 |
| Herpesvirus 8, human | 9 | 173 | 3009 | 19,440,134 | 19,443,143 | 0.05 | 1.55E-04 | 319.5 | 2.49E-20 |
| Macular degeneration | 7 | 175 | 7925 | 19,435,218 | 19,443,143 | 0.04 | 4.08E-04 | 94.4 | 2.06E-12 |
| von Hippel–Lindau disease | 5 | 177 | 1744 | 19,441,399 | 19,443,143 | 0.03 | 8.97E-05 | 306.3 | 9.10E-12 |
| Lymphangioleiomyomatosis | 4 | 178 | 569 | 19,442,574 | 19,443,143 | 0.02 | 2.93E-05 | 751 | 3.29E-11 |
| Endothelial cells | 7 | 175 | 15,420 | 19,427,723 | 19,443,143 | 0.04 | 7.93E-04 | 48.5 | 2.05E-10 |
| Carcinoma, renal cell | 7 | 175 | 16,035 | 19,427,108 | 19,443,143 | 0.04 | 8.25E-04 | 46.6 | 2.68E-10 |
| Exudates and transudates | 6 | 176 | 8354 | 19,434,789 | 19,443,143 | 0.03 | 4.30E-04 | 76.7 | 2.75E-10 |
| Hypoxia-inducible factor 1, alpha subunit | 5 | 177 | 4123 | 19,439,020 | 19,443,143 | 0.03 | 2.12E-04 | 129.6 | 6.57E-10 |
| Vascular malformations | 3 | 179 | 311 | 19,442,832 | 19,443,143 | 0.02 | 1.60E-05 | 1030.5 | 4.11E-09 |
| Telangiectasia, hereditary hemorrhagic | 4 | 178 | 1929 | 19,441,214 | 19,443,143 | 0.02 | 9.92E-05 | 221.5 | 4.25E-09 |
| Activin receptors, type II | 3 | 179 | 346 | 19,442,797 | 19,443,143 | 0.02 | 1.78E-05 | 926.3 | 5.65E-09 |
| Hemangioendothelioma, epithelioid | 3 | 179 | 459 | 19,442,684 | 19,443,143 | 0.02 | 2.36E-05 | 698.2 | 1.31E-08 |
| Vascular endothelial growth factors | 5 | 177 | 7692 | 19,435,451 | 19,443,143 | 0.03 | 3.96E-04 | 69.4 | 1.44E-08 |
| Angiogenic proteins | 3 | 179 | 482 | 19,442,661 | 19,443,143 | 0.02 | 2.48E-05 | 664.9 | 1.52E-08 |
| POEMS syndrome | 3 | 179 | 486 | 19,442,657 | 19,443,143 | 0.02 | 2.50E-05 | 659.4 | 1.56E-08 |
| Osteolysis, essential | 3 | 179 | 541 | 19,442,602 | 19,443,143 | 0.02 | 2.78E-05 | 592.4 | 2.15E-08 |
| Hemangioma, capillary | 3 | 179 | 665 | 19,442,478 | 19,443,143 | 0.02 | 3.42E-05 | 481.9 | 3.97E-08 |
| Protein kinase inhibitors | 5 | 177 | 10,840 | 19,432,303 | 19,443,143 | 0.03 | 5.58E-04 | 49.3 | 7.82E-08 |
| Interferon-alpha | 5 | 177 | 11637 | 19,431,506 | 19,443,143 | 0.03 | 5.99E-04 | 45.9 | 1.11E-07 |
| Pyrroles | 5 | 177 | 11,893 | 19,431,250 | 19,443,143 | 0.03 | 6.12E-04 | 44.9 | 1.23E-07 |
| Von Hippel–Lindau tumor suppressor protein | 3 | 179 | 1082 | 19,442,061 | 19,443,143 | 0.02 | 5.56E-05 | 296.2 | 1.70E-07 |
| Hypoxia-inducible factor 1 | 3 | 179 | 1746 | 19,441,397 | 19,443,143 | 0.02 | 8.98E-05 | 183.6 | 7.10E-07 |
| Endothelial growth factors | 4 | 178 | 7866 | 19,435,277 | 19,443,143 | 0.02 | 4.05E-04 | 54.3 | 1.12E-06 |
| Sarcoma, kaposi | 4 | 178 | 8163 | 19,434,980 | 19,443,143 | 0.02 | 4.20E-04 | 52.3 | 1.30E-06 |
| Linkage disequilibrium | 4 | 178 | 8690 | 19,434,453 | 19,443,143 | 0.02 | 4.47E-04 | 49.2 | 1.66E-06 |
| Rare diseases | 3 | 179 | 2370 | 19,440,773 | 19,443,143 | 0.02 | 1.22E-04 | 135.2 | 1.77E-06 |
| Collagen type XVIII | 2 | 180 | 202 | 19,442,941 | 19,443,143 | 0.01 | 1.04E-05 | 1057.7 | 1.80E-06 |
| Receptors, vascular endothelial growth factor | 3 | 179 | 2497 | 19,440,646 | 19,443,143 | 0.02 | 1.28E-04 | 128.4 | 2.06E-06 |
| Lipoid proteinosis of urbach and wiethe | 2 | 180 | 246 | 19,442,897 | 19,443,143 | 0.01 | 1.27E-05 | 868.5 | 2.67E-06 |
| Retinal vessels | 4 | 178 | 9999 | 19,433,144 | 19,443,143 | 0.02 | 5.14E-04 | 42.7 | 2.88E-06 |
| Stem cell transplantation | 4 | 178 | 10,093 | 19,433,050 | 19,443,143 | 0.02 | 5.19E-04 | 42.3 | 2.99E-06 |
| Receptors, CXCR4 | 3 | 179 | 3315 | 19,439,828 | 19,443,143 | 0.02 | 1.70E-04 | 96.7 | 4.80E-06 |
| Dacarbazine | 3 | 179 | 3542 | 19,439,601 | 19,443,143 | 0.02 | 1.82E-04 | 90.5 | 5.84E-06 |
MeSH, medical subject headings; t+, number of papers annotated with the MeSH in the sample PMIDs; t−, number of papers not annotated with the MeSH in the sample PMIDs; b+, number of papers annotated with the MeSH in whole PMIDs; b−, number of papers not annotated with the MeSH in whole PMIDs; WholeSize, whole PMIDs; t-ratio, proportion in the sample PMIDs; b-ratio, proportion in the whole PMIDs; t-ratio/b-ratio, value relative to the enrichment of the MeSH term; and P-value, Hypergeometric test value.
Initial classification of angiogenesis-related rare diseases
When this analysis was carried out, the Orphanet web contained 5781 rare diseases. This means that a 3% of all the described rare diseases can be considered as A-RDs.
In the present work, the distribution of A-RDs within the subsets A, B, C as a function of the classification range pointed by Orphanet (for those which have a classification in this database) was analyzed. This initial classification was based on the tight physio-pathological relationship of cancer with angiogenesis 4–9, their relevant clinical and pharmacological applications 7–9 and the bibliographic relevance of the Boolean descriptor “cancer AND angiogenesis”. As expected, the greatest subset is that of rare oncologic diseases (subset A), which amounted up to 81 A-RDs. However, the subset C (A-RDs without cancerous phenotype) was almost as big, containing 76 non-oncologic A-RDs. Finally, the subset B (A-RDs with cancerous phenotype only in some of their features) contained 23 diseases, most of them increasing the risk of developing a cancer. Table2 show the lists of A-RDs in each of the three subsets, their relationship with bibliographic references of the supplementary material and the number of genes and drugs associated to each A-RD.
Table 2.
Angiogenesis-related rare diseases
| n° | References | Orpha ID | Rare disease | Orpha genes | Orpha drugs |
|---|---|---|---|---|---|
| Subset A (rare oncologic diseases) | |||||
| 1 | [1, 136] | 519 | Acute myeloid leukemia (ARD)* | 19 | 33 |
| 2 | [59] | 213772 | Adenocarcinoma of the cervix uteri | 0 | 0 |
| 3 | [128] | 1501 | Adrenocortical carcinoma | 0 | 1 |
| 4 | [47] | 163699 | Alveolar soft-part sarcoma | 2 | 0 |
| 5 | [114] | 142 | Anaplastic thyroid carcinoma | 0 | 1 |
| 6 | [52] | 86886 | Angioimmunoblastic T-cell lymphoma | 0 | 0 |
| 7 | 11 | 98731 | Arteriovenous fistula | 0 | 0 |
| 8 | [23, 26, 68, 79, 105, 143] | 211266 | Arteriovenous malformation | 0 | 0 |
| 9 | [51] | 157980 | Bladder Cancer | 0 | 0 |
| 10 | [125, 134] | 223727 | Bone sarcoma | 7 | 0 |
| 11 | [83] | 3395 | Brain tumor (ARD)* | 17 | 0 |
| 12 | [165] | 97287 | Bronchial endocrine tumor | 0 | 0 |
| 13 | [26] | 137667 | Capillary malformation-arteriovenous malformation | 1 | 0 |
| 14 | [45, 112, 143, 152] | 164 | Cerebral cavernous malformations | 0 | 0 |
| 15 | [99] | 86829 | Chronic neutrophilic leukemia | 0 | 0 |
| 16 | [61] | 99970 | Dedifferentiated liposarcoma | 0 | 0 |
| 17 | [61] | 31112 | Dermatofibrosarcoma protuberans | 2 | 0 |
| 18 | 15 | 141209 | Diffuse lymphatic malformation | 0 | 0 |
| 19 | [29, 138, 167] | 877 | Endocrine tumor | 2 | 2 |
| 20 | [29] | 100092 | Enteropancreatic endocrine tumor | 0 | 2 |
| 21 | [103] | 99871 | Eosinophilic granuloma | 0 | 0 |
| 22 | [65, 98, 139, 162] | 157791 | Epithelioid hemangioendothelioma | 0 | 0 |
| 23 | [147, 149] | 99976 | Esophageal adenocarcinoma | 0 | 0 |
| 24 | [149] | 99977 | Esophageal squamous cell carcinoma | 1 | 0 |
| 25 | [61, 134] | 319 | Ewing sarcoma | 5 | 0 |
| 26 | [51, 146, 164] | 733 | Familial adenomatous polyposis | 1 | 4 |
| 27 | [64] | 523 | Familial leiomyomatosis | 1 | 0 |
| 28 | 20 | 99361 | Familial medullary thyroid carcinoma | 0 | 0 |
| 29 | [39, 42, 47, 92, 104, 159, 160] | 151 | Familial renal cell carcinoma (ARD)* | 11 | 22 |
| 30 | [70, 135] | 63443 | Gastric cancer | 2 | 5 |
| 31 | [61, 97] | 44890 | Gastrointestinal stromal tumor | 2 | 5 |
| 32 | [5, 36, 83] | 360 | Glioblastoma | 8 | 33 |
| 33 | [5, 83] | 182067 | Glial tumor (ARD)* | 9 | 26 |
| 34 | [45] | 83454 | Glomuvenous malformation | 1 | 1 |
| 35 | [34] | 99915 | Granulosa cell malignant tumor | 0 | 0 |
| 36 | [184] | 58017 | Hairy cell leukemia | 0 | 2 |
| 37 | [76] | 2126 | Hemangiopericytoma | 0 | 0 |
| 38 | [123, 137] | 88673 | Hepatocellular carcinoma (ARD)* | 2 | 14 |
| 39 | [141] | 227535 | Hereditary breast cancer | 0 | 0 |
| 40 | [94, 107] | 29072 | Hereditary pheochromocytoma-paraganglioma syndrome | 6 | 0 |
| 41 | [166, 167] | 97279 | Insulinoma | 0 | 3 |
| 42 | [87, 118, 144, 158, 162, 169, 172, 177, 179, 180, 181, 185, 186] | 33276 | Kaposi's sarcoma | 0 | 5 |
| 43 | 22 | 213807 | Leiomyosarcoma of the cervix uteri | 0 | 0 |
| 44 | 22 | 213625 | Leiomyosarcoma of the corpus uteri | 0 | 0 |
| 45 | [111] | 65285 | Lhermitte-Duclos disease (ARD)* | 1 | 0 |
| 46 | [141] | 524 | Li-Fraumeni syndrome | 2 | 1 |
| 47 | [173] | 69078 | Liposarcoma | 1 | 2 |
| 48 | [24, 75] | 168811 | Malignant peritoneal mesothelioma | 0 | 0 |
| 49 | [67, 112] | 3148 | Malignant Schwannoma | 0 | 0 |
| 50 | 6 | 98292 | Mastocytosis | 1 | 1 |
| 51 | [20, 165] | 1332 | Medullary thyroid carcinoma | 1 | 3 |
| 52 | [56] | 97338 | Melanoma of soft part | 2 | 0 |
| 53 | [24, 75] | 50251 | Mesothelioma | 0 | 3 |
| 54 | [55, 77, 140, 166, 178, 184] | 29073 | Multiple myeloma | 2 | 23 |
| 55 | 1 | 52688 | Myelodysplastic syndromes | 2 | 10 |
| 56 | [61] | 99967 | Myxoid liposarcoma | 1 | 0 |
| 57 | [80] | 209989 | Non-papillary transitional cell carcinoma of the bladder | 0 | 1 |
| 58 | [134] | 668 | Osteosarcoma | 2 | 5 |
| 59 | [59] | 213504 | Ovarian adenocarcinoma | 0 | 7 |
| 60 | [142] | 2800 | Paget disease extramammary | 0 | 0 |
| 61 | [165] | 217074 | Pancreatic carcinoma | 7 | 21 |
| 62 | [94] | 717 | Pheochromocytoma and secreting paraganglioma (ARD)* | 8 | 1 |
| 63 | [14, 66, 115] | 2905 | POEMS syndrome | 0 | 0 |
| 64 | [87, 169, 172, 177, 180] | 48686 | Primary effusion lymphoma | 0 | 0 |
| 65 | [87, 144, 169, 172, 180, 181] | 99923 | Primary effusion lymphoma associated with HIV infection | 0 | 0 |
| 66 | [59] | 213528 | Rare adenocarcinoma of the breast | 0 | 0 |
| 67 | [74] | 180250 | Rare breast cancer (ARD)* | 12 | 0 |
| 68 | [12, 28, 39, 42, 47, 57, 63, 84, 92, 104, 122, 159, 160, 182] | 217071 | Renal cell carcinoma (ARD)* | 12 | 24 |
| 69 | [61] | 69077 | Rhabdoid tumor | 2 | 0 |
| 70 | [165] | 70573 | Small cell lung cancer | 0 | 4 |
| 71 | [9, 22, 56, 97, 125, 173] | 3394 | Soft tissue sarcomas | 20 | 10 |
| 72 | [162] | 210584 | Spindle cell hemangioma | 0 | 0 |
| 73 | [93] | 67037 | Squamous cell carcinoma of head and neck (ARD)* | 4 | 5 |
| 74 | [131] | 99868 | Thymic carcinoma | 0 | 0 |
| 75 | [131] | 3398 | Thymic epithelial tumor | 0 | 0 |
| 76 | [131] | 100100 | Thymic tumor | 0 | 0 |
| 77 | [131] | 99867 | Thymoma | 0 | 0 |
| 78 | [133] | 1063 | Tufted angioma | 0 | 0 |
| 79 | [52] | 86885 | Unspecified peripheral T-cell lymphoma | 0 | 4 |
| 80 | [85, 88, 159] | 39044 | Uveal melanoma | 0 | 1 |
| 81 | [61] | 99971 | Well-differentiated liposarcoma | 0 | 0 |
| Subset B (A-RDs with cancerous phenotype only in some of their features) | |||||
| 1 | [111] | 109 | Bannayan-Riley-Ruvalcaba syndrome (ARD)* | 1 | 0 |
| 2 | [89, 184] | 521 | Chronic myeloid leukemia (ARD)* | 3 | 12 |
| 3 | [69] | 53721 | Cobb syndrome | 0 | 0 |
| 4 | [60, 109] | 191 | Cockayne syndrome | 5 | 0 |
| 5 | [95] | 2414 | Congenital pulmonary lymphangiectasia | 0 | 0 |
| 6 | [111] | 201 | Cowden syndrome (ARD)* | 3 | 0 |
| 7 | 17 | 324 | Fabry disease | 1 | 3 |
| 8 | [15,16, 73, 118] | 73 | Gorham-Stout disease | 0 | 0 |
| 9 | [35, 110] | 90308 | Klippel-Trenaunay syndrome (ARD)* | 1 | 0 |
| 10 | [90] | 389 | Langerhans cell histiocytosis | 0 | 1 |
| 11 | [174] | 79383 | Lymphedema (ARD)* | 4 | 0 |
| 12 | [50] | 2451 | Mucocutaneous venous malformations (ARD)* | 1 | 1 |
| 13 | [166, 167] | 652 | Multiple endocrine neoplasia type 1 | 2 | 2 |
| 14 | [89, 148] | 824 | Myelofibrosis with myeloid metaplasia | 1 | 2 |
| 15 | [2, 61] | 636 | Neurofibromatosis type 1 (ARD)* | 3 | 2 |
| 16 | [53] | 2869 | Peutz-Jeghers syndrome | 1 | 0 |
| 17 | [91] | 42775 | PHACE syndrome | 0 | 0 |
| 18 | [89] | 729 | Polycythemia vera | 1 | 5 |
| 19 | [174] | 77240 | Primary lymphedema | 2 | 0 |
| 20 | [8,13,23, 45, 79, 105, 143] | 774 | Rendu-Osler-Weber disease (ARD)* | 3 | 1 |
| 21 | [37, 88] | 3205 | Sturge-Weber syndrome | 0 | 0 |
| 22 | [12, 29, 39, 42, 84, 88, 92, 122, 130, 161, 170, 182] | 892 | Von Hippel–Lindau disease (ARD)* | 1 | 2 |
| 23 | [187] | 913 | Zollinger-Ellison syndrome | 1 | 3 |
| Subset C (A-RDs without cancerous phenotype) | |||||
| 1 | [25] | 93585 | Acquired thrombotic thrombocytopenic purpura due to anti-ADAMTS 13 antibodies | 0 | 1 |
| 2 | [54] | 79126 | Acute interstitial pneumonia | 0 | 0 |
| 3 | [19, 27, 32, 40, 43, 48, 62, 88, 176] | 279 | Age-related macular degeneration | 6 | 1 |
| 4 | [58, 126] | 803 | Amyotrophic lateral sclerosis (ARD)* | 17 | 10 |
| 5 | [35, 110] | 2346 | Angio-osteohypertrophic syndrome (ARD)* | 2 | 0 |
| 6 | [25] | 2134 | Atypical hemolytic uremic syndrome (ARD)* | 6 | 3 |
| 7 | [154] | 3453 | Autoimmune polyendocrinopathy type 1 | 1 | 0 |
| 8 | [46] | 117 | Behcet disease | 0 | 2 |
| 9 | 21 | 131 | Budd-Chiari syndrome | 0 | 0 |
| 10 | [30, 120] | 36258 | Buerger's disease | 0 | 0 |
| 11 | [152] | 136 | CADASIL syndrome | 1 | 0 |
| 12 | [31, 179, 185] | 160 | Castleman disease | 0 | 2 |
| 13 | [154] | 178029 | Central diabetes insipidus | 1 | 0 |
| 14 | [68] | 98044 | Central nervous system malformation | 0 | 0 |
| 15 | [40] | 179 | Chorioretinopathy, Birdshot type | 1 | 0 |
| 16 | [155] | 2137 | Chronic autoimmune hepatitis | 0 | 1 |
| 17 | [117] | 183 | Churg-Strauss syndrome | 0 | 1 |
| 18 | [40, 109] | 190 | Coats disease | 1 | 0 |
| 19 | [78] | 2041 | Coronary arterial fistulas | 0 | 0 |
| 20 | [6, 103, 133] | 206 | Crohn disease | 5 | 0 |
| 21 | [116] | 137698 | Cytomegalovirus disease in patients with impaired cell mediated immunity deemed at risk | 0 | 1 |
| 22 | [68] | 97339 | Dural sinus malformation | 0 | 0 |
| 23 | [40] | 40923 | Eales disease | 0 | 0 |
| 24 | [165] | 99889 | Ectopic Cushing syndrome | 0 | 1 |
| 25 | [72] | 199323 | Endophthalmitis | 0 | 0 |
| 26 | [183] | 337 | Fibrodysplasia ossificans progressiva (ARD)* | 1 | 0 |
| 27 | [45] | 2092 | Focal dermal hypoplasia | 1 | 0 |
| 28 | 3 | 221126 | Fowler syndrome | 1 | 0 |
| 29 | 17 | 355 | Gaucher disease | 2 | 5 |
| 30 | 17 | 77260 | Gaucher disease, type 2 | 1 | 4 |
| 31 | 17 | 77261 | Gaucher disease, type 3 | 1 | 4 |
| 32 | [154] | 95509 | Granulomatous hypophysitis | 0 | 0 |
| 33 | [121] | 855 | Hashimoto struma | 1 | 0 |
| 34 | [90] | 158032 | Hemophagocytic syndrome | 14 | 1 |
| 35 | [132, 152] | 85458 | Hereditary cerebral hemorrhage with amyloidosis | 2 | 0 |
| 36 | [132] | 100006 | Hereditary cerebral hemorrhage with amyloidosis, Dutch type | 0 | 0 |
| 37 | [157] | 422 | Idiopathic and/or familial pulmonary arterial hypertension (ARD)* | 3 | 17 |
| 38 | 21 | 69665 | Intrahepatic cholestasis of pregnancy | 2 | 0 |
| 39 | [100] | 2778 | Juvenile chronic recurrent multifocal osteomyelitis | 0 | 0 |
| 40 | [82] | 2331 | Kawasaki disease | 0 | 1 |
| 41 | [156] | 1571 | Knobloch syndrome (ARD)* | 1 | 0 |
| 42 | [101, 163] | 530 | Lipoid proteinosis (ARD)* | 1 | 0 |
| 43 | [7, 108] | 538 | Lymphangioleiomyomatosis | 2 | 0 |
| 44 | 17 | 93448 | Lysosomal storage disease with skeletal involvement | 22 | 0 |
| 45 | [86] | 101338 | Mediterranean spotted fever | 0 | 0 |
| 46 | [168] | 54370 | Membranoproliferative glomerulonephritis | 1 | 0 |
| 47 | [10, 127, 145] | 565 | Menkes disease | 1 | 0 |
| 48 | [68, 106] | 2573 | Moyamoya disease | 1 | 0 |
| 49 | [87, 144, 169, 172, 177, 180] | 93686 | Multicentric Castleman disease | 0 | 0 |
| 50 | [176] | 94058 | Neovascular glaucoma | 1 | 0 |
| 51 | [129] | 649 | Norrie disease | 1 | 0 |
| 52 | [25] | 447 | Paroxysmal nocturnal hemoglobinuria | 0 | 2 |
| 53 | 17 | 85212 | Perinatal-lethal Gaucher disease | 1 | 0 |
| 54 | 18 | 563 | Peripartum cardiomyopathy | 0 | 0 |
| 55 | [40] | 758 | Pseudoxanthoma elasticum | 1 | 0 |
| 56 | [4, 113, 157] | 182090 | Pulmonary arterial hypertension | 0 | 3 |
| 57 | [96, 153] | 199241 | Pulmonary capillary hemangiomatosis | 0 | 2 |
| 58 | [113] | 71198 | Rare pulmonary hypertension | 0 | 1 |
| 59 | [38] | 60032 | Recurrent respiratory papillomatosis | 0 | 0 |
| 60 | [40, 102, 171, 176] | 90050 | Retinopathy of prematurity | 1 | 2 |
| 61 | [33] | 49041 | Retroperitoneal fibrosis | 0 | 0 |
| 62 | [86] | 102021 | Rickettsiae disease | 0 | 0 |
| 63 | [17, 82] | 797 | Sarcoidosis | 1 | 2 |
| 64 | [81, 124] | 801 | Scleroderma | 0 | 3 |
| 65 | [166] | 36426 | Stevens-Johnson syndrome | 0 | 0 |
| 66 | [99] | 3243 | Sweet syndrome | 0 | 0 |
| 67 | [116, 119, 154] | 536 | Systemic lupus erythematosus | 2 | 1 |
| 68 | [81] | 90291 | Systemic sclerosis | 0 | 3 |
| 69 | [135] | 93573 | Thrombotic microangiopathy | 0 | 0 |
| 70 | [25, 151] | 54057 | Thrombotic thrombocytopenic purpura | 1 | 5 |
| 71 | [25] | 90038 | Typical hemolytic uremic syndrome | 0 | 1 |
| 72 | [117] | 52759 | Vasculitis | 0 | 0 |
| 73 | [49, 109, 129] | 98668 | Vitreoretinopathy (ARD)* | 14 | 0 |
| 74 | [86, 151, 175] | 903 | Von Willebrand disease | 1 | 3 |
| 75 | [150] | 51636 | WHIM syndrome | 1 | 0 |
| 76 | [82, 127, 145] | 905 | Wilson disease | 1 | 4 |
ARD: angiogenic rare disease.
Since Orphanet identifies genes listed in its database such as those considered to be involved in the pathophysiology of rare diseases and because this information is extracted from the scientific literature and cross-validated, genes directly related to angiogenesis and A-RDs can be associated (i.e. those genes linked to A-RDs in the systematic review and in the same way recovered with the Gene Ontology GO term called angiogenesis; GO:0001525). In this context, we consider that diseases recovered in this way can be called “angiogenic rare diseases” and they can be retrieved from those 180 rare diseases related in some way with angiogenesis. These 27 angiogenic rare diseases are also highlighted in Table2.
The most commonly used classification of diseases is WHO ICD-10 (http://www.who.int/classifications/icd/en). Tables S2–S4 (Supplementary material) list A-RDs corresponding to subsets A, B, and C, respectively, indicating their ICD-10 when available. It is noteworthy that as many as 83 A-RDs (47 in subset A, 6 in subset B and 30 in subset C) have currently no ICD code assigned.
Genes and drugs associated to angiogenesis-related rare diseases
We manually collected 244 entrances identified as genes in Orphanet. To study the relative distribution of the set of extracted and annotated genes in the list of A-RDs, and also to know their representation in the three initial subsets (A, B, C), three categories of A-RDs where established: those currently associated to no gene, those associated to one gene, and those with two or more genes associated. The number of genes in each A-RD is highlighted in Table2. Furthermore, Tables S5 and S6 (Supplementary material) list all the A-RDs associated to a single or to more than a gene, respectively, identifying the corresponding related genes by their Orpha gene IDs.
It is interesting to note that this relationship among genes and A-RDs is not symmetrical. Tables3 and 4 list the whole set of 186 genes associated to one A-RD and 58 genes associated to two or more A-RDs, respectively, identifying the corresponding A-RDs by their Orpha rare disease IDs. Figure2 shows the distribution of the annotated genes grouped in two categories (those associated to one A-RD and those associated to two or more A-RDs in the whole set and the three subsets of A-RDs.
Table 3.
Genes associated to a single angiogenesis-related rare disease
| n′ | Genes | Rare disases Orpha ID |
|---|---|---|
| 1 | ACTA2 | 2573 (C) |
| 2 | ATF1 | 97338 (A) |
| 3 | ACVR1 | 337 (C) |
| 4 | ADAMTS13 | 54057 (C) |
| 5 | AP3B1 | 158032 (C) |
| 6 | ARMS2 | 279 (C) |
| 7 | APP | 85458 (C) |
| 8 | ALS2 | 803 (C) |
| 9 | ALK | 3395 (A) |
| 10 | ANG | 803 (C) |
| 11 | (TATCCGGAGGGCTCGCCATGCTGCT) | 94058 (C) |
| 12 | AVP | 178029 (C) |
| 13 | ARSB | 93448 (C) |
| 14 | AGA | 93448 (C) |
| 15 | ATG16L1 | 206 (C) |
| 16 | ATP8B1 | 69665 (C) |
| 17 | ATP7A | 565 (C) |
| 18 | ATP7B | 905 (C) |
| 19 | ABCA4 | 279 (C) |
| 20 | ABCB4 | 69665 (C) |
| 21 | ABCC6 | 758 (C) |
| 22 | AIRE | 3453 (C) |
| 23 | BEST1 | 98668 (C) |
| 24 | BLOC1S3 | 158032 (C) |
| 25 | BMPR2 | 422 (C) |
| 26 | BARD1 | 180250 (A) |
| 27 | BRIP1 | 180250 (A) |
| 28 | BCR | 521 (B) |
| 29 | BRCA1 | 180250 (A) |
| 30 | CDH1 | 63443 (A) |
| 31 | CREB3L1 | 3394 (A) |
| 32 | CREB3L2 | 3394 (A) |
| 33 | Cathepsin A—CTSA | 93448 (C) |
| 34 | CEBPA | 519 (A) |
| 35 | CD46 | 2134 (C) |
| 36 | CXCR4 | 51636 (C) |
| 37 | CHGB | 803 (C) |
| 38 | COL2A1 | 98668 (C) |
| 39 | COL9A1 | 98668 (C) |
| 40 | COL11A1 | 98668 (C) |
| 41 | COL11A2 | 98668 (C) |
| 42 | CR1 | 536 (C) |
| 43 | C3 | 2134 (C) |
| 44 | CFB | 2134 (C) |
| 45 | CFI | 2134 (C) |
| 46 | CBFB | 519 (A) |
| 47 | CDKN1B | 652 (B) |
| 48 | CDKN2A | 217074 (A) |
| 49 | CST3 | 85458 (C) |
| 50 | CTLA4 | 536 (C) |
| 51 | DAO | 803 (C) |
| 52 | DEK | 519 (A) |
| 53 | DTNBP1 | 158032 (C) |
| 54 | ENG | 774 (B) |
| 55 | ETV6 | 519 (A) |
| 56 | ERCC1 | 191 (B) |
| 57 | ERCC2 | 191 (B) |
| 58 | ERCC5 | 191 (B) |
| 59 | ERCC6 | 191 (B) |
| 60 | ERCC8 | 191 (B) |
| 61 | EXT1 | 223727 (A) |
| 62 | ECM1 | 530 (C) |
| 63 | FCGR3B | 855 (C) |
| 64 | FLVCR2 | 2211 (C) |
| 65 | FBLN5 | 279 ((C) |
| 66 | FIG 4 | 803 (C) |
| 67 | FLT3 | 519 (A) |
| 68 | FOXC2 | 79383 (B) |
| 69 | FOXO1 | 3394 (A) |
| 70 | FZD4 | 98668 (C) |
| 71 | FUCA1 | 93448 (C) |
| 72 | GALNS | 93448 (C) |
| 73 | GLA | 324 (B) |
| 74 | GLB1 | 93448 (C) |
| 75 | GLMN | 83454 (A) |
| 76 | GNS | 93448 (C) |
| 77 | GUSB | 93448 (C) |
| 78 | HMCN1 | 279 (C) |
| 79 | HGSNAT | 93448 (C) |
| 80 | HPS1 | 158032 (C) |
| 81 | HPS3 | 158032 (C) |
| 82 | HPS4 | 158032 (C) |
| 83 | HPS5 | 158032 (C) |
| 84 | HPS6 | 158032 (C) |
| 85 | HTRA1 | 279 (C) |
| 86 | HYAL1 | 93448 (C) |
| 87 | IDS | 93448 (C) |
| 88 | IDUA | 93448 (C) |
| 89 | IGHG1 | 29073 (A) |
| 90 | ING1 | 67037 (A) |
| 91 | ING3 | 67037 (A) |
| 92 | INSM1 | 877 (A) |
| 93 | IRF4 | 29073 (A) |
| 94 | IL10 | 206 (C) |
| 95 | IL23R | 206 (C) |
| 96 | LRP5 | 98668 (C) |
| 97 | LYST | 158032 (C) |
| 98 | HLA-A | 179 (C) |
| 99 | HLA-DRB1 | 797 (C) |
| 100 | MAN2B1 | 93448 (C) |
| 101 | MANBA | 93448 (C) |
| 102 | MECOM | 52688 (A) |
| 103 | MKL1 | 519 (A) |
| 104 | MANF | 217074 (A) |
| 105 | MUTYH | 63443 (A) |
| 106 | MLL | 519 (A) |
| 107 | MYH11 | 519 (A) |
| 108 | MYST3 | 519 (A) |
| 109 | GNPTAB | 93448 (C) |
| 110 | GNPTG | 93448 (C) |
| 111 | NAGLU | 93448 (C) |
| 112 | NF1 | 636 (B) |
| 113 | NEFH | 803 (C) |
| 114 | NME1 | 3395 (A) |
| 115 | SGSH | 93448 (C) |
| 116 | NR1H3 | 803 (C) |
| 117 | NR2E3 | 98668 (C) |
| 118 | NPM1 | 519 (A) |
| 119 | NUP214 | 519 (A) |
| 120 | NUP98 | 519 (A) |
| 121 | NOD2 | 206 (C) |
| 122 | OPTN | 803 (C) |
| 123 | PAX3 | 3394 (A) |
| 124 | PAX7 | 3394 (A) |
| 125 | PHOX2B | 3395 (A) |
| 126 | PON1 | 803 (C) |
| 127 | PON2 | 803 (C) |
| 128 | PON3 | 803 (C) |
| 129 | PTCH2 | 3395 (A) |
| 130 | PRF1 | 158032 (C) |
| 131 | PRPH | 803 (C) |
| 132 | PLCE1 | 99977 (A) |
| 133 | PDGFRA | 44890 |
| 134 | PORCN | 2092 (C) |
| 135 | KCNJ13 | 98668 (C) |
| 136 | PRLR | 180250 (A) |
| 137 | PML | 519 (A) |
| 138 | PSAP | 355 (C) |
| 139 | RAB27A | 158032 (C) |
| 140 | RAD51 | 180250 (A) |
| 141 | RAD51C | 180250 (A) |
| 142 | RB1 | 668 (A) |
| 143 | RARA | 519 (A) |
| 144 | RS1 | 98668 (C) |
| 145 | RMST | 3394 (A) |
| 146 | RPS14 | 52688 (A) |
| 147 | RNF135 | 636 (B) |
| 148 | RBM15 | 519 (A) |
| 149 | RUNX1 | 521 (B) |
| 150 | RUNX1T1 | 519 (A) |
| 151 | SETX | 803 (C) |
| 152 | STK11 | 2869 (B) |
| 153 | SET | 519 (A) |
| 154 | NEU1 | 93448 (C) |
| 155 | SMAD9 | 422 (C) |
| 156 | SLC17A5 | 93448 (C) |
| 157 | SLC22A4 | 206 (C) |
| 158 | SLC44A4 | 93448 (C) |
| 159 | SOX18 | 79383 (B) |
| 160 | SUMF1 | 93448 (C) |
| 161 | SOD1 | 803 (C) |
| 162 | SUFU | 3395 (A) |
| 163 | SUZ12 | 636 (B) |
| 164 | SS18 | 3394 (A) |
| 165 | SSX1 | 3394 (A) |
| 166 | SSX2 | 3394 (A) |
| 167 | SSX2IP | 3394 (A) |
| 168 | SSX2B | 3394 (A) |
| 169 | STX11 | 158032 (C) |
| 170 | STXBP2 | 158032 (C) |
| 171 | TARDBP | 803 (C) |
| 172 | TEK | 2451 (B) |
| 173 | TSPAN12 | 98668 (C) |
| 174 | THBD | 2134(C) |
| 175 | TSC1 | 538 (C) |
| 176 | TSC2 | 538 (C) |
| 177 | TNFRSF10B | 67037 (A) |
| 178 | UNC13D | 158032 (C) |
| 179 | ABL1 | 521 (B) |
| 180 | VAPB | 803 (C) |
| 181 | VCAN | 98668 (C) |
| 182 | KRAS | 217074 (A) |
| 183 | MYCN | 3395 (A) |
| 184 | VWF | 903 (C) |
| 185 | WT1 | 3394 (A) |
| 186 | ZBTB16 | 519 (A) |
(A), (B), (C) correspond to subset A, B and C respectively.
Table 4.
Genes associated to two or more angiogenesis-related rare diseases
| n° | Genes | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|---|
| Rare diseases Orpha ID | ||||||||
| 1 | PTEN | 3395 (A) | 180250 (A) | 182067 (A) | 67037 (A) | 201 (B) | 109 (B) | 65285 (A) |
| 2 | FUS | 3394 (A) | 519 (A) | 803 (C) | 69078 (A) | 99967 (A) | ||
| 3 | SDHD | 180250 (A) | 717 (A) | 29072 (A) | 201 (B) | 877 (A) | ||
| 4 | TP53 | 3395 (A) | 182067 (A) | 360 (A) | 217074 (A) | 524 (A) | ||
| 5 | CHEK2 | 180250 (A) | 223727 (A) | 524 (A) | 668 (A) | |||
| 6 | EWSR1 | 3394 (A) | 223727 (A) | 319 (A) | 97338 (A) | |||
| 7 | GBA | 355 (C) | 77260 (C) | 77261 (C) | 85212 (C) | |||
| 8 | NDP | 98668 (C) | 190 (C) | 649 (C) | 90050 (C) | |||
| 9 | SDHB | 180250 (A) | 717 (A) | 29072 (A) | 201 (B) | |||
| 10 | TFE3 | 3394 (A) | 217071 (A) | 151 (A) | 163699 (A) | |||
| 11 | VHL | 217071 (A) | 151 (A) | 717 (A) | 892 (B) | |||
| 12 | CFH | 279 (C) | 2134 (C) | 54370 (C) | ||||
| 13 | DMBT1 | 3395 (A) | 182067 (A) | 360 (A) | ||||
| 14 | EGFR | 3395 (A) | 182067 (A) | 360 (A) | ||||
| 15 | FH | 217071 (A) | 151 (A) | 523 (A) | ||||
| 16 | GLTSCR1 | 3395 (A) | 182067 (A) | 360 (A) | ||||
| 17 | GLTSCR2 | 3395 (A) | 182067 (A) | 360 (A) | ||||
| 18 | GLI1 | 3395 (A) | 182067 (A) | 360 (A) | ||||
| 19 | LRRN2 | 3395 (A) | 182067 (A) | 360 (A) | ||||
| 20 | YEATS4 | 3395 (A) | 182067 (A) | 360 (A) | ||||
| 21 | OGG1 | 217071 (A) | 151 (A) | |||||
| 22 | ACVRL1 | 422 (C) | 774 (B) | |||||
| 23 | APC | 3395 (A) | 733 (A) | |||||
| 24 | ASPSCR1 | 3394 (A) | 163699 (A) | |||||
| 25 | AGGF1 | 2346 (C) | 90308 (B) | |||||
| 26 | BRCA2 | 180250 (A) | 217074 (A) | |||||
| 27 | CTNNB1 | 3395 (A) | 88673 (A) | |||||
| 28 | COL1A1 | 3394 (A) | 31112 (A) | |||||
| 29 | COL18A1 | 98668 (C) | 1571 (C) | |||||
| 30 | DIRC1 | 217071 (A) | 151 (A) | |||||
| 31 | DIRC2 | 217071 (A) | 151 (A) | |||||
| 32 | ETV4 | 223727 (A) | 319 (A) | |||||
| 33 | ETV1 | 223727 (A) | 319 (A) | |||||
| 34 | FLT4 | 79383 (B) | 77240 (B) | |||||
| 35 | FLCN | 217071 (A) | 151 (A) | |||||
| 36 | FHIT | 217071 (A) | 151 (A) | |||||
| 37 | FLI1 | 223727 (A) | 319 (A) | |||||
| 38 | GJC2 | 79383 (B) | 77240 (B) | |||||
| 39 | HSPBAP1 | 217071 (A) | 151 (A) | |||||
| 40 | JAK2 | 824 (B) | 729 (B) | |||||
| 41 | MET | 217071 (A) | 88673 (A) | |||||
| 42 | MEN1 | 652 (B) | 913 (B) | |||||
| 43 | NOTCH3 | 136 (C) | 95509 (C) | |||||
| 44 | PRCC | 217071 (A) | 151 (A) | |||||
| 45 | PALB2 | 180250 (A) | 217074 (A) | |||||
| 46 | PDGFB | 3394 (A) | 31112 (A) | |||||
| 47 | RASA1 | 2346 (C) | 137667 (A) | |||||
| 48 | RET | 717 (A) | 1332 (A) | |||||
| 49 | RNF139 | 217071 (A) | 151 (A) | |||||
| 50 | SMAD4 | 217074 (A) | 774 (B) | |||||
| 51 | SDHAF2 | 717 (A) | 29072 (A) | |||||
| 52 | SDHA | 717 (A) | 29072 (A) | |||||
| 53 | SDHC | 717 (A) | 29072 (A) | |||||
| 54 | SMARCA4 | 3394 (A) | 69077 (A) | |||||
| 55 | SMARCB1 | 3394 (A) | 69077 (A) | |||||
| 56 | TMEM127 | 717 (A) | 29072 (A) | |||||
| 57 | ERG | 223727 (A) | 319 (A) | |||||
| 58 | KIT | 44890 (A) | 98292 (A) | |||||
(A), (B), (C) correspond to subset A, B and C respectively.
Fig 2.
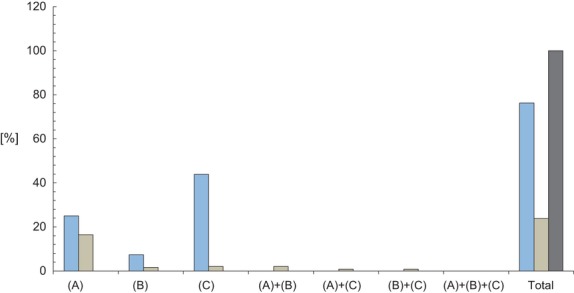
Relative distribution of genes associated to angiogenesis-related rare disease. Percentages of genes belonging to described subsets of A-RDs: A (A-RDs with cancerous phenotype in all their features), B (A-RDs with cancerous phenotype only in some of their features) and C (A-RDs without cancerous phenotype). Horizontal-axis represents the distribution of genes in the different subsets. Color code: blue, genes related to one A-RD; brownish, genes related to two or more A-RDs; dark grey: total genes related to A-RDs.
In the case of drugs, in this study 285 entrances were manually identified in Orphanet database as orphan drugs. As in the case of genes, three categories of A-RDs where established: those currently associated to no drug, those associated to one drug, and those with two or more drugs associated. The number of drugs in each A-RD is highlighted in Table2. Furthermore, Tables S7 and S8 (Supplementary material) list all the A-RDs associated to a single or to more than a drug, respectively, identifying the corresponding related drugs by their Orpha drug IDs. As in the case of genes, the relationship A-RDs-drugs is also asymmetrical. Tables S9 and S10 list the whole set of 198 drugs associated to one A-RD and 87 drugs associated to two or more A-RDs, respectively, identifying the corresponding A-RDs by their Orpha rare disease IDs. Figure3 shows the distribution of the annotated drugs grouped in two categories (those associated to one A-RD and those associated to two or more A-RDs) in the whole set and the three subsets of A-RDs.
Fig 3.
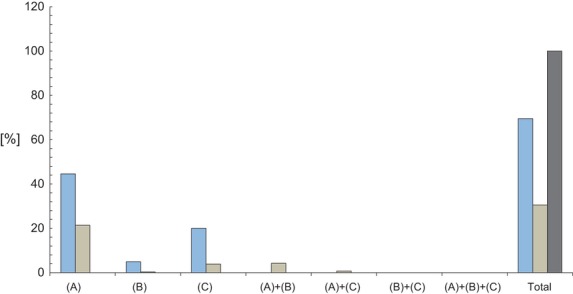
Relative distribution of Orphan drugs associated to angiogenesis-related rare disease. Percentages of drugs belonging to described subsets of A-RDs: A (A-RDs with cancerous phenotype in all their features), B (A-RDs with cancerous phenotype only in some of their features) and C (A-RDs without cancerous phenotype). Horizontal-axis represents the distribution of drugs in the different subsets. Color code: blue, drugs related to one A-RD; brownish, drugs related to two or more A-RDs; dark grey: total drugs related to A-RDs.
Discussion of the literature search and orphanet analysis
Bibliographic systematic review
Our bibliographic systematic review (see Fig.1) has allowed us to select 187 references (see bibliography in Supplementary material) and 180 A-RDs (that is, 180 rare diseases related with the neovascularization process, see Table2A–C). This systematic review has also made possible the extraction of a great amount of data in an exhaustive and trustworthy manner. In the present work, these extracted data could be synthetized, validated and interrelated through the use of a reproducible methodology, based in PRISMA statements 20,21 (see PRISMA checklist in Table S1 and PRISMA flow diagram in Figure S1). When we recovered those A-RDs that are cited three or more times in our set of selected references (29 of the 180 A-RDs), we found that in most cases vascular diseases (or malformations) or angioproliferative disorders were retrieved, with the exceptions of Multicentric castleman disease (OPRPHA93686), Multiple myeloma (ORPHA29073), Primary effusion lymphoma associated with HIV infection (ORPHA99923) and Primary effusion lymphoma (ORPHA48686). Renal cell carcinoma (ORPHA217071) was the angioproliferative disease more times referenced (in fact, it is mentioned in 14 of the 187 selected references included in supplementary material).
On the other hand, a text mining analysis has been used as an automatic and independent validation of the results achieved in the systematic review. We can observe that the enriched MeSH terms of the 187 bibliographic references, obtained from the systematic review, are notably related to angiogenesis and pathological conditions (see Table1). These results of the text mining analysis confirm that our selection of the bibliography has consistent information related to angiogenesis and biological process involved in the pathogenesis. Under these conditions, more than a half of PMIDs linked to MeSH terms related to pathologies (i.e. Neovascularization, Pathologic, Herpesvirus 8, Macular Degeneration, Pathologic-von Hippel–Lindau Disease, lymphangioleiomyomatosis, Renal Cell Carcinoma, Hereditary Hemorrhagic Telangiectasia, Hemangioendothelioma, POEMS Syndrome, Essential osteolysis, Capillary Hemangioma Kaposi Sarcoma, Rare Diseases and Lipoid proteinosis of Urbach and Wiethe) match with those PMIDs that are linked to MeSH terms related to angiogenesis (Angiogenesis inhibitors, Vascular Endothelial Growth Factor A, Thalidomide, Vascular Endothelial Growth Factors, Angiogenic Proteins, Vascular Endothelial Growth Factor Receptors). It is interesting to note that only three PMIDs are indicated for the term called “Rare disease”, which highlights the lack of identification with this type of disease in the scientific literature (see Table1).
Initial classification of angiogenesis-related rare diseases
Orphanet has its own classification of rare diseases, based on a hierarchy of descriptive categories. These categories are pathological descriptions based in published scientific data and information provided by experts in the field. These pathological descriptions are referred to as manifestations along the tables presented in this work. The close relationship between angiogenesis and tumor progression was used for the initial classification of A-RDs into three differentiated subsets (see Table2). The analysis of this classification confirmed that this procedure gave rise to a consistent separation. The dominant subset was “rare neoplasias” (subset A), as expected. Concerning A-RDs included in subset B, they are mostly vascular malformations (as angiomas and hemangiomas), as well as dysplasias and hamartomas. They are non neoplastic pathologies associated to an increased risk to develop tumors. Finally, subset C included a whole array of non-tumoral rare diseases tightly related with (and, in many cases, dependent on) angiogenesis.
On the other hand, the systematic review of the collected data allowed us to detect a number of incoherencies in the Orphanet classification of rare diseases. Some of them are highlighted here:
Cerebral cavernous malformations (ORPHA164) are rare brain vascular malformations that are also named as brain cavernous angioma. The term “angioma” showed concurrence with this and other search terms included in the three subsets, in spite of the fact that an angioma is not a tumor 24. Our analysis led to include cerebral cavernous malformations (ORPHA164) within subset A, although the Orphanet classification for this disease remained undefined (“This disease will be assigned to a classification in the near future”). Furthermore, the clinical description of this disease in Orphanet was unclear enough as to make possible its inclusion in any of the other subsets (either B or C). On the other hand, ICD-10 codes this disease as Q28.3 (“Congenital malformations, deformations and chromosomal abnormalities”), as it is also the case for other angiomas (glomuvenous malformation (ORPHA83454), among others).
POEMS syndrome (ORPHA2905) is a rare paraneoplasia that only presented concurrency with search terms in some of their classifications. However, it is a multisystemic disease in most of the cases associated to osteosclerotic myeloma. This is the main reason why it was finally included into subset A, although there were also reasons to include it into subset B. ICD-10 codes it within R16 (“Symptoms, signs and abnormal clinical and laboratory findings, not elsewhere classified”).
Chronic myeloid leukemia (ORPHA521) appears included in subset B (Table2) because at the moment in which this systematic review and analysis was carried out Orphanet offered four general categories of classification (namely, classification of hematopoietic and lymphoid tumors, classification of disease with platelet number and function anomalies, Orphanet classification of rare hematological diseases and Orphanet classification of rare tumors) for this disease, one of which (namely, classification of disease with platelet number and function anomalies) had no concurrence with search terms. However, updates arranged in Orphanet after our systematic review and analysis offer only two general classification for this disease (namely, Orphanet classification of rare hematological diseases and Orphanet classification of rare tumors), which would be consistent with the inclusion of this disease within subset A. In fact, ICD-10 codes this disease as C92.1 (“Neoplasms”).
These previously commented cases and others reveal the importance of establishing correct and unambiguous denominations to these diseases in databases such as Orphanet. A systematic, unambiguous classification and the actual way in which the information is documented and transmitted can help physicians during health assistance. We have also detected that ICD-10 have no code assigned for 46% (83 out of 180) of the A-RDs listed (see Tables S2–S4 in Supplementary material). The announced release of ICD-11 (which will include a systematic classification based on worldwide accepted clinical criteria) should standardize the classification of rare diseases. This undoubtedly will make it easier the documentation procedure and the curation of the collected information concerning this kind of vaguely described disorders.
It should be mentioned that several classifications of rare diseases available in Orphanet at the moment of carrying out this systematic review were deleted from Orphanet website in more recent actualizations of the site. This was the case of Orphanet classification of red cell diseases, classification of hematopoietic and lymphoid tumors and classification of disease with platelet number and function anomalies. The recent emergence of a new category in the Orphanet classification: Inherited cancer-predisposing syndrome (ORPHA140162) should also be mentioned.
As mentioned, from the 180 selected A-RDs, these 27 highlighted with an asterisk in Table2 can be considered as angiogenic rare diseases. Three of them have same genetic and molecular profiles closely related to angiogenesis: Angio-osteohypertrophic syndrome (ORPHA2346), Neurofibromatosis type I (ORPHA636) and Rendu-Osler-Weber disease (ORPHA774). Orphanet also includes information about this. The other angiogenic rare diseases are pathologies whose genetic and molecular profiles are related to certain aspects of angiogenesis such as VEGF expression, anti-angiogenic therapies or related with vascular proliferation process, among others. In many of these cases further investigation is required. A problem arises with Orpha IDs that are classified in Orphanet as diseases that group other diseases. Under these conditions, since no distinction is made by Orphanet and only in some cases it is indicated correctly (i.e. “This term does not characterize a disease but a group of diseases. To learn about the diseases included under this term, search for in the Classifications menu”), we think that many other diseases could be retrieved. In the case of A-RDs that group other diseases, Orphanet does not only list those genes that are being shared among all diseases belonging to that category, but it also contains all the genes that are within that class of diseases. In this context, no genetic profile is identified in these categories of classification. As mentioned, a classification made in this way is useful when grouping features around a category of diseases and allows a proper allocation of patient, but it says little about its genetic and molecular profiles. No information is displayed in the summary of almost a third of these A-RDs and only two of these that group other diseases are identified (i.e. An Orphanet summary for this disease is currently under development. However, other data related to the disease are accessible from the Additional Information menu located on the right side of this page). The case of Brain tumor (ORPHA3395) is clearly a kind of pathology that groups other diseases. In Oprhanet it is identified as: “This disease will be assigned to a classification in the near future”. For this disease, currently no list of genes, and no classification is indicated in Orphanet.
Another problem for systematic studies is also the way in which Orphanet updates and corrects its database. For instance, up to four angiogenic rare diseases were outdated in the course of our systematic review. Glioma, now called Glial tumor (ORPHA182067), does not remain associated with the gene called PTEN, which in its original link (MIN ID605 691) is identified as a tumor suppressor. Oligodendroglial tumor (ORPHA46484) is linked in UniProt (ID60484) too and it is just a subcategory of glial tumor. In addition, two genes are listed for Brain tumor: PTEN and CTNB1. On the other hand, two genes (PML and RBM15) were listed for Acute myeloid leukemia (ORPHA519). Currently, the symbol RBM15 (RNA binding motif protein 15) does not appear in Orphanet database. In Uniprot (ID Q96T37), Acute megacaryoblastic leukemia (ORPHA518) is listed as an Orphan disease.
Genes and drugs associated to angiogenesis-related rare diseases
Figure4 shows frequence distributions of A-RDs taking into account the number of genes or the number of drugs to which they are related (information abstracted from Tables4 and S5–S10. As Figure4 shows, approximately half of A-RDs are linked to no gene and no drug. The highest percentage of A-RDs linked to two or more genes or drugs corresponds to subset A. This reflects the actual current state of knowledge in this research area, which—as expected—is much higher in neoplastic A-RDs (those included in subset A).
Fig 4.
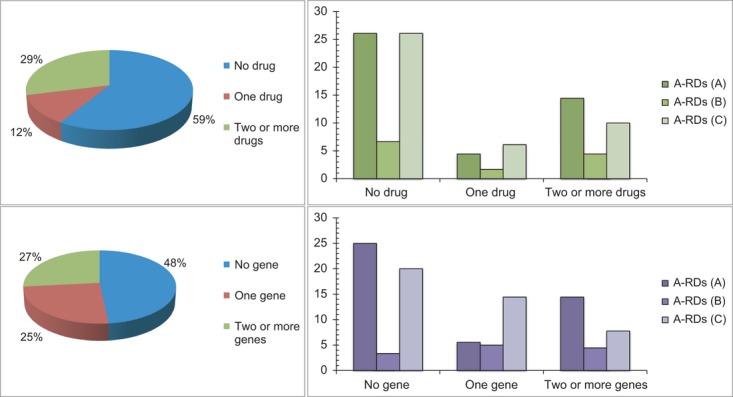
Relative distribution of angiogenesis-related rare diseases associated to genes and Orphan drugs. Left panels. Up, the percentages of A-RDs associated to drugs are shown. Down, the percentages of the three described subsets associated to genes are shown. Subsets: A (angiogenesis-related rare diseases with cancerous phenotype in all their features), B (cancer-related, angiogenesis-related rare diseases with cancerous phenotype only in some of their features) and C (angiogenesis-related rare diseases without cancerous phenotype). Right panels. Up, the percentages of A-RDs associated to drugs are shown. Down, the percentages of the same three A-RDs groups described above, here associated to genes.
Lysosomal storage disease with skeletal involvement (ORPHA93448) is the A-RD linked to the highest number of genes (22), but at the same time is linked to no drug. On the other hand, both Acute myeloid leukemia (ORPHA519) and Glioblastoma (ORPHA360) are the two A-RDs with the highest number of linked drugs (33 in each case), being the third and the 12th respectively according to their linked genes (see Table2 and Table S6).
As Figure2 shows, most of the genes are linked to only one A-RD. The majority of those genes linked to two or more A-RDs in fact are related with A-RDs of the subset A. A similar situation is found in the case of drugs (Fig.3).
Phosphatase and tensin homolog (mutated in multiple advanced cancers 1)—PTEN is one of the genes with more links to A-RDs (Table4).
It is noteworthy that Orphanet updated versions used when this systematic review and analysis was carried out identified the entrance Antisense Oligonucleotide (TATCCGGAGGGCTCGCCATGCTGCT) as both a gene (which is obviously false) and as a drug (see Table3 and Table S9). In both cases, this entrance was linked to an only A-RD: to Neovascular glaucoma (ORPHA94058) in Table3 and to Retinopathy of prematurity (ORPHA90050) in Table S9. We have maintained both entrances according to the results yielded by our systematic methodology. This highlights that this systematic approach allows for a more correct curation of the collected data. In more recent updates of Orphanet some of the detected error have been corrected.
Concluding remarks
The obvious great current interest in angiogenesis and in rare diseases demanded a systematic review of the current state of knowledge in the Boolean intersection of these two research areas, namely, the still not well defined set of angiogenesis-related rare diseases. The present review report contributes to satisfy such a demand.
Current technology allows researchers to have access to great databases containing overwhelming amounts of data. Furthermore, there are a number of biocomputational tools that makes it easier the access, extraction and analysis of biological data contained in databases and they can enhance the molecular knowledge emerged from the systematic review results. We claim that the critical use of such tools makes possible and affordable to carry out systematic reviews of the current state of knowledge of specific research areas. These systematic reviews are aimed to add value to the classical bibliographical reviews thanks to their potential to extract new emergent information. The present systematic review on angiogenesis-related rare diseases can be considered an initial contribution in this way.
Herein we propose a simple classification of angiogenesis-related rare diseases trying to discriminate the tight relationships of those linked to cancer.
We have detected that both the Orphanet classification of rare diseases and the WHO International Classification of Diseases ICD-10 urgently require deep revisions and updates.
Much more research is urgently needed for the identification of genes and drugs related to many angiogenesis-related rare diseases and for the exploration of new diagnostic, prognostic and therapeutic procedures.
Orphanet database is a useful resource for all scientists and physicians engaged with rare diseases, as well as for patients’ associations. However, form a scientific point of view, Orphanet still lacks of an actual semantic classification of rare disease. Furthermore, a deep upgrade of the scientific information contained in Orphanet is urgently required.
This systematic review of available literature on angiogenesis rare diseases has ordered and connected the currently available but up to now dispersed wealth of information on the topic, as reflected by the full set of accompanying figures and tables. This information will be useful for those interested in knowing the full set of described angiogenesis-related rare diseases, and what genes and drug treatments a specific angiogenesis-related rare disease shares with other ones. In particular, this information can be a very useful start point for new, deep reviews focused on concrete angiogenic rare diseases.
Taking the ordered information contained in the accompanying figures and tables of the present work as a starting point, functional enrichment and network analysis tools could be used in the near future to make predictions of new gene targets, drugs and/or treatments for some rare diseases based in their shared spectra with the full set of angiogenesis-related rare diseases herein reviewed.
Acknowledgments
Our experimental work is supported by grants SAF 2011-26518, PS09/02216 and TRACE PT2008-0145 (Spanish Ministry of Science and Innovation, ISCIII and FEDER), Fundación Ramón Areces, and PIE CTS-3759, CVI-6585 and funds from group BIO-267 (Andalusian Government and FEDER). The ‘CIBER de Enfermedades Raras’ is an initiative from the ISCIII (Spain). The funding agencies had no role in study design, the research work carried out and the writing of this manuscript. ARP is a FPU fellow (Ministry of Education, Spain). Proyecta Científica assisted in the management of scientific data.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All the five authors were personally involved bibliographic research and discussion of collected data as well as in the design of the manuscript contents. L.R.C. carried out the primary bibliographic search. A.R.P. was involved in the text-mining analysis with SciMiner. A.R.Q. and F.S.J. critically reviewed the first draft of the manuscript and contributed to its definitive version. M.A.M. supervised the whole procedures and wrote the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. PRISMA flow diagram of the bibliographic systematic review.
Table S1. PRISMA checklist
Table S2. Analysis of the angiogenesis-related rare disease in subset A according to International Classification of Diseases
Table S3. Analysis of the angiogenesis-related rare disease in subset B according to International Classification of Diseases
Table S4. Analysis of the angiogenesis-related rare disease in subset C according to International Classification of Diseases
Table S5. Angiogenesis-related rare diseases associated to a single gene
Table S6. Angiogenesis-related rare diseases associated to two or more genes
Table S7. Angiogenesis-related rare diseases associated to a single drug
Table S8. Angiogenesis-related rare diseases associated to two or more drugs
Table S9. Drugs associated to a single angiogenesis-related rare disease
Table S10. Drugs associated to two or more angiogenesis-related rare diseases
Data S1. Selected bibliographic references related to the 187 angiogenesis-related rare diseases covered by this work.
References
- Ayme S, Schmidtke J. Networking for rare diseases: a necessity for Europe. Bundesgesundheitsblatt-Gesund. 2007;50:1477–83. doi: 10.1007/s00103-007-0381-9. [DOI] [PubMed] [Google Scholar]
- Kaiser J. Human genetics. Affordable ‘exomes’ fill gaps in a catalog of rare diseases. Science. 2010;330:903. doi: 10.1126/science.330.6006.903. [DOI] [PubMed] [Google Scholar]
- Aymé S, Rath A, Bellet B. WHO international classification of diseases (ICD) revision process: incorporating rare diseases into the classification scheme: state of art. Orphanet J Rare Dis. 2010 . doi: 10.1186/1750-1172-5-S1-P1. [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Leung DW, Cachianes G, Kuang WJ, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Quesada AR, Muñoz-Chápuli R, Medina MA. Anti-angiogenic drugs: from bench to clinical trials. Med Res Rev. 2006;26:483–530. doi: 10.1002/med.20059. [DOI] [PubMed] [Google Scholar]
- Medina MA, Muñoz-Chápuli R, Quesada AR. Challenges of antiangiogenic cancer therapy: trials and errors, and renewed hope. J Cell Mol Med. 2007;11:374–82. doi: 10.1111/j.1582-4934.2007.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada AR, Medina MA, Alba E. Playing only one instrument may be not enough: limitations and future of the antiangiogenic treatment of cancer. BioEssays. 2007;29:1159–68. doi: 10.1002/bies.20655. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor and age-related macular degeneration: from basic science to therapy. Nat Med. 2010;16:1107–11. doi: 10.1038/nm1010-1107. [DOI] [PubMed] [Google Scholar]
- Dispenzieri A. POEMS syndrome. 2007;21:285–99. doi: 10.1016/j.blre.2007.07.004. Blood Rev. [DOI] [PubMed] [Google Scholar]
- Chee CE, Dispenzieri A, Gertz MA. Amyloidosis and POEMS syndrome. Expert Opin Pharmacother. 2010;11:1501–14. doi: 10.1517/14656561003769874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg DA, Jin KL. From angiogenesis to neuropathology. Nature. 2005;438:954–9. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Reyes-Palomares A, Montañez R, Real-Chicharro A, et al. Systems biology metabolic modeling assistant: an ontology-based tool for the integration of metabolic data in kinetic modeling. Bioinformatics. 2009;25:834–5. doi: 10.1093/bioinformatics/btp061. [DOI] [PubMed] [Google Scholar]
- Montañez R, Medina MA, Solé RV, et al. When metabolism meets topology: reconciling metabolite and reaction networks. BioEssays. 2010;32:246–56. doi: 10.1002/bies.200900145. [DOI] [PubMed] [Google Scholar]
- Ranea JAG, Morilla I, Lees JG, et al. Finding the “dark matter” in human and yeast protein network prediction and modelling. PLoS Comput Biol. 2010 doi: 10.1371/journal.pcbi.1000945. . doi: 10.1371/journal.pcbi.1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009 . doi: 10.1371/journal.pmed.1000097. [PMC free article] [PubMed] [Google Scholar]
- Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009 doi: 10.1371/journal.pmed.1000100. . doi: 10.1371/journal.pmed.1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renear AH, Palmer CL. Strategic reading, ontologies, and the future of scientific publishing. Science. 2009;325:828–32. doi: 10.1126/science.1157784. [DOI] [PubMed] [Google Scholar]
- Hur J, Schuyler AD, States DJ, et al. SciMiner: web-based literature mining tool for target identification and functional enrichment analysis. Bioinformatics. 2009;25:838–40. doi: 10.1093/bioinformatics/btp049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blann AD, Ramcharan KS, Stonelake PS, et al. The angiome: a new concept in cancer biology. J Clin Pathol. 2011;64:637–43. doi: 10.1136/jcp.2011.088948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PRISMA flow diagram of the bibliographic systematic review.
Table S1. PRISMA checklist
Table S2. Analysis of the angiogenesis-related rare disease in subset A according to International Classification of Diseases
Table S3. Analysis of the angiogenesis-related rare disease in subset B according to International Classification of Diseases
Table S4. Analysis of the angiogenesis-related rare disease in subset C according to International Classification of Diseases
Table S5. Angiogenesis-related rare diseases associated to a single gene
Table S6. Angiogenesis-related rare diseases associated to two or more genes
Table S7. Angiogenesis-related rare diseases associated to a single drug
Table S8. Angiogenesis-related rare diseases associated to two or more drugs
Table S9. Drugs associated to a single angiogenesis-related rare disease
Table S10. Drugs associated to two or more angiogenesis-related rare diseases
Data S1. Selected bibliographic references related to the 187 angiogenesis-related rare diseases covered by this work.