

Abstract
During testicular germ cell differentiation, the structure of nuclear chromatin dynamically changes. The following describes a method designed to preserve the three-dimensional chromatin arrangement of testicular germ cells found in mice; this method has been termed as the three-dimensional (3D) slide method. In this method, testicular tubules are directly treated with a permeabilization step that removes cytoplasmic material, followed by a fixation step that fixes nuclear materials. Tubules are then dissociated, the cell suspension is cytospun, and cells adhere to slides. This method improves sensitivity towards detection of subnuclear structures and is applicable for immunofluorescence, DNA, and RNA fluorescence in situ hybridization (FISH) and the combination of these detection methods. As an example of a possible application of the 3D slide method, a Cot-1 RNA FISH is shown to detect nascent RNAs. The 3D slide method will facilitate the detailed examination of spatial relationships between chromatin structure, DNA, and RNA during testicular germ cell differentiation.
Keywords: Basic Protocol, Issue 83, Chromatin, Germ cells, Sex chromosomes, Testis, Meiotic sex chromosome inactivation, Postmeiotic sex chromatin
Introduction
In testes, germ cells differentiate from diploid spermatogonia through meiosis into mature, haploid spermatozoa. During this process, the nuclear chromatin structure of germ cells is continuously and dynamically remodeled. Surface spreads are commonly used for the cytological examination of individual spermatogenic cells. A prevailing method of surface spreads employs hypotonic treatment by which individual spermatogenic cells are spread and flattened1. These conditions are optimal for detailed analysis of meiotic chromosomes. Chromosomal features such as synaptic status and recombination foci are easily observed using this method. However, the hypotonic treatment disrupts subnuclear chromatin architecture, thus this technique is not suitable for structural analysis of nuclei. Consequently, an improved method has been designed to preserve the three-dimensional chromatin structure of testicular germ cells. This method has been termed as the three-dimensional (3D) slide method (Figure 1). The 3D slide method has enabled the detection of nascent RNA localization in nuclei because it was initially optimized to examine gene expression and chromatin states during testicular germ cell differentiation by RNA fluorescence in situ hybridization (FISH)2,3. This 3D method is also applicable to the combination of immunofluorescence, DNA, and RNA FISH. Additionally, this method has aided in the discovery of postmeiotic sex chromatin (PMSC), a silent compartment of the sex chromosomes found in postmeiotic spermatids2.
The 3D slide method was originally optimized through the combination of two essential steps of slide preparation commonly used for nuclear staining: the fixation step designed to fix nuclear materials and the permeabilization step intended to remove cytoplasmic materials in order to improve the accessibility of staining reagents, such as antibodies and FISH probes. As previously described in another publication3, it has been determined that the permeabilization step must precede the fixation step in order to obtain optimal results with low cytoplasmic background. In this 3D slide method, the permeabilization step and subsequent fixation step are performed directly on seminiferous tubules and are followed by the mechanical dissociation of germ cells with forceps prior to cytospinning onto slides. An alternative method for RNA FISH of spermatogenic cells was developed in another laboratory4. In this method, consistent with the 3D method, the permeabilization step must precede the fixation step in order to obtain optimal results of RNA FISH.
The following protocol describes the 3D slide method and provides an example of a possible application, Cot-1 RNA FISH for the detection of nuclear nascent RNAs. Cot-1 DNA consists of repetitive elements in the genome. Here, a Cot-1 DNA probe is hybridized to an intron and UTR of the nascent transcripts, thereby detecting the transcriptionally active regions in nucleus5,6. 3D slides are versatile and can be applied to a combination of immunofluorescence, DNA and RNA FISH techniques in order to obtain detailed examination of spatial relationships between chromatin architecture.
Protocol
1. 3D Slide Preparation
- Prepare the following reagents:
- CSK buffer: 100 mM NaCl, 300 mM sucrose, 10 mM PIPES, 3 mM MgCl2. Adjust pH to 6.8 with 1 M NaOH. Store the CSK buffer at 4 °C.
- CSK buffer with 0.5% Triton X-100: Add 200 μl of Triton X-100 to 40 ml of CSK buffer. Mix the solution using magnetic stirrer until Triton X-100 is completely dissolved. Store the CSK buffer with 0.5% Triton X-100 at 4 °C.
- 4% Paraformaldehyde (PFA)–PBS, pH 7.4: Dissolve 4 g of PFA in approximately 70-80 ml of water. In order to expedite the dissolving process, add approximately 20 μl of 4 N NaOH and keep the solution in a water bath at 60 °C until the PFA has completely dissolved and the solution is transparent. This process takes approximately 1 hr. After PFA has completely dissolved, add 10 ml of 10x PBS. Adjust the final volume to 100 ml with water and cool down the solution to room temperature (25 °C). Use freshly prepared solution for each new experiment. Note: Obtain institutional permission for all animal work and adhere to relevant animal care guidelines.
Euthanize male mice. Using forceps and scissors, excise the testis from the mouse. Place the testis in PBS (or RPMI 1640). After the removal of testis, quickly carry out the following steps 1.3-1.4 in order to prevent the degradation of RNA.
Rupture the testis on a small Petri dish and remove the tunica albuginea. Unravel the seminiferous tubules in PBS on ice using forceps.
Using forceps, transfer several tubules (several pieces of tubules approximately 5-10 mm in length) into one well of a 4-well dish containing 500 ml of CSK buffer + 0.5% Triton X-100 on ice, and incubate for 6 min. Overexposure to CSK buffer may result in the disruption of cells.
Using forceps, transfer all the tubules to one well of a 4-well dish containing 4% PFA-PBS solution. This step can be preformed without a stereomicroscope, but optionally it can be used. Incubate for 10 min at room temperature. Simultaneously begin preparing 12 cytospin chambers during the incubation periods.
Using forceps, transfer all the tubules to one well of a 4-well dish containing PBS. Incubate for 5 min at room temperature.
On the backside of a glass slide, transfer all the tubules into 30 μl of PBS (the top of the slide should be avoided due to cationic charge). Using the tips of two forceps, tear the tubules to pieces. Chop the tubules in a horizontal direction by clipping tubules between tips of two forceps and pulling the forceps horizontally. Continue with this step for approximately 10-20 sec.
Mix the suspension by pipetting using a P20 pipette.
Transfer the suspension to a microcentrifuge tube, dilute with PBS to approximately 1.3 ml. Apply 100 μl of suspension to each of the 12 cytospin chambers (1.2 ml total). At this step, the concentration of cells does not need to be determined.
Cytospin the samples at 2,000 rpm for 10 min at room temperature. After cytospinning, dry slides on lab bench for a few minutes at room temperature. After slides have been allowed to dry completely for few minutes, go to the next step.
Using a Coplin jar containing PBS, wash the slides for 5 min at room temperature.
Transfer slides to a Coplin jar containing 70% ethanol and wait at least 2 min prior to initiation of RNA FISH. Slides are stable for a few weeks in 70% ethanol at 4 °C. For long-term storage, dehydrate slides using serial treatment with 80% and 100% ethanol in Coplin jars for 2 min each and air-dry completely dehydrate the slides. Store at -80 °C in a slide box.
2. Cot-1 DNA Probe Preparation by Random Priming
- Prepare the following reagents:
- Hybridization buffer: Mix the proceeding components to prepare 900 ml of hybridization buffer stock; this solution will be used to stock probes. This hybridization buffer stock allows for the addition of 10% volume of RNase inhibitor solution (Ribonucleoside Vanadyl Complex) prior to hybridization.
Component Amount (μl) Final concentration Formamide 500 50% (v/v) 50% Dextran 200 10% (w/v) 20x SSC 100 2x 1% BSA 100 0.1% - 20x SSC: Dissolve 88.2 g of sodium citrate and 175.3 g of NaCl in approximately 800 ml of water. Adjust the pH to 7.0 using a few drops of 1 M HCl. Add water to adjust the final volume to 1 L. Sterilize by autoclaving. After preparation, 20x SSC can be stored at room temperature.
- 50% (w/v) Dextran: Stir water and slowly add 50 g of dextran sulfate. Keep stirring overnight at room temperature. Add water to adjust final volume to 100 ml. Store at room temperature.
- Add the following constituents listed below to a PCR tube. Using a PCR machine, mix and incubate the solution for 5 min at 95 °C.
Component Amount per reaction (μl) Final Cot-1 DNA: 1 mg/ml 1 1 μg Random 9mer primer (from kit) 10 Water 27 Centrifuge briefly and place on ice.
- Add the following components. Mix and incubate for 30 min at 37 °C using a PCR machine. (For details of the kit used see the table of reagents and equipment).
Component Amount per reaction (μl) Final concentration Mixture prepared from step 2.2 38 5x nucleotide buffer (from kit) 9.2 Cy3-dUTP (1 mM) 0.8 16 μM Klenow (from kit) 2 Add 2 μl of stop buffer (from kit) to discontinue the reaction.
Purify the stopped reaction using microspin columns.
Add 50 μg (5 μl) of herring sperm DNA (50 fold excess of template Cot-1 DNA) to the purified fraction.
Measure the total volume using a pipette. Add 0.7x volume of 100% ethanol and 0.1x volume of 3 M sodium acetate (pH 5.2), mix well, and centrifuge for 15 min at maximum speed (17,000 x g) using a microcentrifuge at 4 °C.
Remove the liquid, and add 200 μl of 70% ethanol, mix well and centrifuge for 5 min at maximum speed at 4 °C.
Remove the excess liquid. Dry the precipitate for 10-30 min at room temperature.
Dissolve the pellet in 20 μl of hybridization buffer stock. Because the pellet is often difficult to dissolve, pipette repeatedly. As an alternate option, a vortex mixer can also be used to efficiently dissolve the pellet. Probes can be kept at -20 °C until use.
3. Cot-1 RNA FISH
- Add the components listed below to a PCR tube. Using a PCR machine, incubate for 10 min at 80 °C, followed by a preannealing step of approximately 10-30 min at 42 °C. Keep at 42 °C until hybridization. Ribonucleoside Vanadyl Complex is optional; it can be replaced with water.
Component Amount per reaction (μl) Final Cot-1 DNA probe (50 ng/μl) 2 100 ng Ribonucleoside Vanadyl Complex (200 mM) 2 20 mM Hybridization buffer added to 20 Dehydrate slides in a Coplin jar containing 80% ethanol for 2 min, followed by a Coplin jar containing 100% ethanol for 2 min. Dry slides completely on lab bench at room temperature.
Preheat a tip box chamber (filled with 2-3 cm of water) in a 42 °C incubator in advance. Place the slide on the tip box chamber. Briefly centrifuge the preannealed probes and pipette directly onto the dehydrated slides. Avoid making bubbles. Gently cover the slide with a cover slip.
Hybridize for 6 hr to overnight (14-15 hr) at 42 °C.
Prepare wash solutions in Coplin jars (two jars with both 2x SSC and 50% formamide, and two jars with 2x SSC). Preheat at 42 °C for 30 min in a water bath.
Use a razor blade to remove the cover slip from the edge of the slide. Avoid scratching samples. Wash slides in a Coplin jar containing 2x SSC and 50% formamide for 5 min at 45 °C. Repeat this step once.
Wash slides in a Coplin jar containing 2x SSC for 5 min at 45 °C. Repeat this step once.
Add 20 μl of DAPI-mounting media to the slides, and cover with a cover slip. Gently press cover glass to remove extra mounting media. Blot extra mounting media with cleaning tissue. The slides are ready for microscopy evaluation.
Representative Results
A representative result of a 3D slide is shown in Figure 2. In this experiment, Cot-1 RNA FISH was performed to detect nascent transcription together with immunostaining using anti CBX1 and γH2AX antibodies. To combine RNA FISH and immunostaining, immunostaining was performed first, followed by RNA FISH as described in3. CBX1 is a heterochromatin protein that localizes to pericentromeric heterochromatin and silent sex chromosomes in meiosis called an XY body (or sex body). γH2AX is a phosphorylated form of histone variant H2AX and localizes on the XY body. In this picture, Cot-1 signals accumulate on euchromatic regions, but are excluded from pericentromeric heterochromatin and the XY body.
During surface spread preparation, treatment with hypotonic solution swells nuclei and physically extends subnuclear structures. This prevailing method for meiotic study is best suited for capturing all meiotic chromosomes in a single z-plane1. To clarify the difference between the 3D slide method and surface spreads after hypotonic treatment, representative pictures of pachytene spermatocytes of each experiment are shown using the same magnification using an epifluorescent microscope (Figures 2A and 2B). In surface spreads treated with hypotonic solution, the three-dimensional chromatin structure is disrupted and all meiotic chromosome axes can be captured within a single z-plane. 3D slide preparation, however, preserves the intact chromatin structure.
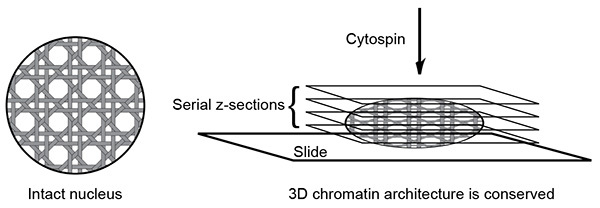 Figure 1.Schematic of 3D slide method. The 3D slide method can preserve the three-dimensional chromatin structure. Due to the three-dimensional nature of the slide, data acquisition with multiple z-section is required to cover a nucleus.
Figure 1.Schematic of 3D slide method. The 3D slide method can preserve the three-dimensional chromatin structure. Due to the three-dimensional nature of the slide, data acquisition with multiple z-section is required to cover a nucleus.
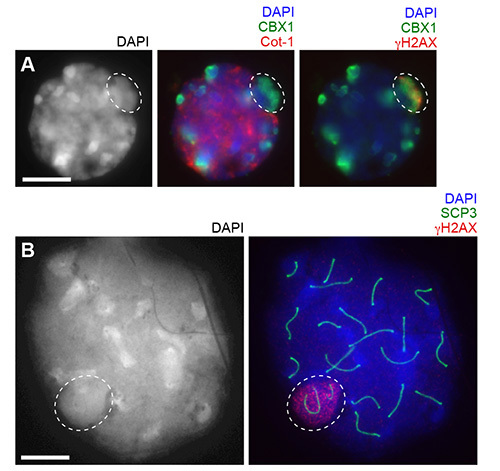 Figure 2.Comparison between the 3D slide method and surface spreads after hypotonic treatment at the same magnification. (A) Using the 3D slide, Cot-1 RNA FISH (cy3) and immunostaining with anti CBX1 (fitc) and γH2AX (cy5) antibodies are performed. A pachytene spermatocyte with a single z-section is shown. (B) Using the surface spreads, immunostaining with anti SCP3 and γH2AX antibodies are performed. A pachytene spermatocyte is shown. Circles: XY body. Bars: 10 μm.
Figure 2.Comparison between the 3D slide method and surface spreads after hypotonic treatment at the same magnification. (A) Using the 3D slide, Cot-1 RNA FISH (cy3) and immunostaining with anti CBX1 (fitc) and γH2AX (cy5) antibodies are performed. A pachytene spermatocyte with a single z-section is shown. (B) Using the surface spreads, immunostaining with anti SCP3 and γH2AX antibodies are performed. A pachytene spermatocyte is shown. Circles: XY body. Bars: 10 μm.
Discussion
In 3D slide preparation, the permeabilization step precedes the fixation step. These steps are performed directly on seminiferous tubules, thereby preserving the three-dimensional chromatin structure. An alternate option to preserving chromatin structure for RNA/DNA FISH is to perform the permeabilization step and the fixation step simultaneously. This alternate technique has been performed in marsupial germ cells and in mouse preimplantation embryos7,8. However, if the fixation step precedes the permeabilization step, it may give rise to high cytoplasmic background. Therefore, the critical determinant of slide optimization for RNA and DNA FISH is dependent upon the order of the permeabilization and the fixation steps. The duration of the permeabilization step is also an important factor for the optimization of slide preparation. Six minutes is the general condition for this method. However, it can be adjusted depending on variation of the materials used.
The preservation of the three-dimensional chromatin structure is a feature of the 3D slide that distinctly separates it from surface spread slides. This advantage differs greatly from that of chromosome spread preparation. The 3D slide method preserves three-dimensional chromatin structure, but requires data acquisition with multiple z-sections to encompass an entire nucleus. A single z-section cannot capture all signals of a nucleus. Thus, the necessity for multiple z-sections is a major limitation to this method. To capture a nucleus in a single z-section, surface spreads have an advantage. Therefore, the 3D method and the surface spread preparation have distinct advantages and mutually compensate the features of each other for the study of meiosis and spermatogenesis.
The 3D slide method can be applied to all cell types found within the testicles, including spermatogonia, round spermatids, and Sertoli cells. This method has also been applied to examine the chromatin compaction of the sex chromosomes at the onset of meiotic sex chromosome inactivation, and to examine epigenetic modifications on PMSC10. In terms of future applications, this method may be applied to other tissues or cells. If so, it is recommended that the permeabilization step precede the fixation step. Alternatively, the permeabilization step and the fixation step can be performed simultaneously.
Disclosures
The author has nothing to disclose.
Acknowledgments
I thank Jeannie T. Lee for supervision of this project when I was in the Lee laboratory and Tyler Broering for editing the manuscript. This work was supported by the Basil O’Connor Starter Scholar Award from the March of Dimes Foundation and the NIH Grant GM098605.
References
- Peters AH, Plug AW, van Vugt MJ, de Boer P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 1997;5:66–68. doi: 10.1023/a:1018445520117. [DOI] [PubMed] [Google Scholar]
- Namekawa SH, et al. Postmeiotic sex chromatin in the male germline of mice. Curr Biol. 2006;16:660–667. doi: 10.1016/j.cub.2006.01.066. [DOI] [PubMed] [Google Scholar]
- Namekawa SH, Lee JT. Detection of nascent RNA, single-copy DNA and protein localization by immunoFISH in mouse germ cells and preimplantation embryos. Nat Protoc. 2011;6:270–284. doi: 10.1038/nprot.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevaiah SK, Costa Y, Turner JM. Using RNA FISH to study gene expression during mammalian meiosis. Methods in molecular biology. 2009;558:433–444. doi: 10.1007/978-1-60761-103-5_25. [DOI] [PubMed] [Google Scholar]
- Hall LL, et al. An ectopic human XIST gene can induce chromosome inactivation in postdifferentiation human HT-1080 cells. Proc Natl Acad Sci U S A. 2002;99:8677–8682. doi: 10.1073/pnas.132468999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh KD, Lee JT. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature. 2003;426:857–862. doi: 10.1038/nature02222. [DOI] [PubMed] [Google Scholar]
- Namekawa SH, VandeBerg JL, McCarrey JR, Lee JT. Sex chromosome silencing in the marsupial male germ line. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9730–9735. doi: 10.1073/pnas.0700323104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Payer B, Huynh KD, Jaenisch R, Lee JT. Two-step imprinted X inactivation: repeat versus genic silencing in the mouse. Mol Cell Biol. 2010;30:3187–3205. doi: 10.1128/MCB.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijima Y, et al. MDC1 directs chromosome-wide silencing of the sex chromosomes in male germ cells. Genes Dev. 2011;25:959–971. doi: 10.1101/gad.2030811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin HS, et al. RNF8 regulates active epigenetic modifications and escape gene activation from inactive sex chromosomes in post-meiotic spermatids. Genes Dev. 2012;26:2737–2748. doi: 10.1101/gad.202713.112. [DOI] [PMC free article] [PubMed] [Google Scholar]