

Abstract
AMP-activated protein kinase (AMPK) is a stress-activated kinase that functions as a cellular fuel gauge and master metabolic regulator. Recent investigation has elucidated novel molecular mechanisms of AMPK regulation and important biological actions of the AMPK pathway that are highly relevant to cardiovascular disease. Activation of the intrinsic AMPK pathway plays an important role in the myocardial response to ischemia, pressure overload and heart failure. Pharmacologic activation of AMPK shows promise as a therapeutic strategy in the treatment of heart disease. The purpose of this review is to assess how recent discoveries have extended and in some cases challenged existing paradigms, providing new insights into the regulation of AMPK, its diverse biological actions and therapeutic potential in the heart.
Keywords: Protein kinases, cardiac metabolism, myocardial ischemia, cardioprotection, heart failure
AMP-activated protein kinase (AMPK) has gained attention over the last decade as a “cellular fuel gauge”1 and “super metabolic regulator”.2 The AMPK pathway orchestrates the cellular response to a variety of stresses in the heart, regulating metabolism, organelle function and cell growth. The discovery of AMPK stemmed from early biochemical studies showing that acetyl-CoA carboxylase (ACC) and HMG-CoA reductase activities were regulated by phosphorylation in the liver. ACC synthesizes malonyl-CoA and is an important regulator of fatty acid synthesis, while HMG-CoA reductase is a key step in cholesterol biosynthesis. ACC phosphorylation was found to be modulated by the concentration of adenine nucleotides.3 The discovery that the protein kinases for ACC and HMG-CoA co-purified and were regulated by AMP led to the first recognition of AMPK.4 These early discoveries set the stage for the cloning and molecular characterization of AMPK and then early pioneering studies of AMPK in the heart.5-7
Recent investigation has elucidated novel biological actions of AMPK that are highly relevant to cardiovascular disease. With the advent of new pharmacologic approaches to activate AMPK in a specific manner, the possibility of targeting this pathway for the treatment of human disease is emerging. The purpose of this review is to assess how recent investigation has extended and in some cases challenged existing paradigms, providing new insights into the regulation of AMPK, its biological actions and therapeutic potential in the heart.
Molecular structure and physiology of AMPK
The molecular structure and mechanisms regulating AMPK activation are important to understanding its physiologic function and to the development of innovative strategies to activate the kinase. AMPK is a heterotrimeric complex consisting of a catalytic alpha (α) subunit and regulatory gamma (γ) and beta (β) subunits (Figure 1). The α-subunit contains the AMPK serine-threonine kinase domain, which has a typical activating residue within the catalytic cleft (Thr172). Phosphorylation of this amino acid by upstream kinases is essential for AMPK activity and its phosphorylation status is often used as an indicator of the activation state of the kinase.8 The α-subunit also includes an auto-inhibitory domain (AID) (residues 313-335) and conformational changes induced by AMP binding to the γ-subunit, relieve auto-inhibition of the complex.9, 10 An “α hook” region (residues 360-394) interacts with the γ-subunit nucleotide binding site when AMP is bound, leading to a change in the configuration of the complex, which promotes activation of the kinase domain.11
Figure 1. Molecular structure of the AMPK complex.
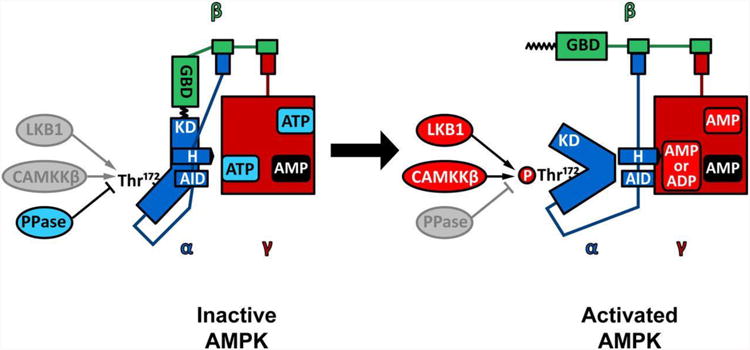
The AMPK complex is composed of a catalytic subunit (α, blue) and two regulatory subunits (β, green and γ, red). The α subunit contains a serine-threonine kinase domain (KD), which is highly activated by phosphorylation of the Thr172 residue in the catalytic cleft by upstream kinases (LKB1, CAMKKβ). AMPK is maintained in an unphosphorylated inactive state by the interaction of kinase domain with an autoinhibitory domain (AID) and with the myristoylated N-terminus of the regulatory β subunit. AMP interaction with the nucleotide binding sites in the γ-subunit induces a conformational change in the heterotrimeric complex via an α hook domain, that relieves the autoinhibition by the AID and promotes phosphorylation of the Thr172 residue. The catalytic cleft of activated AMPK is in a closed conformation, which protects phosphorylated Thr172 from being dephosphorylated by protein phosphatases (PPase).
The β-subunits were initially thought to function as a bridge between the α-catalytic and the γ- regulatory subunits. However, β-subunits also contain a functional glycogen-binding domain (GBD) and glycogen appears to regulate the activity of the kinase.12 The GBD binds best to glycogen with a single glucose α1-6 branch,13 which inhibits AMPK activation by upstream kinases.14
The γ-subunits contain four potential nucleotide-binding sites termed CBS1-4,15 due to their structural homology to binding domains in cystathionine beta-synthetase (CBS). They are also more simply referred to as sites 1-4.16 Recent crystallographic studies show that only sites 1, 3, and 4 bind nucleotides in the mammalian enzyme. Site 1 is a high affinity site, which mediates the allosteric activation of the complex and binds only AMP and ATP.17 Site 3 is a lower affinity site that binds AMP, ADP and ATP and influences the phosphorylation state of Thr172 by upstream kinases and phosphatases. Site 4 binds AMP tightly in a non-exchangeable manner.17
Molecular mechanisms of AMPK activation
The activity of AMPK is primarily determined by the cellular energy state, which is reflected in the ratio of AMP (and ADP) to ATP. Cellular stress leads to the breakdown of ATP to ADP and the subsequent production of AMP through the action of adenylate kinase (2 ADP → AMP + ATP). To a lesser extent AMP is also generated by the cleavage of pyrophosphate from ATP and via de novo purine biosynthesis. AMP content is also determined in part by its degradation by AMP-deaminase and 5′-nucleotidase and inhibition of 5′-nucleotidase is emerging as a novel strategy to activate AMPK.18
Cellular AMP is highly protein bound and the concentration of free AMP in the heart is normally in the low μM range.19 ATP is present in much higher concentrations than AMP, but the predominant form of cellular ATP, Mg++ATP, has a lower binding affinity for AMPK than AMP or ADP, enabling the latter nucleotides to competitively bind to the complex.17
According to the conventional paradigm, AMP is the primary activator of AMPK, but recent studies have challenged this concept. ADP is present in higher concentrations than AMP and under mild stress conditions appears to be a more important regulator of site 3, which induces a conformational change that modulates phosphorylation of the Thr172 site by upstream protein kinases and phosphatases.11 Under more intensive stress, AMP levels rise and allosterically activate the complex through binding to site 1. This previously under-appreciated role of ADP led to brief discussions about renaming the kinase, “ADP-activated protein kinase”, but AMP is the primary allosteric regulator of the complex and the original terminology has managed to prevail.
AMPK phosphorylation and other post-translational modifications
Phosphorylation of the Thr172 site markedly increases the activity of AMPK and is determined by the balance of action of upstream kinases and protein phosphatases. In the heart, the liver kinase B1 (LKB1) is the major upstream AMPK kinase. Each of the AMPK subunits contains two or more isoforms and phosphorylation of the complexes containing the predominant α2 isoform is entirely dependent on LKB1 during ischemia.20 In contrast, the kinase responsible for phosphorylating α1 containing AMPK complexes in the heart has yet to be identified.20
The calcium-calmodulin activated protein kinase kinase β (CaMKKβ) is a potential alternative upstream kinase that has an important role to activate AMPK in the brain and other non-cardiac cells.21 However, CaMKKβ is expressed in much lower amounts in cardiomyocytes and its role in the heart is still not well understood. Transforming growth factor-β–activated protein kinase-1 (TAK1) is known to phosphorylate SNF1, the yeast homolog of the mammalian AMPK α-subunit.22 Although TAK1 is present in the heart and is activated during ischemia, it appears to modulate LKB1 activity rather than directly phosphorylate AMPK.23
Protein phosphatases also have a critical role in regulating Thr172 phosphorylation. Both protein phosphatase 2A (PP2A) and PP2C dephosphorylate AMPK in cell free in vitro assays.24 AMP binding to the γ subunit inhibits the action of PP2C to dephosphorylate Thr172 in vitro.25 Alterations in protein phosphatase expression modulate AMPK activation in the heart, for example, increased PP2C expression decreases AMPK activity in the rodent cardiac lipotoxicity model.26 PP2A also dephosphorylates AMPK, and elevated serum fatty acids stimulate PP2A activity and decrease AMPK phosphorylation in endothelial cells.27 However, at the current time, there is limited understanding of the specific phosphatases, let alone which of their isoforms are physiologically responsible for dephosphorylating Thr172 and maintaining the low basal activity of AMPK in the normal heart.
AMPK activation is also modulated by phosphorylation at other sites. The α1 subunits are phosphorylated on Ser173, Ser485 and Ser497, while the β1 subunits are phosphorylated on Ser24.28 Phosphorylation of Ser173 by protein kinase A blunts the phosphorylation of Thr172 by upstream AMPK kinases.28 Ser485 in α1 and the corresponding Ser491 in α2 subunits are phosphorylated by both Akt and protein kinase A, inhibiting activation at the Thr172 site.29 Phosphorylation of Ser485 or Ser491 is responsible for the effect of high insulin concentrations or constitutively active Akt to blunt AMPK activation in the heart.30
In addition to phosphorylation, recent evidence indicates that AMPK undergoes post-translational acetylation of its α subunits. The acetylation state of AMPK is determined by the reciprocal actions of the acetylase p300 and the histone deacetylase 1 (HDAC1).31 Deacetylation of AMPK promotes its interaction with upstream LKB1, which phosphorylates AMPK and stimulates its activation.31 Interestingly, LKB1 itself is also regulated by acetylation, and the deacetylated form of LKB1 more readily leaves the nucleus and binds to its partner STRAD in the cytoplasm, forming the active LKB1 complex.32 These findings indicate that acetylation is a potentially important determinant of the activity of the LKB1-AMPK pathway, although there is no information on the extent to which this mechanism is operative in the heart.
AMPK subcellular localization
Subcellular localization is an important determinant of cell signaling events and targeting of protein kinases to subcellular domains provides selectivity to specific substrates. Although AMPK has traditionally been considered to be a cytosolic enzyme, evidence is emerging that it may also be targeted to the nucleus and specific membrane domains.
AMPK has well-established functions in the nucleus where it phosphorylates transcription factors, histone proteins and histone deacetylase enzymes.33 The α2, but not the α1 isoform, of the AMPK catalytic subunit has a nuclear localization signal,34 although both α isoforms contain a nuclear export sequence.35 Nuclear translocation of AMPK requires phosphorylation at the Thr172 site,34 indicating that only activated AMPK goes to the nucleus. Exercise causes AMPK activation and nuclear translocation of AMPK complexes containing α2 subunits in skeletal muscle,36 but there are no published data yet demonstrating cardiac AMPK translocation to the nucleus.
Localization of AMPK to specific membrane domains is potentially another important determinant of its action. Myristoylation of AMPK at the Gly2 site of its β-subunits serves to localize the AMPK complex to membranes.37 Interestingly, myristoylation also increases the ability of AMP to allosterically activate AMPK and to promote phosphorylation of the Thr172 site by upstream kinases,37 suggesting possible preferential activation of AMPK at membrane sites. Additional mechanisms may indirectly localize AMPK to membrane domains. For instance, AMPK binds to LKB1, which co-localizes with E-cadherin in adherens junctions in polarized epithelial cells.38 The targeting of AMPK to specific membrane domains is difficult to study in cardiomyocytes, but could be an important determinant of its biological activity to regulate ion channels, membrane associated signaling proteins, cell polarity and cell junction formation in the heart.
AMPK signaling may also be localized to specific cytosolic domains through interacting with scaffold proteins. Scaffold proteins insulate kinase pathways from surrounding signaling cascades or alternatively promote the interaction of various signaling pathways. Interestingly, AMPK interacts with the TAK1–binding protein 1 (TAB1),39 a scaffold protein better known for its role in mediating TAK-1 activation and p38 MAPK autophosphorylation. The interaction with TAB-1 appears to be functionally important in the heart, where impaired AMPK activity decreases p38 MAPK recruitment to TAB1 and blunts p38 activation during ischemia.39
AMPK expression and turnover
Although previous attention has focused primarily on the molecular mechanisms responsible for the acute activation of AMPK complexes, the expression and turnover of AMPK, upstream LKB1 and their component subunits are also regulated. In the heart, AMPK activity increases soon after birth, contributing to the metabolic switch from carbohydrate to fatty acid metabolism.40 The total expression of AMPK does not appear to change significantly at birth in the mouse,41 but there is a developmental shift in the expression pattern of specific AMPK subunit isoforms in the heart.41, 42 There are 3 genes (PRKAG1, PRKAG2 and PRKAG3) encoding the γ subunit isoforms (γ1, γ2, and γ3).43 Transcript levels of the γ1 isoform increase while γ3 content decreases during the development of the embryonic mouse heart.42 The physiologic significance of these expression patterns is uncertain and prior studies have demonstrated that AMPK activity in the adult heart is primarily determined by complexes containing the ubiquitously expressed γ1 isoform.43
The γ2 subunit expression pattern is particularly important in the heart because mutations in the human PRKAG2 gene cause a cardiomyopathy, as subsequently discussed. The PRKAG2 gene encoding the γ2 isoform has 4 alternative transcripts: γ2a (long form), γ2b (short form), γ2c, and a recently discovered γ2-3B (the product of a variant transcript that starts in exon 3B).42 Of the γ2 isoforms, γ2-3B and γ2b (short form) predominate in both human and mouse hearts and their expression increases during development.42 Interestingly, the expression of γ2-3B is relatively restricted to heart, however, it is not known whether it has distinctive physiologic functions.
AMPK expression is also altered during pressure overload and after the development of heart failure. Aortic banding leads to upregulation of α2, β2, and γ2 isoforms in the rodent heart.41, 44 In contrast, hearts explanted from patients with ischemic or non-ischemic cardiomyopathy, demonstrate increased expression of the α1, β1 and γ2c isoforms.41 These differential isoform responses are of interest, although their functional significance is not understood; whether they represent species differences or distinct responses to various types of heart failure is also unknown.
The molecular mechanisms regulating AMPK subunit expression have not been defined. However, recent studies suggest that miRNAs may play a role in regulating the upstream LKB1 complex and thus would modulate AMPK pathway activation. Specifically, miR-451 regulates the expression of the LKB1 binding partner MO25 α, a protein that is required for LKB1 activity, in glioma cells.45 Further study is needed to determine whether the extent to which the expression of AMPK or LKB1 in the heart is regulated by miRNAs, transcriptional regulators or both.
Protein degradation is also emerging as an important determinant of AMPK subunit levels. Free AMPK subunits are relatively labile when not incorporated into the heterotrimeric AMPK complex. For instance, when inactivated or “kinase-dead” α catalytic subunits are expressed in the hearts of transgenic mice, they compete with native α subunits for incorporation into the AMPK heterotrimeric complex and the remaining free α subunits are degraded.46 Recent studies show that AMPK subunits are degraded by ubiquitin-dependent proteosome activity, based on findings that cell death-inducing DFF45-like effector A (cidea) knockout mice have high levels of α, β and γ subunits in adipose tissue.47 Whether a ubiquitin-dependent proteasome mechanism of AMPK degradation is operative in the heart is not yet known.
Stimuli activating AMPK
Heart AMPK activity is increased by a wide array of stimuli, including pathologic and physiologic stress, hormones and cytokines, and drugs (Figure 2). One of the best-studied pathologic stimuli for AMPK activation in the heart is ischemia.48 AMPK is activated by both severe no flow ischemia48 and partial ischemia46 in isolated perfused rodent hearts, as well as during the regional ischemia that accompanies coronary ligation in vivo.49, 50 AMPK activation is rapid and generally sustained during ischemia.51
Figure 2. Regulators of AMPK activity.
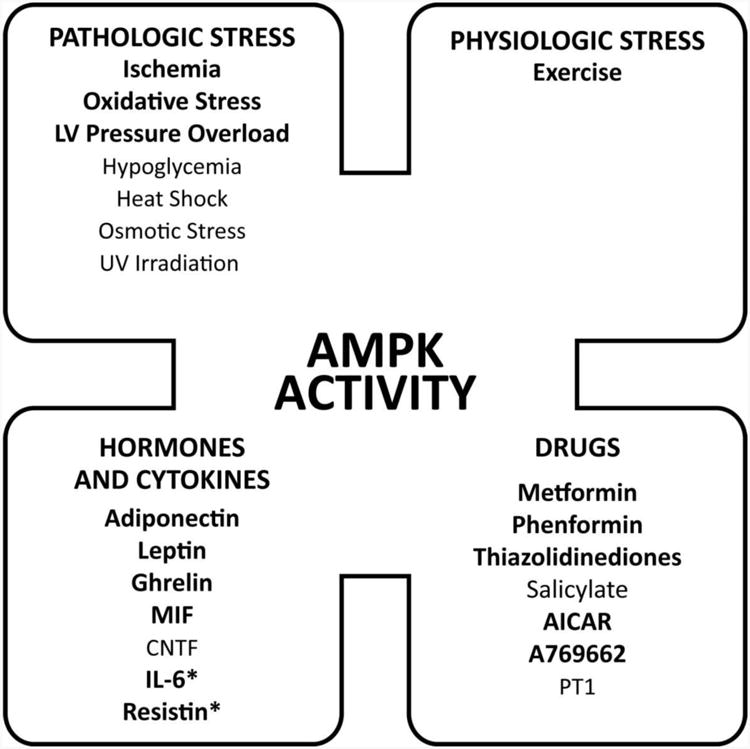
AMPK activity is modulated by several physiologic, pathologic and pharmacologic factors. Factors that are known to regulate heart AMPK are highlighted in bold, while inhibitors of heart AMPK activity are designated by asterisks and interventions that have been shown only in non-cardiac cells are in italics.
Oxidative stress also induces AMPK activation. Hydrogen peroxide activates AMPK in isolated cardiomyocytes,52 although the mechanisms involved remain uncertain. In endothelial cells, oxidative stress triggers peroxynitrite formation53, which is reported to increase AMPK activation through a PKC zeta/LKB1- dependent mechanism.54 In non-excitable cells, an additional mechanism mediating AMPK activation by oxidative stress involves the activation of calcium-release activated calcium channels with subsequent calcium-induced activation of CaMKK β and downstream AMPK.55 In contrast, there is also evidence that oxidative stress might sometimes inactivate the AMPK pathway in the intact heart. In the spontaneously hypertensive rat model, LKB1 is inactivated via formation of lipid peroxidation 4-hydroxy-2-nonenal (HNE)-conjugates, leading to decreased activation of AMPK.56 Thus, oxidative stress might have variable effects on the AMPK pathway, depending on the degree and duration of stress and the specific stimulus inducing oxidative stress.
Pressure overload stress increases AMPK subunit expression, but also the activation state of both α1 and α2 complexes in the heart.44, 57, 58 The metabolic consequences of AMPK activation include increased rates of cardiac glucose transport44 and glycolysis.59 Increased expression of several metabolic genes during pressure overload is also AMPK-dependent.60 However, as previously noted, not all forms of LV pressure overload activate the AMPK pathway in vivo56 and it is also noteworthy that agonists, which induce hypertrophy in neonatal cardiomyocytes, do not increase AMPK activity.61
The primary physiological stress that activates AMPK is exercise, in both skeletal muscle62 and the heart.63, 64 The activation of heart AMPK during exercise is associated with stimulation of downstream GLUT4 translocation and ACC phosphorylation,63 mechanisms that promote the metabolism of glucose and fatty acids, respectively. The degree of activation of α2 containing complexes appears greater than that of α1 complexes,63 consistent with the reported higher sensitivity of α2 complexes to changes in AMP concentration in vitro.65 However, the precise mechanisms responsible for AMPK activation during exercise are not well-defined.63 It is interesting that increased afterload does not appear to activate AMPK in the perfused rat heart,66 raising the possibility that additional factors other than simply cardiac work are required for AMPK activation during exercise.
Additional diverse pathologic stimuli are known to activate AMPK in non-cardiac cells, including hypoglycemia, osmotic stress, heat shock and UV irradiation (Figure 2). Whether these mechanisms are operative or important in the intact heart is less certain. Although nutrient deprivation classically activates AMPK, it is interesting that high concentrations of palmitate or oleate appear to activate AMPK in isolated perfused hearts.67 These findings do not conform with the notion of AMPK as a “fuel gauge”, and have been construed as a feed-forward mechanism to facilitate fatty acid oxidation. One alternative explanation is that high fatty acid concentrations might induce oxidative stress leading to AMPK activation in the heart.
Hormones and cytokines in the activation of AMPK
The traditional paradigm of AMPK as a energy stress-activated kinase has expanded to include what might be considered an outer shell of regulation by endocrine, paracrine and autocrine mechanisms (Figure 2). A diverse array of hormones and cytokines modulate AMPK activity in non-cardiac cells, including adiponectin,68 leptin,69 resistin,70 ghrelin,71 interleukin-672 and ciliary neurotrophic factor.73 The specific mechanisms mediating these effects on AMPK are largely unknown.
In the heart, adiponectin is perhaps the best-studied AMPK-activating hormone. AMPK contributes to adiponectin's cardioprotective effects during ischemia74 and inhibitory effects on cardiac hypertrophy after aortic banding.68 The canonical AdipoR1 and R2 receptors that typically mediate adiponectin action are expressed in the heart,75 but interesting recent findings indicate that the glycoprotein T-cadherin might be the critical receptor for adiponectin binding and AMPK activation in cardiomyocytes.76
The anti-obesity hormone leptin also modulates AMPK activity in a variety of tissues, increasing fatty acid oxidation in skeletal muscle69, while also interestingly inhibiting AMPK in the hypothalamus which decreases food intake.77 Leptin also appears to demonstrate AMPK-dependent actions in the heart. Cardiac specific leptin receptor knock-out mice have impaired activation of AMPK after myocardial infarction, which is associated with impaired glycolytic metabolism, increased apoptosis and adverse LV remodeling.78 In addition, ischemic post-conditioning and AMPK activation are impaired in hearts of leptin deficient ob/ob mice subjected to ischemia/reperfusion,79 indicating a modulating effect of leptin on AMPK activation in the heart.
Pro-inflammatory cytokines also modulate AMPK activation. In the heart, short-term IL-6 infusions appear to reduce AMPK α subunit content and activation.80 Mice fed a high fat diet have elevated plasma levels of IL-6, which may contribute to the downregulation of AMPK seen in this model.80 In contrast, interleukin-6 (IL-6) has a specific autocrine-paracrine effect in skeletal muscle to augment AMPK activation,72 while intravenous infusion of IL-6 has little effect on muscle AMPK activity.81
Macrophage migration inhibitory factor (MIF) is a master-regulator of inflammatory cytokines, and curiously is highly expressed in cardiomyocytes. Hypoxia increases cardiac MIF expression, via a hypoxia-inducible factor-1 α dependent mechanism,82 and ischemia triggers MIF secretion.83 Endogenous cardiac MIF has an important autocrine-paracrine action to modulate AMPK activation during ischemia and hypoxia in the heart.83 Extracellular MIF activates AMPK via its cell surface receptor CD74 with subsequent activation of the signal transducer CD44.83 MIF knockout mice have impaired heart AMPK activation and are more susceptible to ischemic injury.83 These observations have potential clinical relevance based on the observation that a common polymorphism in the MIF gene promoter leads to diminished MIF release and AMPK activation in response to hypoxia in human cells.83 In addition, heart MIF expression is decreased with aging and appears to be responsible for diminished AMPK activation during ischemia in old mice.84 These findings raise the possibility that older patients or those with the low expression MIF gene promoter polymorphism may be at increased risk for ischemic injury and might be more likely to benefit from therapeutics targeting AMPK activation.
Pharmacologic AMPK activators
A number of pharmacologic compounds have been identified that activate AMPK (Figure 2). The first one recognized was the nucleoside 5-aminoimidazole-4-carboxyamide-1-beta-D-ribofuranoside (AICAR), which is taken up by cells and phosphorylated to the AMP-mimetic “ZMP”. AICAR increases AMPK activity in several tissues including the heart.85 However, AICAR is also an adenosine-regulating agent that was developed as “Acadesine” to protect the heart during cardiac surgery, 86 although it is unclear whether the doses administered were adequate to activate AMPK in the human heart. Although AICAR is used extensively for experimental studies, its complex pharmacology leads to variable degrees of AMPK activation and its off target effects render it less than ideal as a drug for clinical studies.
Metformin activates AMPK in various cell types, including cardiomyocytes.87 Metformin inhibits complex 1 of the mitochondrial electron transport chain, which leads to an increase in the AMP to ATP ratio.88 Recent data suggest that metformin might have an additional effect to inhibit AMP deaminase activity,89 which would increase intracellular AMP concentrations and activate AMPK indirectly. Metformin also activates heart AMPK when administered to mice, and has a potent cardio-protective action in the post-ischemic mouse heart.90 However, metformin is now recognized to have important AMPK-independent effects, such as its well-known action to suppress hepatic glucose production in type 2 diabetes.91 A second important class of diabetes drugs, the thiazolidinediones, also activate AMPK in the heart.26 However, it is not clear that they activate AMPK in the human heart at clinically prescribed doses and caution is warranted in terms of attributing their clinical cardiovascular effects to AMPK activation.
The lack of specificity of existing drugs has spurred an interest in the development of more direct and specific AMPK activators. Initial screens yielded compounds that interact directly to activate the AMPK complex. The thienopyridone compound A769662 interacts with β1 subunits and activates AMPK through allosteric mechanisms and by increasing phosphorylation of the Thr172 site.92 In the heart, A769662 preconditions the heart through an AMPK-dependent mechanism,50 as discussed below. Recently, the compound PT1 was shown to activate AMPK through interaction with the α1 subunit at the Glu96 and Lys156 sites that are adjacent to the auto-inhibitory domain.93 PT1 promotes phosphorylation of Thr172 and activates AMPK in an AMP-independent fashion in L6 myotubes.93 Although both of these novel compounds have relatively low potency, they serve as prototypes for the development of additional agents.
Two commonly used non-diabetes drugs have also been reported to activate AMPK. Statins activate AMPK in endothelial cells and AMPK appears to mediate their effect to promote nitric oxide production and angiogenesis.94 The administration of atorvastatin to mice stimulates AMPK activation in both in the aorta and myocardium, although at doses that far exceed those prescribed clinically.94 A recent study demonstrates that salicylate also activates AMPK in non-cardiac cells, through binding to the same β subunit site that interacts with A769662.95 The salicylate concentration required to activate AMPK was in upper range of levels anticipated with high dose aspirin treatment. Although potentially relevant to high dose aspirin therapy in patients with inflammatory disease, these results have limited relevance to the traditionally low doses of aspirin prescribed to patients with cardiovascular disease.
AMPK activation by drugs, hormones and cytokines can involve subtle increases in the cellular ratio of AMP to ATP that are difficult to detect. AMP is present in low concentration and direct measures of the biologically active non-protein bound free AMP concentration are not feasible in intact cells. One innovative approach to determining whether drugs or hormones activate AMPK through alterations in the cellular AMP content, is by testing them in cell lines that express AMPK complexes containing mutant isoforms that are insensitive to AMP.96 This approach shows AMP-dependence for many compounds, but A769662 and salicylate activate AMPK in an AMP-independent fashion.96
Additional strategies are under investigation to activate AMPK by decreasing AMP degradation. Knockdown of the AMP-degrading enzyme 5′-nucleotidase increases the AMP/ATP ratio, enhancing AMPK phosphorylation and the downstream activation of ACC and glucose transport in both myotubes and mouse skeletal muscle.18 Although these results are of interest, the efficacy of inhibiting 5′-nucleotidase in the heart is not yet established and the potential sequelae of altering nucleotide metabolism warrant further consideration.
AMPK inhibitors
Pharmacologic inhibitors are useful reagents to probe the function of protein kinases in both cells and intact animals. Unfortunately, like many kinase inhibitors, the existing AMP inhibitors lack specificity. The most widely used inhibitor, compound C, inhibits AMPK in vitro with an IC50 of 0.1-0.2 μM.97 However, even at a concentration of 1 μM, which is lower than typically used for most cell based experiments, compound C inhibits multiple other protein kinases, such as extracellular signal-regulated kinase 8, Src kinase and MAP kinase-interacting serine/threonine-protein kinase 1.97 This lack of specificity makes it highly desirable to use more specific genetic approaches to inactivate the AMPK pathway rather than to rely primarily on pharmacologic results.
Metabolic pathway regulation by AMPK
AMPK activation coordinates the regulation of several steps in substrate transport and metabolism, which are critical for energy generation and conservation under times of stress (Figure 3). The metabolic actions of AMPK were instrumental in focusing scientific interest in understanding the AMPK pathway in the heart.5-7 The earliest work on AMPK focused on its role in modulating cardiac fatty acid oxidation.5, 48 AMPK phosphorylation of ACC2 inhibits its activity and the synthesis of malonyl-CoA, a potent inhibitor of carnitine palmitoyltransferase-1 (CPT-1). The decrease in malonyl-CoA levels, relieves the inhibition of CPT-1, which transports fatty acyl-CoA groups into the mitochondrial matrix and is the rate-limiting step for heart fatty acid oxidation.5
Figure 3. AMPK regulation of heart glucose and fatty acid metabolism.
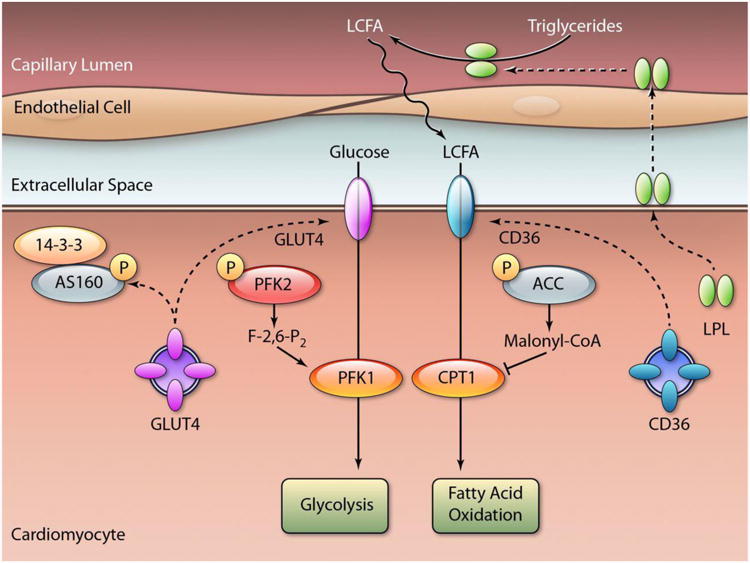
AMPK regulates substrate transporters and the concentrations of allosteric regulators of glycolysis and fatty acid oxidation. Molecules that are phosphorylated by AMPK are designated by the symbol “P”. AMPK phosphorylates the Rab GTPase AS160, which induces GLUT4 glucose transporter translocation to the sarcolemma. AMPK phosphorylates phospho-fructokinase 2 (PFK2), leading to the synthesis of fructose-2,6-bisphosphate, an allosteric activator of PFK1 and glycolysis. AMPK promotes lipoprotein lipase (LPL) translocation from the myocyte sarcolemma to the luminal surface of capillary endothelial cells, where it catalyzes the release of long-chain fatty acids (LCFA) from triglyceride-containing lipoproteins for heart substrate metabolism. Activated AMPK also stimulates CD36 translocation to the sarcolemma increasing cardiomyocyte free fatty acid uptake. AMPK also phosphorylates and inactivates acetyl-coenzyme A carboxylase (ACC) decreasing the concentration of the fatty acid oxidation inhibitor malonyl-CoA. Proteins activated by AMPK are highlighted in red and proteins inactivated by AMPK in grey. (Illustration credit: Ben Smith).
AMPK is rapidly activated by ischemia, but it has no discernible effect to increase fatty acid oxidation during ischemia because of the absence of oxygen.46 However, AMPK activation persists for variable periods of time during reperfusion and plays a role in re-activating fatty acid oxidation early in the post-ischemic heart.5, 46 AMPK also stimulates the myocardial uptake of fatty acids by increasing the translocation of the fatty acid transporter CD36 from intracellular storage membranes to the sarcolemma.98 In addition, AMPK activates lipoprotein lipase, which facilitates fatty acid uptake from triglyceride-containing lipoprotein particles in the heart.99 Thus, activated AMPK has several physiological effects that act in concert to promote fatty acid oxidation.
AMPK also stimulates glucose transport and glycolysis, which are particularly important to the metabolic adaptation of the ischemic heart. Glucose uptake is mediated by glucose transporter (GLUT) 4, and to a lesser extent by GLUT1 in the heart,100 and AMPK increases GLUT4 translocation to the sarcolemma.85 Neither GLUT4, nor GLUT4 containing vesicles, are directly phosphorylated by AMPK. Rather, AMPK phosphorylates Rab GTPase-activating proteins (GAP) that regulate Rab10, which modulates docking and fusion of GLUT4 vesicles with the plasma membrane. The Akt substrate 160 protein is a GAP expressed in heart and regulates the GTP form of Rab10.101 In addition, AMPK may also inhibit endocytosis of GLUT4 in cardiomyocytes, increasing the sarcolemma GLUT4 content and glucose transport.102
AMPK also acts downstream to glucose transport, by indirectly increasing the activity of phosphofructokinase (PFK)-1, the rate-limiting enzyme in glycolysis. Activated AMPK directly phosphorylates and stimulates PFK-2 to synthesize fructose 2,6-bisphosphate, which in turn allosterically activates PFK-1.7 These increases in myocardial glucose transport and glycolysis are important components of the metabolic response to ischemia or hypoxia.103
AMPK regulation of transcription
In addition to the well-established acute metabolic effects of AMPK, AMPK also translocates into the nucleus where it has more prolonged effects on cellular metabolism by regulating gene transcription.60 In the heart, AMPK regulates genes related to mitochondrial energy metabolism, including medium chain acyl-CoA dehydrogenase (MCAD), CPT-1, cytochrome C, and uncoupling protein (UCP)-3.60 These transcriptional effects are mediated in part by activation of the estrogen-related receptor alpha (ERRα) transcription factor.60
AMPK also appears to regulate peroxisome proliferator activated receptor gamma co-activator (PGC)-1α, a critical modulator of cardiac gene expression and mitochondrial biogenesis. AMPK activation increases the expression of PGC-1α in hypoxic cardiomyocytes.104 More detailed mechanisms linking AMPK to PGC-1α and mitochondrial biogenesis have been delineated in skeletal muscle. AMPK increases muscle NAD+ concentrations and the activity of the NAD+-dependent deacetylase sirtuin-1 (SIRT1) during fasting and exercise, and the subsequent deacetylation of PGC-1α promotes its activity.105, 106 How AMPK increases cellular NAD+ concentration is less certain, but may result in part from increased expression of the NAD+ biosynthetic enzyme nicotinamide phosphoribosyltransferase (NAMPT).107 AMPK also directly phosphorylates and activates PGC-1α at Thr177 and Ser538.108 In addition, chronic activation of AMPK also enhances the binding activity of nuclear regulatory factor-1 (NRF-1) and mitochondrial biogenesis in skeletal muscle.109 The extent to which these mechanisms function in the heart is unclear, but would be of interest to better understand mitochondrial biogenesis during growth and cardiac hypertrophy.
AMPK also modulates gene expression through novel mechanisms in non-cardiac cells. AMPK phosphorylates the histone deacetylase (HDAC)5 transcriptional repressor at both the Ser259 and Ser498 sites, which promotes GLUT4 transcription in myotubes.33 Activated AMPK acts as a direct histone kinase, phosphorylating histone 2B on Ser36 and regulating gene transcription110 AMPK also modulates the stability of mRNA transcripts by regulating the RNA-binding protein HuR egress to the cytoplasm.111 Further studies will be needed to determine whether these mechanisms are operative in cardiomyocytes.
AMPK regulation of protein synthesis
AMPK has important actions to inhibit cellular protein synthesis, in line with its role to conserve energy by inhibiting energy-consuming biosynthetic pathways (Figure 4). Under conditions of potentially lethal energy deprivation, there is little need to consume ATP to synthesize long half-life proteins. Amongst the best-characterized targets of AMPK is the eukaryotic elongation factor-2 (eEF2) kinase, which AMPK phosphorylates at Ser398, leading to the phosphorylation and inhibition of downstream eEF2.112 This action may be functionally important in the heart to conserve energy during ischemia50 and to prevent excess hypertrophy during pressure overload.58
Figure 4. Activated AMPK inhibits protein synthesis and increases authophagy.
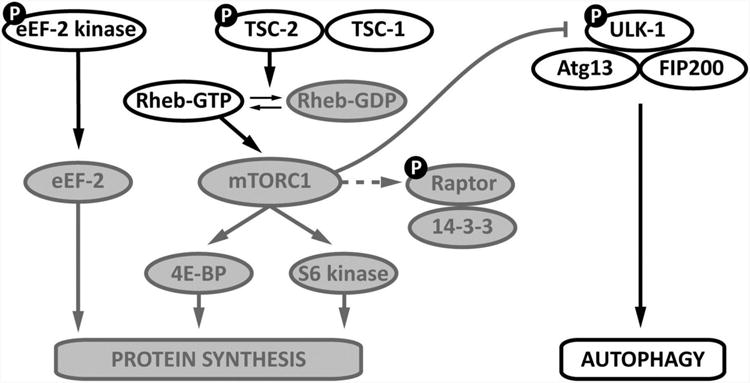
Proteins phosphorylated by AMPK and designated with the symbol “P”. AMPK phosphorylates eukaryotic elongation factor (eEF)-2 kinase, which inhibits eEF-2 activity. AMPK also phosphorylates tuberous sclerosis complex (TSC)-2, which increases the GTP-ase activity of the TSC1-TSC2 complex. AMPK also phosphorylates raptor, which removes it from the mammalian target of rapamycin complex (mTORC) 1 complex. Coordinated TSC2 and raptor phosphorylation result in inactivation of mTOR signaling, which inhibits the function of mTOR in the activation of protein synthesis and inhibition of autophagy. AMPK also phosphorylates Unc-51-like kinase 1 (Ulk1) in the ULK1-Atg13-FIP200 complex to directly promote autophagy. Direct phosphorylation of eEF2 has been shown in the heart, while the other pathways were demonstrated in non-cardiac cells.
Activated AMPK also inhibits the mammalian target of rapamycin (mTOR) pathway. First, AMPK activates the tuberous sclerosis complex (TSC): phosphorylation of TSC2 at Thr1227 and Ser1345 leads to an increase in the TSC1-TSC2 complex “Ras homologue enriched in brain” (Rheb)-GTPase activity and to the downstream inhibition of the mTOR complex 1 (mTORC1).113 In addition, AMPK has a TSC2-independent action to inhibit mTORC1 activity by phosphorylating raptor at Ser722 and Ser792, which induces 14-3-3 binding and dissociation of raptor from mTOR.114 With the caveat that these molecular mechanisms have been elucidated in non-cardiac cells, AMPK appears to be a potent inhibitor of the mTOR pathway and this action could have relevance to modulating cardiac hypertrophy in the setting of LV pressure overload and heart failure.
AMPK regulation of protein degradation and autophagy
AMPK also modulates protein degradation and the turnover of intracellular organelles. AMPK activates the ubiquitin proteasome system and protein degradation in both striated muscle cells115 and the heart.116 In the heart, AMPK increases the expression of the ubiquitin ligases atrogin-1 and the muscle RING finger protein 1 (MuRF1), the latter through activating MEF2.116 Whether MuRF1 upregulation requires PGC-1α activation and how AMPK activates MEF2 in cardiomyocytes is uncertain. It would also appear that there is a complex interplay between AMPK and MEF2, and that activated AMPK counters the effect of MEF2 to promote cardiac hypertrophy.117
In keeping with its function to provide energy during nutritional deprivation, AMPK also promotes autophagy, providing important nutrients from the breakdown of macromolecules and organelles.118 In the ischemic heart, AMPK activation induces autophagy in cardiomyocytes, contributing to their survival.119 In a diabetic mouse heart failure model, autophagy is impaired and metformin treatment enhances autophagic activity leading to the preservation of cardiac function through an AMPK-dependent mechanism.120
The molecular mechanisms responsible for AMPK's stimulation of autophagy are now starting to be unraveled in non-cardiac cells (Figure 5). AMPK inactivates mTORC1, which normally suppresses autophagy when the nutrient supply is adequate. Intriguing recent evidence also indicates that AMPK directly phosphorylates the Unc-51-like kinase (ULK)-1, the mammalian homolog of the yeast kinase autophagy-related 1 (Atg1), which has a critical role in the induction of autophagy.121, 122 AMPK is reported to phosphorylate ULK1 on various sites, including Ser467, Ser555, Thr574, Ser637, Ser317 and Ser777.121, 122 Interestingly, loss of function of either AMPK or ULK1 results in defective mitophagy.121 AMPK appears to bind in a complex that includes ULK-1 and mAtg101, and this interaction is nutritionally–dependent and is inhibited by mTOR phosphorylation of ULK-1 on Ser757.122 Thus, there is a complicated interplay between AMPK, mTORC1 and ULK-1 in regulating autophagy and the specific molecular mechanisms through which AMPK regulates autophagy during ischemia, hypertrophy and heart failure require additional investigation.
Figure 5. Mutations in the human γ2 subunit of AMPK.
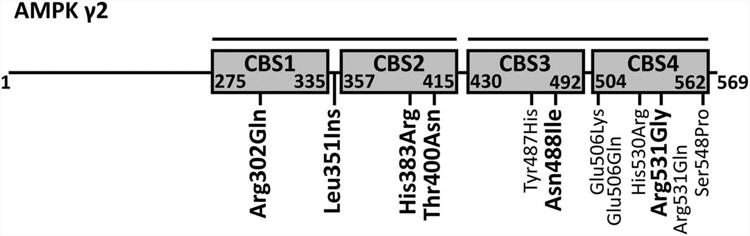
Schematic diagram of the PRKAG2 gene product γ2 isoform highlights the nucleotide binding domains (Bateman domains) and lists mutations with clinical expression curated in the OMIM database (http://www.omim.org/). All mutations cause cardiac hypertrophy and those that cause Wolff-Parkinson-White syndrome are highlighted in bold.
AMPK action during myocardial ischemia-reperfusion
AMPK is activated during ischemia5 and most evidence indicates that intrinsic AMPK activation protects the heart against injury during ischemia-reperfusion.46, 90, 123, 124 Genetic inactivation of heart AMPK by the transgenic expression of a K45R α2 subunit impairs ischemic glucose uptake and ATP homeostasis, and results in poor recovery of LV contractile function, increased cardiac necrosis and greater apoptosis after ischemia-reperfusion.46 Similarly, hearts from mice expressing the KD D157A α2 subunit have reduced glucose uptake and greater ATP depletion during ischemia123 and increased necrosis after coronary occlusion-reperfusion in vivo.90 Hearts from global AMPK α2 knockout mice have reduced glycolysis, greater ATP depletion, more rapid development of contracture during ischemia, as well as impaired metabolism and contractile function after ischemia.125, 126 However, not all studies have documented a protective function of the AMPK pathway during ischemia.127
AMPK inactivation models provide insight into the intrinsic role of the AMPK pathway in the response to ischemia. However, from the clinical perspective, the more important issue is whether pharmacologic AMPK activation represents a potential therapeutic strategy to reduce ischemic injury and evidence to date supports this possibility. Metformin activates AMPK and protects against regional ischemia when administered in vivo in both diabetic and normal mice, before or after ischemia.90 The compound A769662, which directly activates AMPK, protects against ischemic injury during global ischemia in vitro and during regional ischemia in vivo.50 A769662 treatment also enhances ischemic AMPK activation and reduces infarct size in diabetic rat hearts.128 Although pharmacologic agents often have off-target effects, the benefit of metformin and A769662 appears to be specifically mediated by AMPK activation, based on the observation that neither drug has a cardio-protective effect in AMPK-inactivated mouse models.50, 90
AMPK activation has pleiotropic effects in the heart and which mechanisms are essential to the cardio-protective actions of AMPK are still poorly understood. AMPK activators both activate AMPK prior to ischemia and enhance the degree of ischemic AMPK activation,50 and it is difficult to tease apart these actions experimentally. In addition to preserving energy stores,50 modulating the induction of autophagy and other molecular mechanisms might be important. A well-known action of AMPK is to phosphorylate endothelial nitric oxide synthase on Ser1177, which appears to be critical for the cardio-protective effect of metformin,90 but is less important during A769662 treatment.50 Recent evidence suggests that the cardio-protective effect of A769662 might also be mediated in part through inhibition of mitochondrial permeability transition pore opening.128
AMPK is activated by additional cardio-protective interventions, providing indirect support for the hypothesis that it defends against ischemic injury. For example, adiponectin activates AMPK and prevents ischemia-reperfusion injury and apoptosis in mice,74 although adiponectin has additional mechanisms of action, including stimulation of cyclooxygenase-274 and ceramidase. 129 The latter enzyme generates the anti-apoptotic sphingolipid sphingosine-1-phosphate and its activity appears to be independent of the LKB1-AMPK pathway in cardiomyocytes.129 Interestingly, short-term caloric restriction 124 and erythropoietin 130 both increase AMPK activation and have cardio-protective effects that are inhibited by the AMPK inhibitor compound C, although caution is warranted because of the known non-specific effects of this pharmacologic inhibitor97.
There is some concern that persistent AMPK activation during early reperfusion promotes fatty acid oxidation, which can be detrimental in the post-ischemic heart.131 Excessive fatty acid oxidation impairs glucose oxidation via a Randle cycle mechanism, leading to persistent lactate production and intracellular acidosis that triggers sodium-proton exchange and calcium overload via sodium-calcium exchange.131 High plasma fatty acid concentrations occur during the stress of myocardial ischemia and potentially might augment this phenomenon.131 However, in vivo experiments in the AMPK-inactivated mouse models suggest that any potential adverse effects are counter-balanced by protective effects of AMPK activation to reduce myocardial necrosis during ischemia-reperfusion.50, 90
AMPK action in cardiac hypertrophy
Cardiac hypertrophy has an adaptive function to reduce wall tension in the setting of the LV pressure overload that accompanies hypertension or obstructive valvular heart disease. However, pressure overload can lead to pathologic LV remodeling, characterized by hypertrophy exceeding vascular perfusion capacity, interstitial fibrosis, diastolic dysfunction or overt systolic contractile failure. Thus, strategies that limit LV remodeling are potentially important and AMPK activation appears to modulate the myocardial response to pressure overload.
The effects of pro-hypertrophic stimuli are blunted by treatment with pharmacologic agents that activate AMPK. For instance, metformin, AICAR and resveratrol each activate AMPK and diminish hypertrophy induced by either phenylephrine or constitutively-active Akt in neonatal rat cardiomyocytes.61 Adiponectin also decreases agonist-induced cardiomyocyte hypertrophy in vitro in an AMPK-dependent fashion, as does constitutively active AMPK.68 In contrast, the diabetic heart has increased expression of the hormone resistin, which inhibits AMPK activation, worsening hypertrophy in neonatal rat cardiomyocytes.70 Taken together with the known effects of activated AMPK to inhibit protein synthesis, these data support the paradigm of AMPK functioning as an anti-hypertrophic pathway in the cardiomyocyte.
In the intact heart, intrinsic activation of AMPK appears to have an important role in the functional response to pressure overload. Mice with global α2 AMPK deletion demonstrate greater hypertrophy, contractile dysfunction, fibrosis, heart failure and mortality after aortic banding.58 Greater activation of the mTOR/S6-kinase signaling pathway, which is normally inhibited by AMPK, likely contributes to the excess hypertrophy in this model58. Exaggerated cardiac hypertrophy and contractile failure also occur in adiponectin knockout mice, which have reduced AMPK phosphorylation after aortic banding.68 Recent data indicate that AMPK deficiency also exacerbates obesity-induced cardiac hypertrophy and contractile dysfunction.132
Although AMPK restrains the development of pathologic hypertrophy during aortic banding, intrinsic AMPK activation does not appear to regulate normal heart growth. It is striking that none of the mouse models with impaired heart AMPK activation develop cardiac hypertrophy.46, 123 The greater influence of AMPK activation during pressure overload hypertrophy might reflect the fact that AMPK activation is more sustained after banding, as compared to the intermittent AMPK activation associated with exercise during normal growth. AMPK also blocks the hypertrophic response to aortic banding, by blunting the increase in cardiac angiotensin II and endothelin concentrations and inhibiting the nuclear factor-κB and the calcineurin/nuclear factor of activated T cells pathways.57, 133
From a translational perspective, there appears to be an opportunity for therapeutic intervention, since AMPK activity is not maximally increased during pressure overload and can be further augmented by pharmacologic means. Treatment with the AMPK activator AICAR blunts LV hypertrophy induced by aortic banding in rats, although AICAR also decreased blood pressure confounding interpretation of these results.57 Resveratrol has similar beneficial effects on AMPK and hypertrophy in this model, but has pleiotropic effects that might contribute to its action.134 Adiponectin over-expression also activates AMPK in the heart and diminishes hypertrophy after banding in mice.68 Thus, these initial observations support the possibility that AMPK activators might have a role in modulating the hypertrophic and functional consequences of pressure overload in the heart and warrant further study.
AMPK in heart failure
Heart failure is associated with numerous alterations in cardiac substrate and energy metabolism, leading to an interest in understanding the role of AMPK in mediating these changes and the potential benefit of pharmacologically activating AMPK in failing hearts. In the rapid pacing-induced canine heart failure model, both metformin and AICAR treatment significantly diminish contractile dysfunction, apoptosis and fibrosis.135 Both treatments augment AMPK activation in the paced hearts, suggesting that heart failure does not maximally activate the AMPK pathway and that further pharmacologic activation might be beneficial.135 These treatments also improve systemic insulin resistance and increase plasma nitric oxide levels and it is possible that these non-cardiac effects might contribute to their beneficial effects.135 Metformin and AICAR also have additional effects on the heart, the former as a mitochondrial complex 1 inhibitor and the latter as an adenosine regulator. Some caution is warranted because the dose of metformin in experimental studies exceeds the doses used clinically.135 However, recent observational studies suggest that metformin improves the survival of patients with heart failure and type 2 diabetes,136 supporting the possibility that pharmacologic activation of AMPK might be beneficial in heart failure.
AMPK also appears to play a role in preventing heart failure in the mouse coronary occlusion model. Metformin administered at the time of reperfusion augmented AMPK activation, eNOS phosphorylation and PGC-1α expression and reduced LV remodeling 4 weeks after myocardial infarction. 137 AMPK activation had a critical role in mediating metformin's action, based on the observation that metformin had no effect in mice with genetically inactivated AMPK in the heart.137
As evidence accumulates that AMPK activation might be beneficial in preventing heart failure, a number of issues require careful consideration. First, there needs to be a better understanding of whether AMPK activation has benefit in the treatment of established LV dysfunction and heart failure, recognizing that these effects might vary depending on the model. Second, the specific role of AMPK activation needs to be confirmed in order to be certain that observed effects are indeed attributable to AMPK activation; more specific AMPK activators and the use of AMPK-inactivated genetic models will be important in this regard. Finally, the long term therapeutic efficacy and safety of AMPK activators will need to be assessed.
AMPK γ subunit mutations
Mutations in the gene encoding the γ2 subunit (PRKAG2) on chromosome 7q3 are linked to familial hypertrophic cardiomyopathy associated with the Wolf-Parkinson-White (WPW) syndrome.138-140 Unlike the more common forms of hypertrophic cardiomyopathy that are associated with mutations in sarcomeric proteins, the hallmark of the PRKAG2 syndrome is the accumulation of glycogen in cardiomyocytes without myofibrillar disarray.140 PRKAG2 mutations cause cardiac glycogen overload similar to Pompe's or Danon's diseases, but do not cause impaired glycogenolysis.141
PRKAG2 mutations fall within or adjacent to the nucleotide binding domains (Figure 5).138-140 The degree of hypertrophy and associated conduction system disease vary depending on the mutation and the individual. The syndrome of hypertrophic cardiomyopathy and WPW has been linked to mutations at H383R in CBS2,138 T400N in CBS2,140 N488I in CBS3,140 R302Q in CBS1139, 140 and a leucine insertion between CBS1 and CBS2.138 In addition, the R531G mutation in CBS4 has been observed in children with WPW and conduction system disease without cardiac hypertrophy.142
The molecular mechanisms responsible for the cardiac phenotype are still under investigation. Complexes containing γ2-mutations generally have diminished activation by AMP in cell free systems, with reduced AMP dependence (R302Q, H338R) or sensitivity (N488I, R531Q, R531G) in vitro.143, 144 They also appear to have reduced ATP binding which diminishes the inhibitory effects of ATP on AMPK activation.143, 144 Although mouse hearts expressing the N488I143, R531G145 and R302Q146 mutations all demonstrate cardiac glycogen overload, LV hypertrophy and WPW, they are reported to have variable AMPK activity. The N488I hearts have constitutively high AMPK activity,143 while AMPK activity in R531G hearts is normal at 1 week and then decreases following the glycogen accumulation,145 and surprisingly R302Q hearts have low AMPK activity.146 The reason for these disparities in AMPK activity remains uncertain.
The N488I mouse model is the best studied to date and has constitutively high AMPK activity, which appears to be responsible for its phenotype.147 The N488I phenotype is largely reversed when crossed with mice expressing inactivated AMPK in the heart.147 The increase in activity of the N488I complex is due to a high level of Thr172 phosphorylation, but what leads to this phosphorylation is not fully understood. Nonetheless, the high AMPK activity promotes glycogen synthesis, leading to cardiac glycogen overload that is the hallmark of the PRKAG2 syndrome.148
The finding that glycogen synthesis is increased in the PRKAG2 model appears paradoxical from the perspective that activated AMPK phosphorylates and inhibits glycogen synthase in non-cardiac cells.149 However, AMPK stimulation of heart glucose uptake leads to an increase in glucose-6-phosphate concentrations, which both increase substrate availability and allosterically activate glycogen synthase.148 Because AMPK also increases fatty acid oxidation, which inhibits glucose oxidation, glucose-6-phosphate is preferentially shunted into glycogen synthesis in the N488I hearts. These hearts also have increased expression of UDP-glucose pyrophosphorylase, which synthesizes UDP-glucose, the direct substrate for glycogen synthesis,148 although this enzyme is not rate limiting for glycogen synthesis. Thus, increased AMPK activity and glucose uptake in the absence of increased energy demand promotes glycogen accumulation in the N488I model.
The development of the WPW syndrome in patients with PRKAG2 mutations is an example of how altered metabolism affects cardiac electrophysiological development. WPW is characterized by ventricular pre-excitation, via one or more muscle bridges traversing the annulus fibrosis, which “bypass” the AV node and rapidly depolarize the ventricles. These bridges normally undergo apoptosis prior to birth, but in the N488I model, glycogen-filled myocytes are presumably resistant to apoptosis and persist as physiologic bypass tracks that disrupt the annulus fibrosis.143 Consistent with the developmental basis of WPW, post-natal induction of the PRKAG2 mutation in mice fails to induce ventricular pre-excitation, although it does cause LV cardiac glycogen overload.150
Because the PRKAG2 mutations appear to be activating in some cases, this research has raised concerns that AMPK activators might cause cardiac glycogen overload with prolonged use in the clinical setting. However, there are potential biological differences between the cardiac effects of AMPK activating therapy and chronically activating PRKAG2 mutations. The degree of AMPK activation might differ, and the γ mutations might also alter the subcellular distribution of AMPK or its interaction with specific substrates. It is also noteworthy that existing diabetes drugs that activate AMPK do not cause cardiac glycogen overload, although the actual degree to which they activate AMPK in the human heart is not known. Nonetheless, if excess cardiac glycogen accumulation emerges as a clinical problem, then AMPK activators would have to be restricted to use on a short-term basis, or alternative pharmacologic strategies that activate AMPK outside of the heart would have to be considered.
Conclusions
In the last decade, there has been substantial progress in understanding the biological actions of AMPK and a growing appreciation of its importance in the cardiovascular system, but substantial gaps in knowledge remain to be addressed. In discussing many of the recent novel molecular mechanisms responsible for AMPK activation and action, it is readily apparent that most have been discovered in non-cardiac cells. Some of these mechanisms might be ubiquitous, while others might not prove to be operative in the heart.
Activation of AMPK is a highly regulated process that goes beyond changes in AMP and ATP, and role of local autocrine-paracrine factors, upstream kinases including CaMKKβ, and specific protein phosphatases in the heart are all areas that require further investigation. There is also a fundamental lack of understanding of the cell biology of AMPK complexes in the heart with respect to their subcellular localization and action. Although the complex membrane structure and myofibrillar proteins pose technical challenges to studying compartmentalized signaling mechanisms, this is a key area that warrants innovative approaches to unravel. The heterogeneity of cell types in the heart presents an additional challenge to understanding the function of the AMPK pathway in the intact heart, since AMPK might be differentially regulated in endothelial cells, fibroblasts and smooth muscle cells.
Although the acute metabolic actions of AMPK in the heart are fairly well characterized, the regulation of gene expression in the heart by AMPK is in its infancy and further research in this area is needed. Similarly, the degree to which AMPK regulates mitochondrial function, autophagy/mitophagy and electrophysiological properties of the heart, requires additional investigation, particularly in relevant models of myocardial ischemia, pressure overload and heart failure.
The function of specific AMPK subunit isoforms has not yet been delineated in the heart. In particular, whether the individual γ2 variants confer distinct physiologic actions to AMPK complexes is an open issue. Defining the regulation and action of the γ2 variants might provide additional insight into the pathogenesis of the human PRKAG2 syndrome.
The potential translation of basic research on AMPK to the clinical arena is a goal of many ongoing research programs. Since the first discoveries that activated AMPK stimulates glucose transport in skeletal and heart muscles,6, 151, 152 there has been a great deal of interest in the development of highly potent and specific AMPK activators for the treatment of type 2 diabetes. Since diabetic patients often have concomitant cardiovascular disease, understanding the potential cardiovascular benefits and any safety issues associated with AMPK activators would be important.
With regards to the potential clinical applications of AMPK activators in cardiovascular disease, there is still much work that needs to be done. For application to ischemia reperfusion, additional research is clearly needed to better assess the efficacy of AMPK activators administered at the time of reperfusion. Protective effects at the time of reperfusion would enhance the use of AMPK activators as potential adjunct therapy to coronary revascularization in patients with acute myocardial infarction. If long-term therapy with AMPK activators proves feasible, then it would be appropriate to consider their application to the treatment of heart failure. Additional studies would be needed to determine whether AMPK activators prevent the transition to heart failure during LV pressure overload and most importantly whether they might reverse LV dysfunction and adverse remodeling in the setting of established heart failure. Establishing efficacy and safety in large animal models would be essential before AMPK activators would be appropriate for introduction into clinical studies. Nonetheless, existing results to date offer encouragement to further pursue research on AMPK as a novel target for the treatment of cardiovascular disease.
Acknowledgments
Sources of funding: This work was supported in part by grants from the NIH: R01 HL63811 and T32 HL007950.
Non-standard abbreviations
- ACC
acetyl-CoA carboxylase
- AICAR
5-aminoimidazole-4-carboxyamide-1-beta-D-ribofuranoside
- AMPK
AMP-activated protein kinase
- AID
auto-inhibitory domain
- Atg1
autophagy-related 1
- CPT-1
carnitine palmitoyltransferase-1
- CaMKKβ
calcium-calmodulin activated protein kinase kinase β
- Cidea
cell death-inducing DFF45-like effector A
- CBS
cystathionine beta-synthetase
- ERRα
estrogen-related receptor alpha
- eEF2
eukaryotic elongation factor-2
- FOX
forkhead box
- GLUT
glucose transporter
- GBD
glycogen-binding domain
- GAP
GTPase-activating proteins
- HDAC
histone deacetylase
- HNE
4-hydroxy-2-nonenal
- IL-6
interleukin-6
- LKB1
liver kinase B1
- MIF
macrophage migration inhibitory factor
- MCAD
medium chain acyl-CoA dehydrogenase
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- MuRF1
muscle RING finger protein 1
- MEF
myocyte enhancer factor
- NAMPT
nicotinamide phosphoribosyltransferase
- NRF-1
nuclear regulatory factor-1
- PGC
peroxisome proliferator activated receptor gamma co-activator
- PFK
phosphofructokinase
- PP2A
protein phosphatase 2A
- SIRT1
sirtuin-1
- TAB1
TAK1–binding protein 1
- TAK1
transforming growth factor-β–activated protein kinase-1
- TSC
tuberous sclerosis complex
- ULK
Unc-51-like kinase
- UCP
uncoupling protein
- WPW
Wolf-Parkinson-White
Footnotes
Disclosures: LHY served as an unpaid consultant to Merck Pharmaceuticals.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 2.Kemp BE, Stapleton D, Campbell DJ, Chen ZP, Murthy S, Walter M, Gupta A, Adams JJ, Katsis F, Van Denderen B, Jennings IG, Iseli T, Michell BJ, Witters LA. AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans. 2003;31:162–168. doi: 10.1042/bst0310162. [DOI] [PubMed] [Google Scholar]
- 3.Yeh LA, Lee KH, Kim KH. Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. J Biol Chem. 1980;255:2308–2314. [PubMed] [Google Scholar]
- 4.Carling D, Clarke P, Zammit V, Hardie D. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem. 1989;186:129–136. doi: 10.1111/j.1432-1033.1989.tb15186.x. [DOI] [PubMed] [Google Scholar]
- 5.Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. 1995;270:17513–17520. doi: 10.1074/jbc.270.29.17513. [DOI] [PubMed] [Google Scholar]
- 6.Russell RR, 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol Heart Circ Physiol. 1999;277:H643–649. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- 7.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 8.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 9.Pang T, Xiong B, Li JY, Qiu BY, Jin GZ, Shen JK, Li J. Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits. J Biol Chem. 2007;282:495–506. doi: 10.1074/jbc.M605790200. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Jiao ZH, Zheng LS, Zhang YY, Xie ST, Wang ZX, Wu JW. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature. 2009;459:1146–1149. doi: 10.1038/nature08075. [DOI] [PubMed] [Google Scholar]
- 11.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC, Jennings IG, Campbell DJ, Witters LA, Parker MW, Kemp BE, Stapleton D. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol. 2003;13:867–871. doi: 10.1016/s0960-9822(03)00292-6. [DOI] [PubMed] [Google Scholar]
- 13.Koay A, Rimmer KA, Mertens HD, Gooley PR, Stapleton D. Oligosaccharide recognition and binding to the carbohydrate binding module of AMP-activated protein kinase. FEBS Lett. 2007;581:5055–5059. doi: 10.1016/j.febslet.2007.09.044. [DOI] [PubMed] [Google Scholar]
- 14.McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009;9:23–34. doi: 10.1016/j.cmet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kemp BE, Oakhill JS, Scott JW. AMPK structure and regulation from three angles. Structure. 2007;15:1161–1163. doi: 10.1016/j.str.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36:470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 18.Kulkarni SS, Karlsson HK, Szekeres F, Chibalin AV, Krook A, Zierath JR. Suppression of 5′-nucleotidase enzymes promotes AMP-activated protein kinase (AMPK) phosphorylation and metabolism in human and mouse skeletal muscle. J Biol Chem. 2011;286:34567–34574. doi: 10.1074/jbc.M111.268292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frederich M, Balschi JA. The relationship between AMP-activated protein kinase activity and AMP concentration in the isolated perfused rat heart. J Biol Chem. 2002;277:1928–1932. doi: 10.1074/jbc.M107128200. [DOI] [PubMed] [Google Scholar]
- 20.Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur AC, Dutta A, Prescott AR, Vanoverschelde JL, Ashworth A, Jovanovic A, Alessi DR, Bertrand L. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPK{alpha}2 but not AMPK{alpha}1. Am J Physiol Endocrinol Metab. 2006;290:E780–788. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong SP, Momcilovic M, Carlson M. Function of Mammalian LKB1 and Ca2+/Calmodulin-dependent Protein Kinase Kinase {alpha} as Snf1-activating Kinases in Yeast. J Biol Chem. 2005;280:21804–21809. doi: 10.1074/jbc.M501887200. [DOI] [PubMed] [Google Scholar]
- 22.Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 23.Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, Mann DL, Taffet GE, Baldini A, Khoury DS, Schneider MD. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A. 2006;103:17378–17383. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies S, Helps N, Cohen P, Hardie D. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 25.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang MY, Unger RH. Role of PP2C in cardiac lipid accumulation in obese rodents and its prevention by troglitazone. Am J Physiol Endocrinol Metab. 2005;288:E216–221. doi: 10.1152/ajpendo.00004.2004. [DOI] [PubMed] [Google Scholar]
- 27.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–9788. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 28.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. The EMBO journal. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pulinilkunnil T, He H, Kong D, Asakura K, Peroni OD, Lee A, Kahn BB. Adrenergic regulation of AMP-activated protein kinase in brown adipose tissue in vivo. J Biol Chem. 2011;286:8798–8809. doi: 10.1074/jbc.M111.218719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 31.Lin YY, Kiihl S, Suhail Y, Liu SY, Chou YH, Kuang Z, Lu JY, Khor CN, Lin CL, Bader JS, Irizarry R, Boeke JD. Functional dissection of lysine deacetylases reveals that HDAC1 and p300 regulate AMPK. Nature. 2012;482:251–255. doi: 10.1038/nature10804. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–4327. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kazgan N, Williams T, Forsberg LJ, Brenman JE. Identification of a nuclear export signal in the catalytic subunit of AMP-activated protein kinase. Mol Biol Cell. 2010;21:3433–3442. doi: 10.1091/mbc.E10-04-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGee SL, Howlett KF, Starkie RL, Cameron-Smith D, Kemp BE, Hargreaves M. Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes. 2003;52:926–928. doi: 10.2337/diabetes.52.4.926. [DOI] [PubMed] [Google Scholar]
- 37.Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci U S A. 2010;107:19237–19241. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sebbagh M, Santoni MJ, Hall B, Borg JP, Schwartz MA. Regulation of LKB1/STRAD localization and function by E-cadherin. Curr Biol. 2009;19:37–42. doi: 10.1016/j.cub.2008.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, 3rd, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97:872–879. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 40.Makinde AO, Gamble J, Lopaschuk GD. Upregulation of 5′-AMP-activated protein kinase is responsible for the increase in myocardial fatty acid oxidation rates following birth in the newborn rabbit. Circ Res. 1997;80:482–489. doi: 10.1161/01.res.80.4.482. [DOI] [PubMed] [Google Scholar]
- 41.Kim M, Shen M, Ngoy S, Karamanlidis G, Liao R, Tian R. AMPK isoform expression in the normal and failing hearts. J Mol Cell Cardiol. 2012;52:1066–1073. doi: 10.1016/j.yjmcc.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinter K, Grignani RT, Czibik G, Farza H, Watkins H, Redwood C. Embryonic expression of AMPK gamma subunits and the identification of a novel gamma2 transcript variant in adult heart. J Mol Cell Cardiol. 2012 doi: 10.1016/j.yjmcc.2012.05.017. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Coven DL, Miller EJ, Hu X, Young ME, Carling D, Sinusas AJ, Young LH. Activation of AMPK alpha- and gamma-isoform complexes in the intact ischemic rat heart. Am J Physiol Heart Circ Physiol. 2006;291:H1927–1934. doi: 10.1152/ajpheart.00251.2006. [DOI] [PubMed] [Google Scholar]
- 44.Tian R, Musi N, D'Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–1669. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- 45.Godlewski J, Nowicki MO, Bronisz A, Nuovo G, Palatini J, De Lay M, Van Brocklyn J, Ostrowski MC, Chiocca EA, Lawler SE. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell. 2011;37:620–632. doi: 10.1016/j.molcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. The EMBO journal. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kudo N, Gillespie JG, Kung L, Witters LA, Schulz R, Clanachan AS, Lopaschuk GD. Characterization of 5′AMP-activated protein kinase activity in the heart and its role in inhibiting acetyl-CoA carboxylase during reperfusion following ischemia. Biochim Biophys Acta. 1996;1301:67–75. doi: 10.1016/0005-2760(96)00013-6. [DOI] [PubMed] [Google Scholar]
- 49.Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, Young LH. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009;119:3807–3816. doi: 10.1172/JCI39738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, Zaha V, Sakamoto K, Young LH. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:24–32. doi: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baron S, Li J, Russell RR, 3rd, Neumann D, Miller EJ, Tuerk R, Walliman T, Hurley R, Witters LA, Young LH. Dual mechanisms regulating AMP-Activated protein kinase action in the ischemic heart. Circ Res. 2005;96:337–345. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 52.Sartoretto JL, Kalwa H, Pluth MD, Lippard SJ, Michel T. Hydrogen peroxide differentially modulates cardiac myocyte nitric oxide synthesis. Proc Natl Acad Sci U S A. 2011;108:15792–15797. doi: 10.1073/pnas.1111331108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou MH, Hou XY, Shi CM, Nagata D, Walsh K, Cohen RA. Modulation by peroxynitrite of Akt- and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J Biol Chem. 2002;277:32552–32557. doi: 10.1074/jbc.M204512200. [DOI] [PubMed] [Google Scholar]
- 54.Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, Neumann D, Schlattner U, Zou MH. Activation of protein kinase Czeta by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006;281:6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 55.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dolinsky VW, Chan AY, Robillard Frayne I, Light PE, Des Rosiers C, Dyck JR. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation. 2009;119:1643–1652. doi: 10.1161/CIRCULATIONAHA.108.787440. [DOI] [PubMed] [Google Scholar]
- 57.Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, Dong YG. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem. 2007;100:1086–1099. doi: 10.1002/jcb.21197. [DOI] [PubMed] [Google Scholar]
- 58.Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, Xu W, Wiczer B, Bernlohr DA, Bache RJ, Chen Y. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Allard MF, Parsons HL, Saeedi R, Wambolt RB, Brownsey R. AMPK and metabolic adaptation by the heart to pressure overload. Am J Physiol Heart Circ Physiol. 2007;292:H140–148. doi: 10.1152/ajpheart.00424.2006. [DOI] [PubMed] [Google Scholar]
- 60.Hu X, Xu X, Lu Z, Zhang P, Fassett J, Zhang Y, Xin Y, Hall JL, Viollet B, Bache RJ, Huang Y, Chen Y. AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension. 2011;58:696–703. doi: 10.1161/HYPERTENSIONAHA.111.174128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–32779. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 62.Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol Endocrinol Metab. 1999;277:E1–10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- 63.Coven DL, Hu X, Cong L, Bergeron R, Shulman GI, Hardie DG, Young LH. Physiological role of AMP-activated protein kinase in the heart: graded activation during exercise. Am J Physiol Endocrinol Metab. 2003;285:E629–636. doi: 10.1152/ajpendo.00171.2003. [DOI] [PubMed] [Google Scholar]
- 64.Musi N, Hirshman MF, Arad M, Xing Y, Fujii N, Pomerleau J, Ahmad F, Berul CI, Seidman JG, Tian R, Goodyear LJ. Functional role of AMP-activated protein kinase in the heart during exercise. FEBS Lett. 2005;579:2045–2050. doi: 10.1016/j.febslet.2005.02.052. [DOI] [PubMed] [Google Scholar]
- 65.Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334:177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beauloye C, Marsin AS, Bertrand L, Vanoverschelde JL, Rider MH, Hue L. The stimulation of heart glycolysis by increased workload does not require AMP-activated protein kinase but a wortmannin-sensitive mechanism. FEBS Lett. 2002;531:324–328. doi: 10.1016/s0014-5793(02)03552-4. [DOI] [PubMed] [Google Scholar]
- 67.Clark H, Carling D, Saggerson D. Covalent activation of heart AMP-activated protein kinase in response to physiological concentrations of long-chain fatty acids. Eur J Biochem. 2004;271:2215–2224. doi: 10.1111/j.1432-1033.2004.04151.x. [DOI] [PubMed] [Google Scholar]
- 68.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–1389. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 70.Kang S, Chemaly ER, Hajjar RJ, Lebeche D. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J Biol Chem. 2011;286:18465–18473. doi: 10.1074/jbc.M110.200022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem. 2005;280:25196–25201. doi: 10.1074/jbc.C500175200. [DOI] [PubMed] [Google Scholar]
- 72.Kelly M, Keller C, Avilucea PR, Keller P, Luo Z, Xiang X, Giralt M, Hidalgo J, Saha AK, Pedersen BK, Ruderman NB. AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochem Biophys Res Commun. 2004;320:449–454. doi: 10.1016/j.bbrc.2004.05.188. [DOI] [PubMed] [Google Scholar]
- 73.Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D, Kemp BE, Febbraio MA, Steinberg GR. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat Med. 2006;12:541–548. doi: 10.1038/nm1383. [DOI] [PubMed] [Google Scholar]
- 74.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding G, Qin Q, He N, Francis-David SC, Hou J, Liu J, Ricks E, Yang Q. Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes and upregulated by activation of peroxisome proliferator-activated receptor gamma. J Mol Cell Cardiol. 2007;43:73–84. doi: 10.1016/j.yjmcc.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest. 2010;120:4342–4352. doi: 10.1172/JCI43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 78.McGaffin KR, Witham WG, Yester KA, Romano LC, O'Doherty RM, McTiernan CF, O'Donnell CP. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc Res. 2011;89:60–71. doi: 10.1093/cvr/cvq288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bouhidel O, Pons S, Souktani R, Zini R, Berdeaux A, Ghaleh B. Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am J Physiol Heart Circ Physiol. 2008;295:H1580–1586. doi: 10.1152/ajpheart.00379.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ko HJ, Zhang Z, Jung DY, Jun JY, Ma Z, Jones KE, Chan SY, Kim JK. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes. 2009;58:2536–2546. doi: 10.2337/db08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Geiger PC, Hancock C, Wright DC, Han DH, Holloszy JO. IL-6 increases muscle insulin sensitivity only at superphysiological levels. Am J Physiol Endocrinol Metab. 2007;292:E1842–1846. doi: 10.1152/ajpendo.00701.2006. [DOI] [PubMed] [Google Scholar]
- 82.Takahashi M, Nishihira J, Shimpo M, Mizue Y, Ueno S, Mano H, Kobayashi E, Ikeda U, Shimada K. Macrophage migration inhibitory factor as a redox-sensitive cytokine in cardiac myocytes. Cardiovasc Res. 2001;52:438–445. doi: 10.1016/s0008-6363(01)00408-4. [DOI] [PubMed] [Google Scholar]
- 83.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 84.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, Merk M, Zierow S, Bernhagen J, Ren J, Bucala R, Li J. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282–292. doi: 10.1161/CIRCULATIONAHA.110.953208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Russell RR, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol Heart Circ Physiol. 1999;277:H643–H649. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- 86.Mangano DT. Effects of acadesine on myocardial infarction, stroke, and death following surgery. JAMA. 1997;277:325–332. doi: 10.1001/jama.277.4.325. [DOI] [PubMed] [Google Scholar]
- 87.An D, Kewalramani G, Chan JK, Qi D, Ghosh S, Pulinilkunnil T, Abrahani A, Innis SM, Rodrigues B. Metformin influences cardiomyocyte cell death by pathways that are dependent and independent of caspase-3. Diabetologia. 2006;49:2174–2184. doi: 10.1007/s00125-006-0338-9. [DOI] [PubMed] [Google Scholar]
- 88.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem. 2011;286:1–11. doi: 10.1074/jbc.M110.121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 91.Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 93.Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, Shao W, Su MB, Li JY, Nan FJ, Li J. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008;283:16051–16060. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y, Shyy JY. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655–2662. doi: 10.1161/CIRCULATIONAHA.106.630194. [DOI] [PubMed] [Google Scholar]
- 95.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell metabolism. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luiken JJ, Coort SL, Willems J, Coumans WA, Bonen A, van der Vusse GJ, Glatz JF. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes. 2003;52:1627–1634. doi: 10.2337/diabetes.52.7.1627. [DOI] [PubMed] [Google Scholar]
- 99.An D, Pulinilkunnil T, Qi D, Ghosh S, Abrahani A, Rodrigues B. The metabolic “switch” AMPK regulates cardiac heparin-releasable lipoprotein lipase. Am J Physiol Endocrinol Metab. 2005;288:E246–253. doi: 10.1152/ajpendo.00211.2004. [DOI] [PubMed] [Google Scholar]
- 100.Young LH, Renfu Y, Russell RR, Hu X, Caplan MJ, Ren J, Shulman GI, Sinusas AJ. Low-flow ischemia leads to translocation of canine heart GLUT-4 and GLUT-1 glucose transporters to the sarcolemma in vivo. Circulation. 1997;95:415–422. doi: 10.1161/01.cir.95.2.415. [DOI] [PubMed] [Google Scholar]
- 101.Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem. 2008;283:9187–9195. doi: 10.1074/jbc.M708934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang J, Holman GD. Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J Biol Chem. 2005;280:4070–4078. doi: 10.1074/jbc.M410213200. [DOI] [PubMed] [Google Scholar]
- 103.Tian R, Abel ED. Responses of GLUT4-Deficient Hearts to Ischemia Underscore the Importance of Glycolysis. Circulation. 2001;103:2961–2966. doi: 10.1161/01.cir.103.24.2961. [DOI] [PubMed] [Google Scholar]
- 104.Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv Z, Gu Z, Zhang J, Wang J, Zen K, Xiang Y, Wang D, Zhang CY. Hypoxia induces PGC-1alpha expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010;20:676–687. doi: 10.1038/cr.2010.46. [DOI] [PubMed] [Google Scholar]
- 105.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 110.Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang W, Fan J, Yang X, Furer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol. 2002;22:3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- 113.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 114.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krawiec BJ, Nystrom GJ, Frost RA, Jefferson LS, Lang CH. AMP-activated protein kinase agonists increase mRNA content of the muscle-specific ubiquitin ligases MAFbx and MuRF1 in C2C12 cells. Am J Physiol Endocrinol Metab. 2007;292:E1555–1567. doi: 10.1152/ajpendo.00622.2006. [DOI] [PubMed] [Google Scholar]
- 116.Baskin KK, Taegtmeyer H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ Res. 2011;109:1153–1161. doi: 10.1161/CIRCRESAHA.111.252742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu J, Gong NL, Bodi I, Aronow BJ, Backx PH, Molkentin JD. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J Biol Chem. 2006;281:9152–9162. doi: 10.1074/jbc.M510217200. [DOI] [PubMed] [Google Scholar]
- 118.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 120.Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy. 2011;7:1254–1255. doi: 10.4161/auto.7.10.16740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem. 2003;278:28372–28377. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- 124.Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T, Bolli R. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation. 2007;116:2809–2817. doi: 10.1161/CIRCULATIONAHA.107.725697. [DOI] [PubMed] [Google Scholar]
- 125.Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, Horman S, Vaulont S, Hoerter J, Viollet B, Hue L, Vanoverschelde JL, Bertrand L. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am J Physiol Heart Circ Physiol. 2006;291:H2875–2883. doi: 10.1152/ajpheart.01032.2005. [DOI] [PubMed] [Google Scholar]
- 126.Carvajal K, Zarrinpashneh E, Szarszoi O, Joubert F, Athea Y, Mateo P, Gillet B, Vaulont S, Viollet B, Bigard X, Bertrand L, Ventura-Clapier R, Hoerter JA. Dual cardiac contractile effects of the alpha2-AMPK deletion in low-flow ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2007;292:H3136–3147. doi: 10.1152/ajpheart.00683.2006. [DOI] [PubMed] [Google Scholar]
- 127.Folmes CD, Wagg CS, Shen M, Clanachan AS, Tian R, Lopaschuk GD. Suppression of 5′-AMP-activated protein kinase activity does not impair recovery of contractile function during reperfusion of ischemic hearts. Am J Physiol Heart Circ Physiol. 2009;297:H313–321. doi: 10.1152/ajpheart.01298.2008. [DOI] [PubMed] [Google Scholar]
- 128.Paiva MA, Rutter-Locher Z, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, Mocanu MM. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am J Physiol Heart Circ Physiol. 2011;300:H2123–2134. doi: 10.1152/ajpheart.00707.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li XJ, Wang XW, Du YJ. Protective effects of erythropoietin on myocardial infarction in rats: the role of AMP-activated protein kinase signaling pathway. Am J Med Sci. 2011;342:153–159. doi: 10.1097/MAJ.0b013e318210041d. [DOI] [PubMed] [Google Scholar]
- 131.Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Turdi S, Kandadi MR, Zhao J, Huff AF, Du M, Ren J. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol. 2011;50:712–722. doi: 10.1016/j.yjmcc.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chan AY, Dolinsky VW, Soltys CL, Viollet B, Baksh S, Light PE, Dyck JR. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem. 2008;283:24194–24201. doi: 10.1074/jbc.M802869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Juric D, Wojciechowski P, Das DK, Netticadan T. Prevention of concentric hypertrophy and diastolic impairment in aortic-banded rats treated with resveratrol. Am J Physiol Heart Circ Physiol. 2007;292:H2138–2143. doi: 10.1152/ajpheart.00852.2006. [DOI] [PubMed] [Google Scholar]
- 135.Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M, Ito S, Ogai A, Asakura M, Kim J, Minamino T, Takashima S, Sanada S, Sugimachi M, Komamura K, Mochizuki N, Kitakaze M. Metformin prevents progression of heart failure in dogs: role of AMP-activated protein kinase. Circulation. 2009;119:2568–2577. doi: 10.1161/CIRCULATIONAHA.108.798561. [DOI] [PubMed] [Google Scholar]
- 136.Aguilar D, Chan W, Bozkurt B, Ramasubbu K, Deswal A. Metformin use and mortality in ambulatory patients with diabetes and heart failure. Circulation Heart failure. 2011;4:53–58. doi: 10.1161/CIRCHEARTFAILURE.110.952556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gundewar S, Calvert JW, Jha S, Toedt-Pingel I, Ji SY, Nunez D, Ramachandran A, Anaya-Cisneros M, Tian R, Lefer DJ. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009;104:403–411. doi: 10.1161/CIRCRESAHA.108.190918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215–1220. doi: 10.1093/hmg/10.11.1215. [DOI] [PubMed] [Google Scholar]
- 139.Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Ali Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, Bachinski LL, Roberts R, Hassan AS. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823–1831. doi: 10.1056/NEJM200106143442403. [DOI] [PubMed] [Google Scholar]
- 140.Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–362. doi: 10.1172/JCI14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zou L, Shen M, Arad M, He H, Lofgren B, Ingwall JS, Seidman CE, Seidman JG, Tian R. N488I mutation of the gamma2-subunit results in bidirectional changes in AMP-activated protein kinase activity. Circ Res. 2005;97:323–328. doi: 10.1161/01.RES.0000179035.20319.c2. [DOI] [PubMed] [Google Scholar]
- 142.Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, Roberts R. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation. 2001;104:3030–3033. doi: 10.1161/hc5001.102111. [DOI] [PubMed] [Google Scholar]
- 143.Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation. 2003;107:2850–2856. doi: 10.1161/01.CIR.0000075270.13497.2B. [DOI] [PubMed] [Google Scholar]
- 144.Burwinkel B, Scott JW, Buhrer C, van Landeghem FK, Cox GF, Wilson CJ, Hardie DG, Kilimann MW. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet. 2005;76:1034–1049. doi: 10.1086/430840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Davies JK, Wells DJ, Liu K, Whitrow HR, Daniel TD, Grignani R, Lygate CA, Schneider JE, Noel G, Watkins H, Carling D. Characterization of the role of {gamma}2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff-Parkinson-White syndrome. Am J Physiol Heart Circ Physiol. 2006;290:H1942–1951. doi: 10.1152/ajpheart.01020.2005. [DOI] [PubMed] [Google Scholar]
- 146.Sidhu JS, Rajawat YS, Rami TG, Gollob MH, Wang Z, Yuan R, Marian AJ, DeMayo FJ, Weilbacher D, Taffet GE, Davies JK, Carling D, Khoury DS, Roberts R. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation. 2005;111:21–29. doi: 10.1161/01.CIR.0000151291.32974.D5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ahmad F, Arad M, Musi N, He H, Wolf C, Branco D, Perez-Atayde AR, Stapleton D, Bali D, Xing Y, Tian R, Goodyear LJ, Berul CI, Ingwall JS, Seidman CE, Seidman JG. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005;112:3140–3148. doi: 10.1161/CIRCULATIONAHA.105.550806. [DOI] [PubMed] [Google Scholar]
- 148.Luptak I, Shen M, He H, Hirshman MF, Musi N, Goodyear LJ, Yan J, Wakimoto H, Morita H, Arad M, Seidman CE, Seidman JG, Ingwall JS, Balschi JA, Tian R. Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J Clin Invest. 2007;117:1432–1439. doi: 10.1172/JCI30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carling D, Hardie DG. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta. 1989;1012:81–86. doi: 10.1016/0167-4889(89)90014-1. [DOI] [PubMed] [Google Scholar]
- 150.Wolf CM, Arad M, Ahmad F, Sanbe A, Bernstein SA, Toka O, Konno T, Morley G, Robbins J, Seidman JG, Seidman CE, Berul CI. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations. Circulation. 2008;117:144–154. doi: 10.1161/CIRCULATIONAHA.107.726752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol Endocrinol Metab. 1997;273:E1107–1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 152.Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47:1369–1373. doi: 10.2337/diab.47.8.1369. [DOI] [PubMed] [Google Scholar]