

Abstract
Substance P (SP) is involved in the pathophysiology of acute pancreatitis (AP) via binding to its high-affinity receptor, neurokinin-1-receptor (NK1R). An up-regulation of SP and NK1R expression was observed in experimental AP and in caerulein-stimulated pancreatic acinar cells. However, the mechanisms that lead to this up-regulation are not fully understood. In this study, we showed the role of protein kinase C (PKC) in caerulein-induced SP and NK1R production in isolated mouse pancreatic acinar cells. Caerulein (10−7 M) stimulation rapidly activated the conventional PKC-α and novel PKC-δ as observed by the phosphorylation of these molecules. Pre-treatment of pancreatic acinar cells with Gö6976 (1–10 nM) and rottlerin (1–10 μM) inhibited PKC-α and PKC-δ phosphorylation, respectively, but not the other way round. At these concentrations used, PKC-α and PKC-δ inhibition reversed the caerulein-induced up-regulation of SP and NK1R, indicating an important role of PKCs in the modulation of SP and NK1R expression. Further experiments looking into signalling mechanisms showed that treatment of pancreatic acinar cells with both Gö6976 and rottlerin inhibited the activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase (JNK). Inhibition of PKC-α or PKC-δ also affected caerulein-induced transcription factor activation, as represented by nuclear factor-κB and AP-1 DNA-binding activity. The findings in this study suggested that PKC is upstream of the mitogen-activated protein kinases and transcription factors, which then lead to the up-regulation of SP/NK1R expression in caerulein-treated mouse pancreatic acinar cells.
Keywords: PKC, substance P, neurokinin-1-receptor, acute pancreatitis
Introduction
Acute pancreatitis (AP) is a potentially fatal clinical condition caused by a rapid onset inflammation of the pancreas. AP may be induced by injury of pancreatic acinar cells, which are cells that produce digestive enzymes [1, 2]. It is widely thought that premature activation of these digestive enzymes within the pancreas, particularly trypsinogen, leads to auto-digestion of the organ and cause subsequent inflammatory response [2, 3]. Exposure of isolated pancreatic acinar cells with supramaximal doses of caerulein has been reported to mimic many responses that resemble AP, including the production of pro-inflammatory mediators [1]. Mechanisms that regulate the severity of the inflammatory response have not been clearly understood so far.
Neuropeptides have frequently been linked with the progression of AP, those implicated included, but not limited to, ghrelin, galanin and substance P (SP) [4–6]. SP is an 11-amino acid peptide and is the product of the preprotachykinin-A (PPT-A) gene. SP is a well-recognized mediator of neuro-immune interaction and plays a pathogenic role in immune and inflammatory disorders. SP is found in the pancreas and is involved in the mediation of neurogenic inflammation in AP. In earlier reports, the use of PPT-A knockout mice and neurokinin-1-receptor (NK1R) antagonism were found to have a less severe form of AP and its associated lung injury [7, 8]. In mouse macrophages and pancreatic acinar cells, SP was found to promote inflammatory responses by up-regulating pro-inflammatory cytokines and chemokines [9, 10]. Recently, SP and its receptor, NK1R is expressed and have been shown to be up-regulated in caerulein stimulated mouse pancreatic acinar cells [11, 12]. The up-regulation of SP levels was found to mediated via the cholecystokinin (CCK) receptor, CCKA[11]. Although the expression of SP is relatively low in pancreatic acinar cells when compared to neurons, its local effect and a large amount of pancreatic acinar cells in the pancreas might be a significant contributor of SP mediated inflammation in AP.
The molecular mechanism by which caerulein exerts its effects is now becoming clearer. In recent years, the role of protein kinase C (PKC) has been extensively studied in the pathogenesis of AP. There are more than 10 isoforms of PKC divided into three different classes, namely, conventional, novel and atypical PKC. The conventional PKC isoforms α, β and γ are regulated by Ca2+ and diacylglycerol (DAG). On the other hand, novel PKC isoforms δ, ε, ν and θ respond to DAG, but do not involve Ca2+. The atypical PKC isoforms ζ, λ/ι and μ are different as their activation does not need the presence of Ca2+ and DAG [13]. In the pancreatic acinar cells, the conventional PKC-α and novel PKC-δ, ε and atypical ζ isoforms had been identified [14]. In general, the activation of PKC is characterized by the phosphorylation of the molecule, followed by translocation of distinct intracellular compartments in which it performs its specific function [15]. The individual roles of PKCs in the regulation of pancreatic acinar function have been reported in various studies, where PKC-δ is the most studied. PKC-δ is activated and translocated to the plasma membrane and participates in secretion [16], regulates protease activation [17] and also modulates inflammatory molecule expression in the pancreatic acinar cells [18]. The role of conventional PKC-α is less understood, but with CCK treatment stimulates increases in Ca2+ and DAG in the pancreatic acinar cells, a potential role of PKC-α cannot be ruled out [19].
Previously, we have shown the involvement of mitogen-activated protein kinases (MAPKs) ERK1/2 and JNK, and also transcription factors nuclear factor-κB (NF-κB) and AP-1, with caerulein-induced SP/NK1R up-regulation in mouse pancreatic acinar cells [11]. Although different isoforms of PKCs have been associated with downstream signalling of MAPKs and the transcription factors, whether PKCs are associated with caerulein’s gene activating signalling is not yet clear. In this study, we sought to investigate whether caerulein activates PKCs in isolated mouse pancreatic acinar cells and examine their involvement in caerulein-stimulated SP/NK1R up-regulation. We also aim to differentiate the role of conventional PKC-α and novel PKC-δ in our study. To our knowledge, none has investigated whether members of the PKC family regulate the expression of SP and NK1R. Therefore, we investigate the role of PKC in caerulein-induced SP/NK1R expression in an in vitro model of isolated pancreatic acinar cells and study the underlying mechanisms involved.
Materials and methods
Animals and chemicals
All experimental procedures were approved by the Animal Ethics Committee of the National University of Singapore and carried out in accordance with established International Guiding Principles for Animal Research. Swiss mice (male, 25–30 g) were acclimatized in a controlled environment with an ambient temperature of 23°C and a 12:12-hr light–dark cycle. Caerulein, a CCK analogue, was purchased from Bachem California (Torrance, CA, USA). Glucose, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and soybean trypsin inhibitor were obtained from Sigma-Aldrich (St. Louis, MO, USA). Type IV collagenase was purchased from Worthington (Freehold, NJ, USA). Gö6976 and rottlerin, which was widely used as a conventional PKC inhibitor and a PKC-δ inhibitor, respectively, were purchased from Calbiochem (Darmstadt, Germany) [20, 21]. All chemicals were purchased with the highest purity available.
Preparation of pancreatic acini
Pancreatic acini were obtained from mouse pancreas by collagenase treatment as described previously [7]. Briefly, Swiss mice were killed by a lethal dose of sodium pentobarbitone. Fresh pancreas were infused with buffer A (mM: 140 NaCl, 4.7 KCl, 1.13 MgCl2, 1CaCl2, 10 glucose, and 10 HEPES and 0.5 mg/ml soybean trypsin inhibitor, pH 7.3) containing 200 IU/ml Type IV collagenase. The pancreas was then minced and placed in a 50 ml tube with 12 ml of buffer A containing 200 IU/ml Type IV collagenase and incubated in a shaking water bath for 10 min. at 37°C with shaking. To obtain dispersed acini, the digested tissue was passed through small pipette tips. The cells were then passed through a solution of 50 mg/ml bovine serum albumin (BSA) and then washed twice with buffer A before further experiments. The viability of pancreatic acinar cells was determined by trypan blue exclusion assay. Cell preparations with at least 95% viability were used for further experiments.
Treatment of pancreatic acinar cells
Isolated pancreatic acinar cells were treated with caerulein. For time-dependent studies, cells were treated with caerulein (10−7 M) for 2, 5, 15, 30 and 60 min. in a 37°C water bath. For other experiments, cells were pre-treated with either Gö6976 or rottlerin for 45 min. before addition of caerulein (10−7 M). The concentrations used were 1, 5 and 10 nM for Gö6976 and 1, 5 and 10 μM for rottlerin.
SP extraction and detection
Treated pancreatic acinar cells were homogenized in ice-cold SP assay buffer and SP was concentrated by adsorbing on C18 cartridge columns (Bachem) as described previously [8]. The adsorbed peptides were eluted with 1.5 ml of 75% acetonitrile. They were freeze-dried overnight and reconstituted with SP assay buffer. SP content was then determined with an ELISA kit (Bachem) according to manufacturer’s instructions. The results were quantified by spectophotometry at 450 nm. The results were then normalized with DNA content of the acinar cell samples. DNA assay was performed fluorometrically by using Hoechst dye 33256 and calf thymus DNA as a standard [22]. SP expression was corrected as nanograms per microgram of DNA.
Quantitative real time PCR analysis
Total RNA from the pancreatic acinar cells was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. All the steps were done on ice-cold conditions. The integrity of RNA was verified by ethidium bromide staining for the presence of distinct 28S and 18S bands on a 1.2% agarose gel. One microgram (1 μg) of total RNA was reverse transcribed using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) in a total volume of 20 μl. The reaction was commenced at 25°C for 5 min. and 42°C for 30 min., followed by 85°C for 5 min. A total of 2 μl of cDNA was used as a template for PCR amplification by using SYBR-green PCR master mix from Roche Diagnostics (Singapore). The primers were intron spanning to exclude possible genomic DNA contamination. No template controls were performed to ensure that no PCR reagent contamination is present. All reactions were done in duplicates. PCR reaction mix was first subjected to 95°C for 5 min., followed by a 45 cycles of amplification. Each cycle consisted of 95°C for 30 sec., annealing temperature of 57.5°C for 15 sec., and elongation temperature of 72°C for 15 sec. Primer sequence, annealing temperature and product size for the genes tested were indicated in Table 1. β-actin was used as a housekeeping gene to normalize the mRNA expression of PPT-A and NK1R. Expression of PPT-A, NK1R and β-actin was determined using the ‘crossing point (Cp)’ of the sample, where Cp is the point (cycle number) at which the fluorescence of a sample rises above the background fluorescence.
Table 1.
PCR primer sequences, annealing temperatures and product sizes
| Gene (Genbank access no.) | Primer sequence | Annealing temperature | Size (bp) |
|---|---|---|---|
| β-actin (NM_007393) | Sense: 5′-TGTTACCAACTGGGACGACA-3′ Antisense: 5′-GGGGTGTTGAAGGTCTCAAA-3′ | Annealing: 57.5°C | 165 |
| NK1R (NM_009313) | Sense: 5′-GCTGCCCTTCCACATCTTCT-3′ Antisense: 5′-TTCCAGCCCCTCATAATCAC-3′ | Annealing: 57.5°C | 223 |
| PPT-A (NM_009311) | Sense: 5′-CGCGATGCAGAACTACGAAA-3′ Antisense: 5′GCTTGGACAGCTCCTTCATC-3′ | Annealing: 57.5°C | 282 |
Nuclear cell extract preparation and NF-κB/AP-1 DNA-binding activity
Nuclear cell extracts were prepared by using Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA). In brief, cells were washed in ice-cold PBS in the presence of phosphatase inhibitors. After centrifugation, the cell pellets were re-suspended in a hypotonic buffer, treated with detergent and centrifuged at 14,000 ×g for 30 sec. After removing of the cytoplasmic fraction, the pellet (nuclei) was lysed with lysis buffer containing protease inhibitors and nuclear proteins were solubilized in the lysis buffer. Nuclear protein concentrations were determined by Bradford protein assay (Bio-Rad Laboratories, Hercules, CA, USA). The binding of nuclear NF-κB and AP-1 to DNA was measured with ELISA based NF-κB p65 assay kit and AP-1 c-Jun assay kit, respectively (Active Motif). The plates were pre-coated with an unlabelled oligonucleotide containing the consensus-binding site (NF-κB p65: 5′-GGGACTTTCC-3′; AP-1 c-Jun: 5′-TGAGTCA-3′). Nuclear proteins (20 μg) were used for the assay and the results were quantified by spectrophotometry at 450 nm.
Whole cell lysate preparation and Western blot analysis
After treatment of pancreatic acinar cells, they were homogenized on ice in radioimmunoprecipitation assay lysis buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitor cocktail (Sigma-Aldrich). Protein concentrations were determined by the Bradford protein assay. Protein samples (80 μg) were separated by 10% SDS-PAGE gels and electrophoretically transferred to polyvinylidene difluoride membranes. Non-specific binding was blocked by 1 hr incubation of the membranes in 5% non-fat dry milk in PBST (0.05% Tween 20 in PBS). The blots were then incubated overnight with primary antibody against NK1R (Abcam, Cambridge, United Kingdom), phospho-PKC-α (Thr638), phospho-PKC-δ (Thr505), IκBα, phospho-IκBα (Ser32), ERK1/2, phospho-ERK1/2(Thr202/Tyr204), JNK and phospho-JNK (Thr183/Tyr185) (Cell Signaling Technology, Danvers, MA, USA) at 1:1000 dilutions in the buffer containing 2.5% non-fat dry milk in PBST. After which they were washed four times with PBST, and finally incubated for 2 hrs with goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:2000 dilutions in the buffer containing 2.5% non-fat dry milk in PBST. Membranes were washed and then incubated in SuperSignal™ West Pico chemiluminescent substrate (Pierce, Rockford, IL, USA) before exposure to X-ray films (CL-Xposure™; Pierce). In certain experiments, hypoxanthine-guanine phosphoribosyltransferase (HPRT) (Santa Cruz Biotechnology) was applied as an internal control to normalize protein loading. The intensity of bands was quantified using LabWorks™ Image Analysis software (UVP).
Statistical analysis
The data were expressed as the mean ± S.E.M. The significance of changes among groups was evaluated by using anova with a post-hoc Tukey’s test. A P-value ≤0.05 was considered as statistically significant.
Results
Caerulein induces phosphorylation of PKC-α and PKC-δ in mouse pancreatic acinar cells
Caerulein and its analogue CCK are known to activate PKC family in the pancreatic acinar cells. We first examined the time-dependant phosphorylation of PKC-α and PKC-δ in our model of caerulein-induced pancreatic acinar cells. To do this, freshly isolated pancreatic acinar cells were treated with caerulein (10−7 M) for 2, 5, 15, 30 and 60 min. Western blot analysis of the whole cell lysates showed a modest but significant increase in phosphorylation of both PKC-α and PKC-δ. Both PKC-α and PKC-δ are rapidly activated, with significant increases after 2 min. of caerulein incubation. The activation of PKC-α was transient, as the phosphorylated molecules decreased after 1 hr of incubation (Fig. 1B). On the other hand, PKC-δ remained phosphorylated throughout the studied time course (Fig. 1C). HPRT was used as an internal control for this experiment.
Fig 1.

Caerulein induces phosphorylation of PKC-α and PKC-δ in mouse pancreatic acinar cells. Freshly prepared cells were treated with caerulein (10−7 M) for 2, 5, 15, 30 and 60 min. Whole cell lysates were prepared and 50 μg of protein were fractionated on 10% SDS-PAGE gels for Western blot analysis of phospho-PKC-α (Thr638), phospho-PKC-δ (Thr505) and HPRT. (A) Representative blots of phospho-PKC-α and phospho-PKC-δ from six independent experiments. (B) Time response study on PKC-α phosphorylation. (C) Time response study on PKC-δ phosphorylation. Results are expressed as means ± S.E.M. from six independent experiments. *P < 0.05, versus control.
Effect of Gö6976 and rottlerin on PKC-α and PKC-δ phosphorylation
After determining the time course response of PKC-α and PKC-δ, we proceed to investigate the effect of Gö6976 and rottlerin on PKC-α and PKC-δ activation. From the previous data, we chose a stimulation timing of 15 min., as the response at this time is greatest and more stable for both types of studied PKC. Pancreatic acinar cells were pre-treated with Gö6976 (1, 5 and 10 nM) or rottlerin (1, 5 and 10 μM) for 45 min. followed by caerulein (10−7 M) stimulation. Western blot analysis indicate that Gö6976 (IC50= 2.3 nM) showed a concentration-dependent inhibition of PKC-α and PKC-α phosphorylation is completely reversed at a concentration of 10 nM (Fig. 2A). However, Gö6976 does not inhibit PKC-δ at the concentrations used (Fig. 2B). On the other hand, rottlerin (IC50= 3 μM) significantly inhibited PKC-δ phosphorylation even at a low concentration of 1 μM (Fig. 2C). Rottlerin pre-treatment also showed a very small decrease of PKC-α activation when compared to the caerulein-treated group but they are far from statistical significance (Fig. 2D). Treatment of cells with the used inhibitors alone does not significant affect the phosphorylation of PKC-α and PKC-δ.
Fig 2.
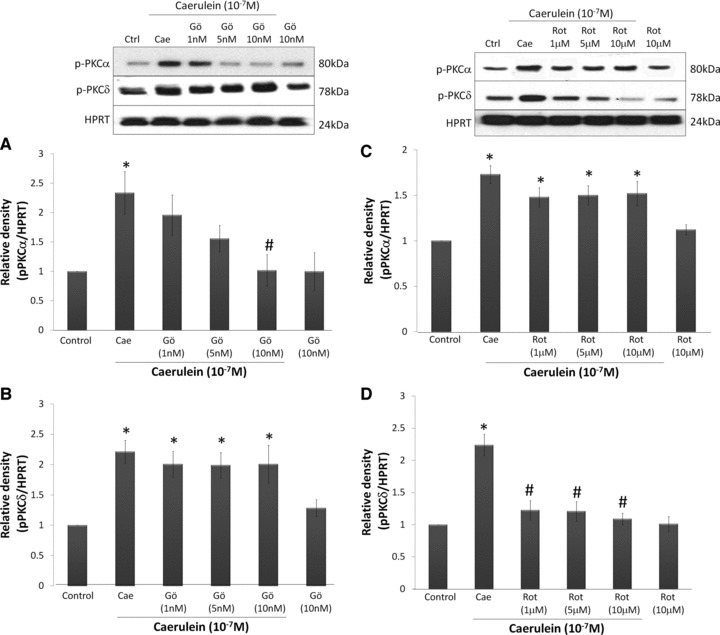
The effect of rottlerin and Gö6976 on PKC-α and PKC-δ phosphorylation. Pancreatic acinar cells were pre-treated with Gö6976 (1–10 nM) or rottlerin (1–10 μM) for 45 min. before caerulein (10−7 M) stimulation for 15 min. Whole cell lysates were prepared and 50 μg of protein were fractionated on 10% SDS-PAGE gels for Western blot analysis of phospho-PKC-α (Thr638), phospho-PKC-δ (Thr505) and HPRT. (A) Effect of Gö6976 on PKC-α phosphorylation. (B) Effect of Gö6976 on PKC-δ phosphorylation. (C) Effect of rottlerin on PKC-δ phosphorylation. (D) Effect of rottlerin on PKC-δ phosphorylation. Results are expressed as means ± S.E.M. from six independent experiments. *P < 0.05, versus control, #P < 0.05 versus caerulein.
PKC-α and PKC-δ are involved in caerulein-induced SP and NK1R up-regulation in mouse pancreatic acinar cells
Our previous studies have shown that caerulein hyperstimulation significantly up-regulated SP peptide expression and its gene, PPT-A, as well as NK1R mRNA expression in mouse pancreatic acinar cells. To examine the involvement of PKC-α and PKC-δ in this up-regulation, we pre-treated cells with Gö6976 and rottlerin. It was previously shown that Gö6976 and rottlerin are specific to PKC-α and PKC-δ, respectively (Fig. 2A–D). Using quantitative real time PCR, caerulein (10−7 M) significantly up-regulated PPT-A and NK1R mRNA expression when compared to the control. When the cells were pre-treated with Gö6976 or rottlerin, the gene expression of PPT-A and NK1R was significantly reduced (Figs 3A, C and 4B, D). In line with the observations for PPT-A, the peptide expression of SP was significantly reversed with pre-treatment of Gö6976 or rottlerin, indicating that PPT-A mRNA expression is one of the factors that mediate SP levels in pancreatic acinar cells (Fig. 3B and D). Previously, we have not shown the protein expression of NK1R in our model of caerulein-treated pancreatic acinar cells. To address this, we have done a time-dependent study to see the effects of caerulein stimulation. There is a slow but steady increase of protein expression observed, with statistically significant up-regulation observed after 2 hrs of caerulein incubation (Fig. 4A). However, the increase is modest (1.8-fold, P= 0.01). Pre-treatment with either Gö6976 or rottlerin inhibited this increased expression of NK1R protein (Fig. 4C and E). Generally, there is a slight concentration dependant inhibitory effect observed for the experiments done, as there was a more complete inhibition when higher concentrations of inhibitors were used.
Fig 3.
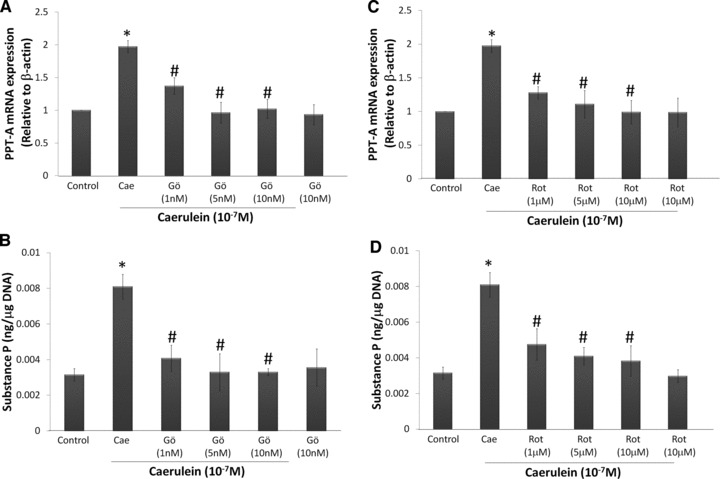
Caerulein stimulates PKC-α and PKC-δ mediated SP gene and protein expression. Freshly prepared pancreatic acinar cells were pre-treated with Gö6976 (1–10 nM) or rottlerin (1–10 μM) for 45 min. before caerulein (10−7 M) stimulation for 60 min. PPT-A mRNA expression was determined with real time PCR and SP levels were determined by a commercially available ELISA kit. PPT-A mRNA expression was normalized with β-actin expression and SP levels was normalized with DNA content in the samples. (A) PPT-A mRNA expression of cells treated with Gö6976. (B) SP expression of cells treated with Gö6976. (C) PPT-A mRNA expression of cells treated with rottlerin. (D) SP expression of cells treated with rottlerin. Results are expressed as means ± S.E.M. from four to six independent experiments. *P < 0.05 versus control, #P < 0.05 versus caerulein.
Fig 4.
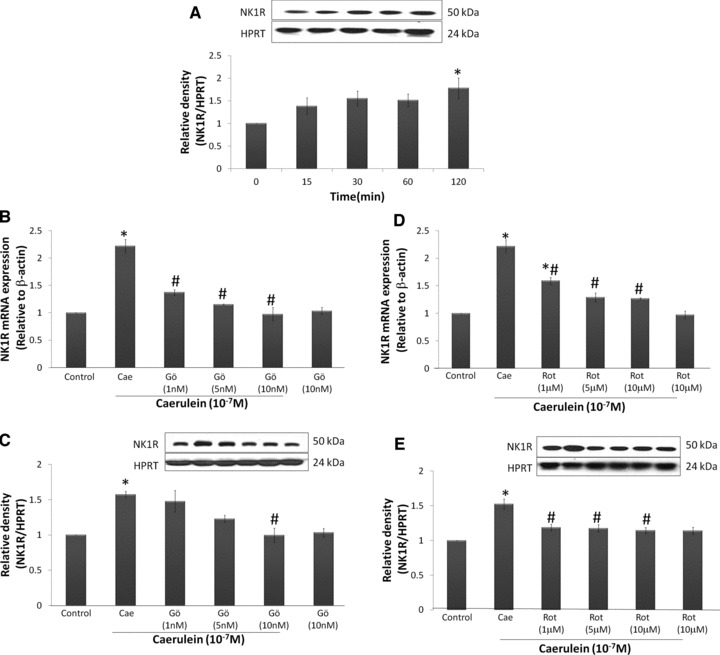
Caerulein stimulates PKC-α and PKC-δ mediated NK1R gene and protein expression. NK1R mRNA expression was determined with real time PCR and protein expression was determined by Western blotting. NK1R mRNA expression was normalized with β-actin expression and protein expression was normalized with HPRT expression in the samples. Freshly prepared pancreatic acinar cells were treated with caerulein (10−7 M) for 0, 15, 30, 60 and 120 min. (A) Time response study of NK1R expression. In other experiments, cells were pre-treated with Gö6976 (1–10 nM) or rottlerin (1–10 μM) for 45 min. before caerulein (10−7 M) stimulation for 60 min. (mRNA expression) or 120 min. (protein expression). (B) NK1R mRNA expression of cells treated with Gö6976. (C) NK1R protein expression of cells treated with Gö6976. (D) NK1R mRNA expression of cells treated with rottlerin. (E) NK1R protein expression of cells treated with rottlerin. Results are expressed as means ± S.E.M. from four to six independent experiments. *P < 0.05 versus control, #P < 0.05 versus caerulein.
PKC-α and PKC-δ are involved in caerulein-induced ERK1/2 and JNK activation in mouse pancreatic acinar cells
The role of PKCs in caerulein-induced SP/NK1R activation was further studied by looking at possible downstream signalling molecules. Our previous results have shown that the up-regulation of SP and NK1R involves the activation of the MAP kinases ERK1/2 and JNK [11]. To determine if PKC-α and PKC-δ are involved in the activation of ERK and JNK, pancreatic acinar cells were treated with Gö6976 (1–10 nM) or rottlerin (1–10 μM) followed by caerulein stimulation. Western blot analysis of whole cell lysates revealed that ERK1/2 and JNK were significantly activated after stimulated with caerulein. Our results also showed that both Gö6976 and rottlerin inhibited the phosphorylation of ERK1/2 and JNK (Fig. 5A–D). Furthermore, rottlerin treatment strongly inhibits JNK phosphorylation such that the rottlerin-treated groups fell below control levels (Fig. 5D). However, this decrease in JNK phosphorylation was not statistically significant in our study.
Fig 5.
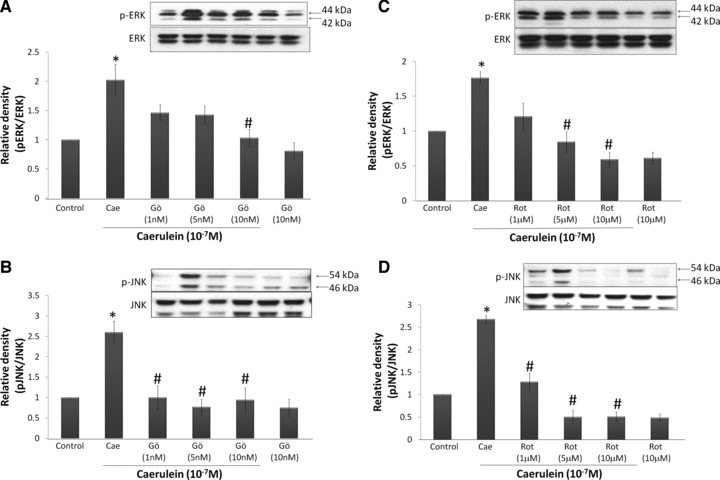
The activation of ERK1/2 and JNK in mouse pancreatic acinar cells is dependent on both PKC-α and PKC-δ. Freshly prepared pancreatic acinar cells were pre-treated with Gö6976 (1–10 nM) or rottlerin (1–10 μM) for 45 min. before caerulein (10−7 M) stimulation for 60 min. Whole cell lysates were prepared and 50 μg of protein were fractionated on 10% SDS-PAGE gels for Western blot analysis of phospho-JNK (Thr183/Tyr185), JNK, phospho-ERK1/2(Thr202/Tyr204) and ERK1/2. (A) ERK1/2 activation of cells treated with Gö6976. (B) JNK activation of cells treated with Gö6976. (C) ERK1/2 activation of cells treated with rottlerin. (D) JNK activation of cells treated with rottlerin. Results are expressed as means ± S.E.M. from four to six independent experiments. *P < 0.05 versus control, #P < 0.05 versus caerulein.
Inhibition of PKC-α and PKC-δ attenuates caerulein induced NF-κB and AP-1 activation in mouse pancreatic acinar cells
We examined the effect of Gö6976 and rottlerin on transcription factor NF-κB and AP-1 in isolated pancreatic acinar cells. Nuclear fractions of treated cells were extracted. DNA-binding activity was performed with an ELISA based method using nuclear fractions. Treatment of caerulein for 60 min. significantly increased NF-κB and AP-1 DNA-binding activity and this increase is attenuated by pre-treatment of cells with Gö6976 or rottlerin (Fig. 6B and C). Due to a small difference observed in NF-κB DNA-binding activity, we used an indirect method, the phosphorylation of IκB, to further confirm NF-κB activation. Whole cells lysates were used to perform Western blot, and the results shown a significant increase in IκB phosphorylation, which was attenuated with Gö6976 or rottlerin pre-treatment (Fig. 6A). The trend of IκB phosphorylation correlated with NF-κB-binding activity, confirming the role of PKC-α and PKC-δ in the activation of transcription factor NF-κB.
Fig 6.

PKC-α and PKC-δ activation is involved in the DNA-binding activity of NF-κB and AP-1. Pancreatic acinar cells were pre-treated with Gö6976 (1–10 nM) or rottlerin (1–10 μM) for 45 min. before caerulein (10−7 M) stimulation for 60 min. Subsequently, nuclear extracts was obtained and AP-1 and NF-κB DNA-binding ability was determined by using a commercially available ELISA kit. Whole cell lysates were prepared for Western blot analysis of phospho-IκB (Ser32) and HPRT. (A) Representative phospho-IκB Western blot results and relative intensity are averages of six independent experiments. (B) NF-κB DNA-binding activity. (C) AP-1 DNA-binding activity. Results are expressed as the means ± SE from six independent experiments. *P < 0.05 versus control, #P < 0.05 versus caerulein.
Discussion
It is becoming clear that the interaction between SP and NK1R is involved in pro-inflammatory reactions in AP. This is clearly shown by knocking out of the PPT-A gene, as well as antagonism of NK1R with pharmacological inhibitors [7, 8]. In both studies, lung and pancreatic injury was less severe and pro-inflammatory molecule levels were decreased. Isolated pancreatic acinar cells, which were widely considered as the initiating point of AP, were also found to express SP/NK1R and were up-regulated upon direct stimulation with caerulein [12]. Therefore, understanding the signalling pathway that leads to the up-regulation SP and NK1R could help to discover ways to control its expression levels. Members of the PKC family, including PKC-α and PKC-δ, have been associated with CCK receptor signalling. However, their role in caerulein-induced SP/NK1R up-regulation has not been explored.
In this study, we identified that both PKC-α and PKC-δ are activated in a time-dependent manner during supramaximal caerulein exposure in murine pancreatic acinar cells. This is shown by a rapid phosphorylation at key activation sites of the molecule. The phosphorylation of PKC-α is transient, as we observed a decrease of phosphorylation around 60 min. of caerulein treatment, when compared with earlier time-points. Caerulein has previously been shown to activate PKC-δ for various cellular responses in pancreatic acinar cells. However, the activation of PKC-α is still not clear in high concentrations of CCK/caerulein used. Satoh et al. did not observe increased PKC-α translocation to the membrane after 30 min. of supramaximal CCK stimulation [23]. On the other hand, Cosen-Binker et al. found significant PKC-α activity after treating pancreatic acinar cells with CCK [24]. Previous studies have shown an excessive rise in Ca2+ levels in caerulein-induced pancreatic acinar cells [25]. Therefore, the Ca2+-dependent activation of PKC-α is very likely to occur. The different incubation times, animal strain used or detection methods might explain discrepancies between studies, and further investigation will be needed to address the issue.
In order to differentiate the role of PKC-α and PKC-δ in caerulein stimulated cells, we employed the use of isozyme specific pharmacological inhibitors. Gö6976 (IC50= 2.3 nM) is a Ca2+-dependent PKC isozyme inhibitor, and therefore it was used to block the actions of PKC-α in our study [20]. Rottlerin (IC50= 3 μM) has some selectivity towards PKC-δ and is widely used to investigate the cellular mechanisms mediated by PKC-δ[16, 18, 21]. In an attempt to avoid inhibition of Gö6976 and rottlerin, we tested the inhibitors with reference to the reported IC50 values. The efficacy and specificity of the two inhibitors was tested and confirmed in our results. Gö6976 showed some inhibition of PKC-α at a concentration of 1 nM and further inhibited PKC-α phosphorylation when the concentration was increased to 10 nM. On the other hand, rottlerin showed a complete inhibition of PKC-δ phosphorylation when a concentration of 1–10 μM was used. At these concentrations used, Gö6976 and rottlerin did not significantly block the other form of studied PKC. This could then be applied to differentiate the role of conventional PKC-α and novel PKC-δ in caerulein-induced SP/NK1R up-regulation in our model. Despite the specificity shown in PKC inhibition, rottlerin has also recently been shown to affect several PKC-independent mechanisms, mostly related to mitochondrial uncoupling pathway [26, 27]. Therefore, it is important to control the concentration of rottlerin used and caution exercised when interpreting the results.
Caerulein (10−7 M) has been shown to regulate gene transcription of PPT-A and NK1R, and also SP protein levels in mouse pancreatic acinar cells [11]. In the current study, a time response study on NK1R protein expression was done to supplement the previous data showing caerulein-induced NK1R gene expression. Western blot analysis of NK1R clearly showed a slow but steady increase of NK1R protein expression in mouse pancreatic acinar cells, and is statistically significant after 2 hrs of caerulein exposure. We then employed the use of selective PKC-α inhibitor Gö6976, and selective PKC-δ inhibitor rottlerin, to observe the role of PKCs on SP/NK1R expression. Our data indicated that pre-treatment with either Gö6976 or rottlerin significantly attenuated caerulein-induced SP up-regulation. Furthermore, real time PCR analysis revealed that mRNA expression of PPT-A was significantly reduced with Gö6976 or rottlerin pre-treatment. Studies done on NK1R expression also showed similar results that pre-treatment of pancreatic acinar cells with either Gö6976 or rottlerin significantly attenuated NK1R mRNA and protein expression. The concentration of Gö6976 and rottlerin required to reverse SP and NK1R up-regulation was also in line with the concentration required to inhibit phosphorylation of PKC-α and PKC-δ. These observations suggested that the PKCs play a role in mediating the gene expression of PPT-A and NK1R, which in turn cause an increase in protein expression of SP and NK1R in the mouse pancreatic acinar cells. Overall, our data showed that activation of both conventional PKC-α and novel PKC-δ is equally important in caerulein-induced SP/NK1R up-regulation in the mouse pancreatic acinar cells.
Evidence from several models demonstrated the relationships between PKCs and MAPKs in the expression of genes. Some suggested that the PKC family, as a whole, activate the MAPK pathway [28]; many others attempted to identify the role of individual PKC isoforms in the activation of MAPKs [29, 30]. There is also evidence that the activation of MAPKs is concurrently activated by more than one PKC isoforms. Sun et al. suggested in a macrophage cell line that SP induced ERK1/2 phosphorylation via activation of both PKC-α, PKC-δ and PKCε[10]. Therefore, there is still much to distinguish between the role of individual PKC isoforms in the activation of MAPKs. In this current study, we used the inhibitors that had been shown to inhibit SP and NK1R to investigate its effect on ERK1/2 and JNK activation. We found that pre-treatment with Gö6976 or rottlerin significantly attenuated caerulein-induced JNK and ERK1/2 phosphorylation, showing that PKC-α and PKC-δ are up-stream of the MAPKs. Our results were consistent with the findings that rottlerin treatment affected the phosphorylation of JNK and ERK1/2 in SP-treated mouse pancreatic acinar cells [18]. Gö6976 or rottlerin treatment was also found to inhibit renin receptor expression and osteoclastogenesis via the PKC-MAPK pathways [31, 32]. We also noticed that there is a consistent, but insignificant decrease of JNK phosphorylation with rottlerin pre-treatment. This could be due to baseline phosphatase activity, which dephosphorylates JNK when PKC kinase activity is inhibited by the drug.
In pancreatic acinar cells, it was suggested that stimulation of CCK receptors could modulate gene expression via activation of several transcription factors, such as NF-κB and AP-1 [33, 34]. In previous studies done on mouse pancreatic acinar cells, we have shown that caerulein-induced up-regulation of SP and NK1R expression were mediated via activation of ERK1/2 and JNK, followed by transcription factors NF-κB and AP-1 [11]. The role of NF-κB was also shown directly by using a specific NF-κB inhibitor, Bay11-7082 [11]. In this study, we found that PKC-α and PKC-δ activation is necessary for the activation of MAPKs, and therefore it is logical to speculate that PKCs are located upstream of NF-κB and AP-1. Both Gö6976 and rottlerin has been shown frequently to inhibit the activation of NF-κB and AP-1 in various models [35–38]. However, there are reports suggesting PKC-independent activation of NF-κB [39, 40]. Results from a study from Holden et al. suggested that the conflicting results observed could be due to different stimulating agents used. In that report, PKC activation was not involved in tumour necrosis factor-α induced NF-κB activation, whereas phorbol 12-myristate 13-acetate induced NF-κB activation was found to require the activation of novel PKCs in pulmonary A549 cells [41]. Our data showed that cells pre-treated with Gö6976 or rottlerin significantly inhibited caerulein stimulated increase of NF-κB and AP-1 DNA-binding activity. Due to the relatively small changes in NF-κB DNA-binding activity observed, we verified the activation of NF-κB by checking the phosphorylation of IκB, which is an important step for removing the inhibition of NF-κB translocation into the nucleus. Western blot results of IκB phosphorylation showed significant results with a greater magnitude, while maintaining a similar response pattern with NF-κB DNA-binding activity. Therefore, we suggest caerulein-induced PKC-α and PKC-δ activation is necessary for the activity of NF-κB and AP-1, which in turn required for SP and NK1R up-regulation.
In conclusion, the results in this current study further demonstrate several key elements leading to the up-regulation of SP and NK1R in the mouse pancreatic acinar cells. Caerulein activates both PKC-α and PKC-δ, which in turn mediate the expression of SP and NK1R at both genetic and protein levels. PKC-α and PKC-δ are also necessary for the activation of ERK1/2 and JNK, followed by transcriptional activity of NF-κB and AP-1. Overall, we did not find a difference in the role of conventional PKC-α and novel PKC-δ, as activation of both molecules are necessary for the cellular responses observed. These results provide a clearer picture on the mechanisms that lead to up-regulation of SP and NK1R, and thus provide the basis for a possible therapy for AP via controlling its expression levels.
Acknowledgments
This work was supported by National Medical Research Council grant R-184-000-156-213. We thank Ms. Mei-Leng Shoon (Department of Pharmacology, National University of Singapore) for technical assistance.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Grady T, Liang P, Ernst SA, et al. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology. 1997;113:1966–75. doi: 10.1016/s0016-5085(97)70017-9. [DOI] [PubMed] [Google Scholar]
- 2.Gorelick FS, Thrower E. The acinar cell and early pancreatitis responses. Clin Gastroenterol Hepatol. 2009;7:S10–4. doi: 10.1016/j.cgh.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pezzilli R. Pharmacotherapy for acute pancreatitis. Expert Opin Pharmacother. 2009;10:2999–3014. doi: 10.1517/14656560903382630. [DOI] [PubMed] [Google Scholar]
- 4.Barreto SG, Carati CJ, Schloithe AC, et al. The combination of neurokinin-1 and galanin receptor antagonists ameliorates caerulein-induced acute pancreatitis in mice. Peptides. 2010;31:315–21. doi: 10.1016/j.peptides.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 5.Dembinski A, Warzecha Z, Ceranowicz P, et al. Ghrelin attenuates the development of acute pancreatitis in rat. J Physiol Pharmacol. 2003;54:561–73. [PubMed] [Google Scholar]
- 6.Bhandari M, Thomas AC, Hussey DJ, et al. Galanin mediates the pathogenesis of cerulein-induced acute pancreatitis in the mouse. Pancreas. 2009;39:182–7. doi: 10.1097/MPA.0b013e3181bdc152. [DOI] [PubMed] [Google Scholar]
- 7.Lau HY, Wong FL, Bhatia M. A key role of neurokinin 1 receptors in acute pancreatitis and associated lung injury. Biochem Biophys Res Commun. 2005;327:509–15. doi: 10.1016/j.bbrc.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 8.Bhatia M, Saluja AK, Hofbauer B, et al. Role of substance P and the neurokinin 1 receptor in acute pancreatitis and pancreatitis-associated lung injury. Proc Natl Acad Sci USA. 1998;95:4760–5. doi: 10.1073/pnas.95.8.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramnath RD, Sun J, Bhatia M. Involvement of SRC family kinases in substance P-induced chemokine production in mouse pancreatic acinar cells and its significance in acute pancreatitis. J Pharmacol Exp Ther. 2009;329:418–28. doi: 10.1124/jpet.108.148684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun J, Ramnath RD, Tamizhselvi R, et al. Role of protein kinase C and phosphoinositide 3-kinase-Akt in substance P-induced proinflammatory pathways in mouse macrophages. FASEB J. 2009;23:997–1010. doi: 10.1096/fj.08-121756. [DOI] [PubMed] [Google Scholar]
- 11.Koh YH, Tamizhselvi R, Bhatia M. Extracellular signal-regulated kinase 1/2 and c-Jun NH2-terminal kinase, through nuclear factor-{kappa}B and activator protein-1, contribute to caerulein-induced expression of substance P and Neurokinin-1 receptors in pancreatic acinar cells. J Pharmacol Exp Ther. 2010;332:940–8. doi: 10.1124/jpet.109.160416. [DOI] [PubMed] [Google Scholar]
- 12.Tamizhselvi R, Moore PK, Bhatia M. Hydrogen sulfide acts as a mediator of inflammation in acute pancreatitis: in vitro studies using isolated mouse pancreatic acinar cells. J Cell Mol Med. 2007;11:315–26. doi: 10.1111/j.1582-4934.2007.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332:281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bastani B, Yang L, Baldassare JJ, et al. Cellular distribution of isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1995;1269:307–15. doi: 10.1016/0167-4889(95)00120-0. [DOI] [PubMed] [Google Scholar]
- 15.Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. J Biochem. 2002;132:663–8. doi: 10.1093/oxfordjournals.jbchem.a003271. [DOI] [PubMed] [Google Scholar]
- 16.Li C, Chen X, Williams JA. Regulation of CCK-induced amylase release by PKC-delta in rat pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G764–71. doi: 10.1152/ajpgi.00111.2004. [DOI] [PubMed] [Google Scholar]
- 17.Thrower EC, Wang J, Cheriyan S, et al. Protein kinase C delta-mediated processes in cholecystokinin-8-stimulated pancreatic acini. Pancreas. 2009;38:930–5. doi: 10.1097/MPA.0b013e3181b8476a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramnath RD, Sun J, Adhikari S, et al. Role of PKC-delta on substance P-induced chemokine synthesis in pancreatic acinar cells. Am J Physiol Cell Physiol. 2008;294:C683–92. doi: 10.1152/ajpcell.00360.2007. [DOI] [PubMed] [Google Scholar]
- 19.Williams JA. Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu Rev Physiol. 2001;63:77–97. doi: 10.1146/annurev.physiol.63.1.77. [DOI] [PubMed] [Google Scholar]
- 20.Martiny-Baron G, Kazanietz MG, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–7. [PubMed] [Google Scholar]
- 21.Gschwendt M, Muller HJ, Kielbassa K, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 22.Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal Biochem. 1980;102:344–52. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 23.Satoh A, Gukovskaya AS, Nieto JM, et al. PKC-delta and -epsilon regulate NF-kappaB activation induced by cholecystokinin and TNF-alpha in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G582–91. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 24.Cosen-Binker LI, Lam PP, Binker MG, et al. Alcohol/cholecystokinin-evoked pancreatic acinar basolateral exocytosis is mediated by protein kinase C alpha phosphorylation of Munc18c. J Biol Chem. 2007;282:13047–58. doi: 10.1074/jbc.M611132200. [DOI] [PubMed] [Google Scholar]
- 25.Okada N, Ohshio G, Tanaka T, et al. Intracellular Ca2+ response of pancreatic acini in cerulein-induced acute pancreatitis in rats. Hepatogastroenterology. 1998;45:840–5. [PubMed] [Google Scholar]
- 26.Tapia JA, Jensen RT, Garcia-Marin LJ. Rottlerin inhibits stimulated enzymatic secretion and several intracellular signaling transduction pathways in pancreatic acinar cells by a non-PKC-delta-dependent mechanism. Biochim Biophys Acta. 2006;1763:25–38. doi: 10.1016/j.bbamcr.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Soltoff SP. Rottlerin is a mitochondrial uncoupler that decreases cellular ATP levels and indirectly blocks protein kinase Cdelta tyrosine phosphorylation. J Biol Chem. 2001;276:37986–92. doi: 10.1074/jbc.M105073200. [DOI] [PubMed] [Google Scholar]
- 28.Lin WN, Luo SF, Lin CC, et al. Differential involvement of PKC-dependent MAPKs activation in lipopolysaccharide-induced AP-1 expression in human tracheal smooth muscle cells. Cell Signal. 2009;21:1385–95. doi: 10.1016/j.cellsig.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida K, Miki Y, Kufe D. Activation of SAPK/JNK signaling by protein kinase Cdelta in response to DNA damage. J Biol Chem. 2002;277:48372–8. doi: 10.1074/jbc.M205485200. [DOI] [PubMed] [Google Scholar]
- 30.Cheng CY, Hsieh HL, Sun CC, et al. IL-1 beta induces urokinase-plasminogen activator expression and cell migration through PKC alpha, JNK1/2, and NF-kappaB in A549 cells. J Cell Physiol. 2009;219:183–93. doi: 10.1002/jcp.21669. [DOI] [PubMed] [Google Scholar]
- 31.Huang J, Siragy HM. Regulation of (pro)renin receptor expression by glucose-induced mitogen-activated protein kinase, nuclear factor-kappaB, and activator protein-1 signaling pathways. Endocrinology. 2010;151:3317–25. doi: 10.1210/en.2009-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tiedemann K, Hussein O, Sadvakassova G, et al. Breast cancer-derived factors stimulate osteoclastogenesis through the Ca2+/protein kinase C and transforming growth factor-beta/MAPK signaling pathways. J Biol Chem. 2009;284:33662–70. doi: 10.1074/jbc.M109.010785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gukovsky I, Reyes CN, Vaquero EC, et al. Curcumin ameliorates ethanol and nonethanol experimental pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2003;284:G85–95. doi: 10.1152/ajpgi.00138.2002. [DOI] [PubMed] [Google Scholar]
- 34.Orlichenko LS, Behari J, Yeh TH, et al. Transcriptional regulation of CXC-ELR chemokines KC and MIP-2 in mouse pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2010;299:G867–76. doi: 10.1152/ajpgi.00177.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kontny E, Kurowska M, Szczepanska K, et al. Rottlerin, a PKC isozyme-selective inhibitor, affects signaling events and cytokine production in human monocytes. J Leukoc Biol. 2000;67:249–58. doi: 10.1002/jlb.67.2.249. [DOI] [PubMed] [Google Scholar]
- 36.Langlet C, Springael C, Johnson J, et al. PKC-alpha controls MYD88-dependent TLR/IL-1R signaling and cytokine production in mouse and human dendritic cells. Eur J Immunol. 2010;40:505–15. doi: 10.1002/eji.200939391. [DOI] [PubMed] [Google Scholar]
- 37.Shin Y, Yoon SH, Choe EY, et al. PMA-induced up-regulation of MMP-9 is regulated by a PKCalpha-NF-kappaB cascade in human lung epithelial cells. Exp Mol Med. 2007;39:97–105. doi: 10.1038/emm.2007.11. [DOI] [PubMed] [Google Scholar]
- 38.Bhatt KH, Pandey RK, Dahiya Y, et al. Protein kinase Cdelta and protein tyrosine kinase regulate peptidoglycan-induced nuclear factor-kappaB activation and inducible nitric oxide synthase expression in mouse peritoneal macrophages in vitro. Mol Immunol. 2010;47:861–70. doi: 10.1016/j.molimm.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 39.Shames BD, Selzman CH, Pulido EJ, et al. LPS-Induced NF-kappaB activation and TNF-alpha release in human monocytes are protein tyrosine kinase dependent and protein kinase C independent. J Surg Res. 1999;83:69–74. doi: 10.1006/jsre.1998.5564. [DOI] [PubMed] [Google Scholar]
- 40.Meichle A, Schutze S, Hensel G, et al. Protein kinase C-independent activation of nuclear factor kappa B by tumour necrosis factor. J Biol Chem. 1990;265:8339–43. [PubMed] [Google Scholar]
- 41.Holden NS, Squires PE, Kaur M, et al. Phorbol ester-stimulated NF-kappaB-dependent transcription: roles for isoforms of novel protein kinase C. Cell Signal. 2008;20:1338–48. doi: 10.1016/j.cellsig.2008.03.001. [DOI] [PubMed] [Google Scholar]