

Abstract
Context:
Pituitary effects of long-term therapy with mifepristone, a glucocorticoid receptor antagonist, in Cushing's disease (CD) patients are not well understood.
Objective:
Our objective was to report changes in ACTH and pituitary magnetic resonance imaging (MRI) findings during long-term use of mifepristone in CD patients.
Design and Setting:
The Study of the Efficacy and Safety of Mifepristone in the Treatment of Endogenous Cushing's Syndrome (SEISMIC) was a 24-week, open-label study of mifepristone, and its long-term extension (LTE) is a multicenter U.S. study.
Patients:
Forty-three CD patients (mean age 45.3 years) were enrolled in SEISMIC with 27 continuing into the LTE study.
Interventions:
Mifepristone (300–1200 mg) was administered once daily.
Main Outcome Measures:
ACTH and pituitary MRI were assessed at baseline and at regular intervals during treatment.
Results:
A ≥2-fold increase in ACTH was observed in 72% of patients treated for a median duration of 11.3 months. The mean peak increase in ACTH was 2.76 ± 1.65-fold during SEISMIC, and mean ACTH concentrations remained stable during the LTE. ACTH was directly correlated with mifepristone dose and declined to near baseline levels after mifepristone discontinuation. Tumor regressed in 2 patients and progressed in 3 patients with macroadenomas. An additional microadenoma was identified after 25 months of treatment after a baseline tumor-negative MRI.
Conclusions:
In the largest prospective study to date, long-term mifepristone treatment increased ACTH in approximately two-thirds of patients with CD. ACTH elevations were observed within the first few weeks of treatment, were dose-dependent, and generally remained stable over time. Corticotroph tumor progression and regression may occur over time, but patients may have significant increases in ACTH levels without evidence of tumor growth.
Cushing's disease (CD) is a serious condition of chronic hypercortisolism caused by an ACTH-secreting pituitary tumor and is associated with increased morbidity and mortality (1, 2). Transsphenoidal surgical resection of the adenoma in patients with CD results in initial cure rates between 65% and 90% (3), yet recurrence after initial surgical remission is reported in up to one-quarter of cases (4). Radiotherapy is sometimes used when surgery alone has been ineffective but often requires months to years to be effective, and medical therapy is needed in the interim (5, 6). Bilateral adrenalectomy (BLA) provides prompt resolution of hypercortisolism but is irreversible and mandates lifelong glucocorticoid and mineralocorticoid replacement (3, 7, 8). Medical therapies that target ACTH or cortisol production or competitively antagonize the glucocorticoid receptor (GR) are primarily used after surgical failure (3, 6, 9).
Therapies that reduce the negative feedback of cortisol at the hypothalamus and pituitary are expected to result in secondary increases in ACTH and cortisol (10). Drugs that reduce cortisol levels such as metyrapone and mitotane have been associated with increases in ACTH (11, 12); interestingly, ACTH increases may be less commonly observed in patients treated with ketoconazole possibly due to an independent effect on ACTH inhibition (13, 14). Castinetti et al (15) reported that the glucocorticoid antagonist mifepristone can result in up to a 3-fold increase in ACTH among CD patients. BLA usually results in complete cortisol deficiency and represents the most dramatic reduction in negative feedback; Assié et al (16) reported that the median ACTH level increased 5-fold in the year after BLA.
Nelson's syndrome, characterized by a rapid enlargement of the pituitary tumor, elevations in ACTH, and hyperpigmentation, is a severe complication caused by absent glucocorticoid negative feedback and occurs in approximately one-fourth of patients after BLA (3, 7, 16). Improved diagnostics and imaging modalities have likely led to a reduced frequency of Nelson's syndrome (16, 17). The role of neoadjuvant radiotherapy in reducing tumor progression is still controversial (17, 18). However, a recent retrospective study suggested that prophylactic stereotactic radiation before BLA could decrease the incidence of Nelson's syndrome (19). Mild corticotroph tumor progression has been reported in one-fourth to one-third of CD patients treated medically with mitotane or ketoconazole (12, 20).
The Study of the Efficacy and Safety of Mifepristone in the Treatment of Endogenous Cushing's Syndrome (SEISMIC) demonstrated that mifepristone, a competitive GR antagonist, improved the metabolic and clinical status in a majority of patients with Cushing's syndrome (21, 22). During the 6-month treatment period of SEISMIC, 62.8% of the 43 patients with CD experienced at least a 2-fold increase in ACTH (21). Study participants who completed SEISMIC were allowed to continue into a long-term extension (LTE) study during which ACTH and serial magnetic resonance imaging (MRI) scans were monitored. We describe the changes in ACTH levels and pituitary MRI findings during long-term mifepristone use in CD patients.
Patients and Methods
Patients
Forty-three patients with CD were enrolled into SEISMIC as previously described (21). The study was approved by the institutional review board at each center and was registered with www.clinicaltrials.gov (NCT00569582 and NCT00936741). All patients provided written informed consent. Twenty-seven of 31 patients with CD who completed the 24-week treatment period of SEISMIC were enrolled into the LTE study after a 6-week off-drug safety evaluation period (Figure 1). The starting dose of mifepristone in SEISMIC was 300 mg once daily (in the morning) with nonforced dose titration in 300-mg increments at day 14, week 6, and week 10 to a maximum dose of 1200 mg once daily; decreases in dose were allowed at the investigator's discretion. The starting dose in the LTE phase was the same as the final dose in SEISMIC for each patient, and dose increases during the LTE were permissible, but 1200 mg was the maximal daily dose. The duration of treatment during the LTE varied based upon the time of enrollment into SEISMIC and ranged from 0.5 to 42 months.
Figure 1.
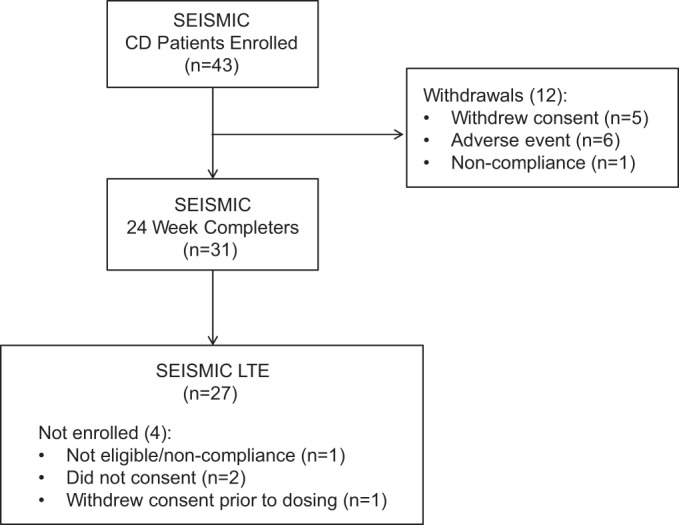
Patient disposition.
Study visits after the screening period in SEISMIC occurred at baseline (day 1), day 14, weeks 6, 10, 16, 20, and 24 and after a 6-week off-drug safety period (6-week follow-up). Entry into the LTE study occurred at or within 2 weeks of the 6-week follow-up visit of SEISMIC and was followed by study visits at months 1 and 3 and then at 3-month intervals.
Assessments
ACTH was monitored during SEISMIC at baseline, day 14, and weeks 6, 10, 16, and 24 and at the 6-week off-drug safety follow-up period, and then every 3 months during the LTE. There was also a 6-week off-drug period at the end of the LTE at which time ACTH was measured. During SEISMIC, sampling for mifepristone trough levels and ACTH levels occurred on the same days. On those days, administration of mifepristone was held until just after samples were taken for trough levels. Blood samples for ACTH measurements were drawn between 7:00 and 9:00 am. During the LTE, the timing between mifepristone administration and ACTH sampling was not specified. Biochemical measurements were conducted in a central laboratory (Quest Diagnostics). ACTH was measured with an immunochemiluminometric assay (Immulite 2000 ACTH; Siemens Medical Solutions Diagnostics); normal range is 5 to 27 pg/mL (1.1–5.9 pmol/L) for females and 7 to 50 pg/mL (1.5–11 pmol/L) for males. Intra-assay and interassay coefficients of variation were 6.7% to 9.5% and 6.1% to 10.0%, respectively. Urinary and salivary cortisol were assayed by liquid chromatography tandem mass spectroscopy (normal ranges, respectively, are 2–42.4 μg/24 hours [5.5–117 nmol/24 hours] and ≤0.09 μg/dL [2.5 nmol/L]); serum cortisol normal range is 4 to 22 μg/dL (110–607 nmol/24 hours). ACTH and serum cortisol were measured between 7:00 and 9:00 am. Blood samples were drawn for trough mifepristone drug concentrations and measured by liquid chromatography tandem mass spectroscopy (lower limit of detection 10 ng/mL) at day 14 and weeks 6, 10, 16, and 24 in SEISMIC and then every 6 months in the LTE.
Pituitary MRI was performed before starting study drug, at weeks 10 and 24, and then every 6 months during the LTE study. Specific MRI acquisition protocols were not prespecified and all imaging studies were read locally at the research sites. All scans fulfilled minimum requirements of a 1.5-T MRI, coronal and sagittal T1 with and without contrast, and a maximum slice thickness of 3 mm through the sella region. If recommended by the local neuroradiologist, dynamic T1-coronal or T2-coronal or -sagittal sequences were added. Investigators reviewed MRIs, documented the findings, and recorded any clinically significant change as adverse events. Forty-one patients had a baseline MRI, and 36 had at least 1 postbaseline MRI. Digitized MRIs for these 36 patients were submitted for central reevaluation, as previously described (23, 24) to the Neurosurgical Department of the University Hospital Erlangen, Germany. The T1-weighted sequences after contrast enhancement were used for comparative analysis. The images were adjusted for grayscale and image amplification in the coronal and sagittal dimensions. After blinding by a radiological technician, each dataset was independently analyzed by 2 senior neurosurgeons on a Siemens Syngo Workstation. Using anatomical landmarks (internal carotid artery, pituitary stalk, optic chiasm, and sphenoid sinus), comparable images were identified. The pituitary gland, stalk, and adenoma (if visible) were identified in the comparison images. If a distinct adenoma was present, the maximum tumor extension was measured in sagittal, coronal, and axial dimensions. The final diameter was a mean value of at least 6 individual measurements (3 by each reviewer).The results were categorized into 7 possible categories: A, no adenoma visible and no change in sellar contents; B, stable adenoma; C, increase in adenoma size (≥2 mm in any dimension); D, adenoma regression; E, progression and regression; F, regression and progression; and G, insufficient data.
Statistics
Data are presented as mean and SD (unless otherwise noted) and are based on the safety population defined as all subjects who received at least 1 dose of study medication. Baseline was defined as the last measurement before the start of study drug in SEISMIC or restart of drug at entry into the LTE. There was no imputation of missing data. In the analyses of mifepristone dose or concentration vs ACTH, comparisons were made at steady state defined as at least 5 days on a stable dose. Statistical significance was tested using Student's t tests (paired and unpaired) and ANOVA with post hoc testing using Fisher's protected least significant difference. Statistical significance was set at P < .05. Correlation was determined using Spearman and Pearson correlation coefficients where indicated. Statistical software used included Microsoft Excel 2010 and StatView version 5.0.1 (SAS Institute).
Results
Patients
The 43 patients (74% female, n = 32) were 45.3 ± 11.5 years of age and had CD for a median of 37 (range 2–159) months. All but 1 patient had undergone previous transsphenoidal pituitary surgery.
Previous adjuvant therapy for CD was used in 58% of patients and included medication only (n = 7), radiation only (n = 7), or both medication and radiation (n = 11). Eighteen had at least 1 course of radiotherapy before study enrollment with a median of 47 (range 1–87) months between the latest radiation treatment and the first dose of mifepristone. One patient had RT after SEISMIC and before entry into the LTE. The most common previous medication used for CD was ketoconazole (n = 16), and there were rare uses of cabergoline, metyrapone, and octreotide. Measures of ACTH and cortisol were elevated at baseline as previously described (21), and values at entry into the LTE were similar to baseline (Table 1). Patients received mifepristone for a median of 11.3 (range 0.5–42) months.
Table 1.
Baseline Biochemistry
| Baseline in SEISMIC | Entry Into SEISMIC LTEa | |
|---|---|---|
| n | 43 | 27 |
| ACTH, pg/mL | 63 ± 51 | 71.6 ± 53.9 |
| 24-h UFC (normal, 2.0–42.4 μg) | 139 ± 137 | 139.3 ± 96.7 |
| Serum cortisol, μg/dL | 21.2 ± 6.0 | 23.5 ± 7.7 |
| Late-night salivary cortisol, μg/dL | 0.29 ± 0.29 | NDb |
Abbreviation: ND, not determined.
Measurements at entry into LTE were after 6-week off-drug period after 24 weeks of SEISMIC study.
Late-night salivary cortisol not measured during LTE.
ACTH and cortisol
Increases in ACTH levels occurred at the first measurement at day 14, plateaued from weeks 10 through 24, and declined to near baseline levels 6 weeks after mifepristone was discontinued at the end of dosing in SEISMIC (Figure 2). The mean peak ACTH value during SEISMIC was 152.2 ± 127.4 pg/mL (2.76 ± 1.65-fold over baseline; P < .0001 vs baseline); the highest ACTH observed was 619 pg/mL (5.7-fold increase over baseline). During the LTE, ACTH levels remained stable on average with mean peak values of 182.8 ± 126.7 pg/mL (P < .0001 vs baseline). The highest value observed during the LTE study was 614 pg/mL in a patient whose peak value during treatment in SEISMIC was 133 pg/mL representing 5.8- and 1.25-fold increases over baseline, respectively. A 2-fold or greater increase in ACTH levels was observed in 72% of patients. Baseline ACTH was not correlated with the fold increase in ACTH, but higher baseline ACTH levels were associated with higher levels during treatment (Pearson r = 0.58, P < .001). There was an apparent decrease in ACTH from month 27 onward (Figure 2); however, it should be noted that ACTH values were available for only 4 subjects at month 36.
Figure 2.

ACTH concentrations during the study. Study visits are shown on the x-axis: B, baseline; 6W FU, 6-week follow-up visit after 6 weeks after discontinuation of mifepristone; entry into LTE occurred at 6W FU after W24 visit. Visits labeled with M indicate visit time on LTE and do not represent cumulative time on mifepristone. M2/3 represents ACTH levels from month 3 or 2 for subjects not having a month-3 visit due to protocol amendment (see text). The small n at the 6W FU visit at the conclusion of the LTE is due to several patients transitioning to commercially available drug.
The mean peak values for cortisol on SEISMIC and in the LTE, respectively, were 40.2 ± 19.3 μg/dL (1.97 ± 1.02-fold increase; P < .0001 vs baseline) and 57.4 ± 20.8 μg/dL (2.85 ± 1.05-fold increase; P < .0001 vs baseline) for serum cortisol and 985.4 ± 1584.9 μg/d (8.46 ± 14.6-fold increase; P < .001 vs baseline) and 1575.9 ± 2250.5 μg/d (16.4 ± 41.9-fold increase; P < .01 vs baseline) for urinary free cortisol (UFC). The average maximal level of late-night salivary cortisol during SEISMIC was 2.6 ± 4.7 μg/dL (10.6 ± 11.96-fold increase; P < .001); salivary cortisol was not measured during the LTE. The increases in ACTH during SEISMIC were correlated with 24-hour UFC, serum cortisol, and late-night salivary cortisol (Figure 3B).
Figure 3.

A, Mifepristone dose and B, ACTH concentration. ACTH increase correlates with cortisol. *, Spearman correlation coefficient r.
Pituitary RT
Although baseline ACTH levels were higher in patients who had previous pituitary radiation (n = 18, 86.1 ± 67.7 pg/mL) compared with those naive to radiation (n = 25, 45.8 ± 25.8 pg/mL, P = .01), there were no statistically significant differences in ACTH levels during mifepristone treatment. When the fold increase over baseline was examined, previous radiation appeared to blunt the rise in ACTH until week 6 [radiation therapy (RT) 1.40 ± 0.45 vs no RT 2.08 ± 0.97 at day 14, P = .01; RT 1.68 ± 0.80 vs no RT 2.34 ± 0.96, P = .03], but not thereafter. A correlation analysis assessing the impact time since RT on ACTH levels was not performed due to the small sample size, the heterogeneity of the radiation treatments, and the wide distribution of time spans between treatments to the first dose of mifepristone.
Mifepristone dose and ACTH concentration
Mifepristone dose was directly correlated with ACTH levels during SEISMIC (P < .001, ANOVA). Compared with ACTH concentrations when patients were not taking mifepristone (64.5 ± 54.4 pg/mL), only steady-state doses of 600, 900, or 1200 mg were associated with statistically significant higher ACTH levels (129.6 ± 94.6 pg/mL, P = .001; 125.0 ± 111.2 pg/mL, P = .008; and 179.9 ± 156.9 pg/mL, P < .0001, respectively) (Figure 3A). Statistically significant higher levels of ACTH were observed at 1200 mg compared with 300 mg (100.9 ± 66.0 pg/mL, P = .02); differences at 600 mg (P = .057) or 900 mg (P = .054) were of borderline statistical significance when compared with the 1200-mg dose (Figure 3A). Analysis of change in ACTH from baseline according to dose resulted in similar findings; the 300-mg dose did not result in a statistically significant change in ACTH from baseline, whereas all other doses did (P < .0001, ANOVA). In log-log regression analyses, ACTH change from baseline was directly correlated with mifepristone concentration (Pearson r = 0.392, P < .0001), and the interval change in ACTH was correlated with the change in mifepristone concentration between visits (Pearson r = 0.294, P < .0001).
MRI findings
Table 2 shows the MRI findings of the 36 study patients with a baseline and postbaseline MRI. Among these 36 patients, treatment duration lasted ≥12 months in 24, ≥18 months in 21, ≥24 months in 20, and ≥30 months in 10 patients. Tumor remained stable in 30 patients (groups A and B) and regressed in 2 patients (group D, 1 microadenoma and 1 macroadenoma). Tumor progression (group C) was observed in 3 patients with macroadenomas (Figure 4, A–C) at 2.5, 6, and 19 months of treatment. The appearance of a 4-mm microadenoma was identified in an additional patient with a tumor-negative MRI (nonvisible) at baseline after 25 months of treatment. Two patients with progression at 2.5 and 19 months had previous RT. The former had a large invasive atypical tumor at baseline that had previously been a silent corticotroph adenoma that transitioned into a functional corticotroph adenoma before study enrollment. Although there was insufficient statistical power to detect significant differences in the pattern of ACTH change over time in these patients compared with those without progression, the ACTH increases in these individuals were not unusual relative to other study participants (Table 3). A graph of ACTH levels over time among patients with tumor progression is available (Supplemental Figure 1). Regression of a macroadenoma occurred after 1 year of treatment in a patient who had pituitary radiation treatment before the study (Figure 4D); complete disappearance of a microadenoma occurred after 24 weeks of treatment in a patient naive to radiation. There were no distinguishing baseline characteristics between those patients whose tumors progressed (n = 4) compared with those that did not progress (n = 30) and those patients whose tumors regressed (n = 2). The changes in ACTH were similar between the groups.
Table 2.
Findings of Central MRI Reading
| Baseline | Progressed (C)a | Stable (A or B)a | Regressed (D)a | |
|---|---|---|---|---|
| Nonvisible | 20 | 1 | 19 | |
| Microadenoma (<10 mm) | 9 | 0 | 8 | 1 |
| Macroadenoma (≥10 mm) | 7 | 3 | 3 | 1 |
Category based on central MRI reading: A, no adenoma visible and no change in sellar contents; B, stable adenoma; C, increase in adenoma size (≥2 mm in any dimension); D, adenoma regression.
Figure 4.
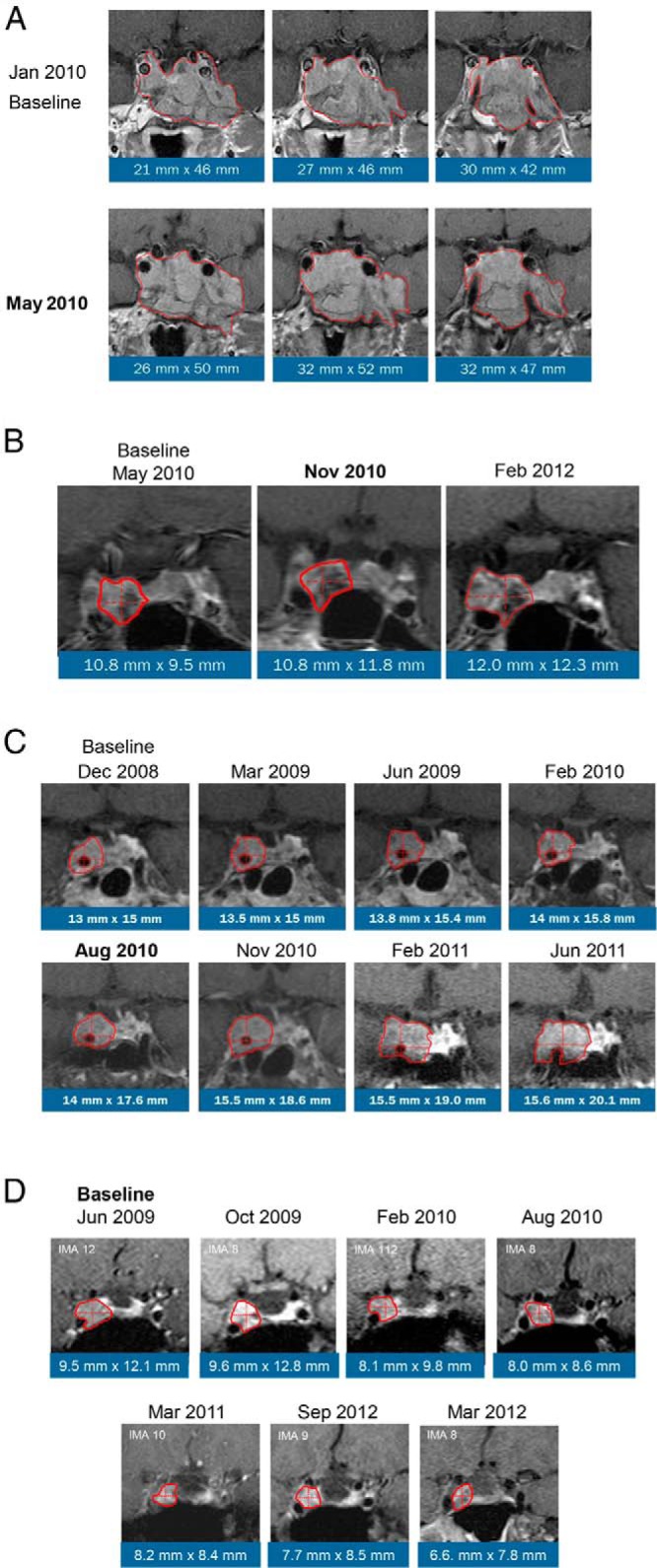
Central image analysis of MRIs that showed macroadenomas with progression or regression. Dates shown in bold indicate first detection of progression. A, Atypical macroadenoma, progression at 2.5 months of treatment; B, macroadenoma progression at 6 months of treatment; C, macroadenoma, progression at 19 months of treatment; D, macroadenoma, regression at 1 year.
Table 3.
Baseline Characteristics of the 4 Patients With Tumor Progression
| Age, y | Sex | CD Duration, mo | Tumor Size, mm | Cavernous Sinus Invasion | Radiation | ACTH, pg/mL (Fold Increase)a |
|---|---|---|---|---|---|---|
| 55 | Female | 118 | Nonvisible | No | No | 102 (3.29) |
| 57 | Female | 84 | 10.8 × 9.5 | Yes | No | 98 (1.24) |
| 52 | Male | 38 | 13 × 15 | Yes | Yes | 149 (3.63) |
| 52 | Male | 134 | 21 × 46 | Yes | Yes | 405 (3.55) |
Fold increase over baseline at point of progression.
Discussion
Glucocorticoids regulate ACTH secretion via a sensitive negative feedback system acting at the level of the hypothalamus and pituitary through the GR. In CD, this negative feedback system is attenuated. Conversely, therapeutic interventions that reduce cortisol levels in CD (eg, BLA and cortisol synthesis inhibitors) reduce negative feedback and result in increased ACTH production. Similarly, antagonism of cortisol action at the GR is also expected to reduce negative feedback and leads to increases in ACTH (25).
Mifepristone is a competitive GR antagonist with substantially greater binding affinity for this receptor than cortisol (26–28); it also exhibits a minor amount of agonist activity (29). Short-term administration of mifepristone to healthy male volunteers resulted in 2- to 8-fold increases of ACTH (30–32); these changes occurred rapidly and resolved soon after discontinuation of mifepristone (30, 32). Previously, the effects of mifepristone on ACTH in CD patients were not well understood. Bertagna et al (33) administered mifepristone (400 mg daily) for 3 days to 5 patients with CD and documented a 1.8-fold increase in lipotropin as a marker of ACTH secretion. Case reports of long-term mifepristone in CD therapy suggest 2- to 3-fold increases of ACTH (15, 34). Furthermore, we previously reported in a 6-month study that ACTH levels increased at least 2-fold in approximately two-thirds of CD patients and that these changes are reversible upon cessation of the study drug (21). In this present study, ACTH elevation during chronic therapy required several weeks to become maximal but did not appear to be progressive with continued treatment for as long as 3.5 years in some patients. Interestingly, one-third of patients experienced ≥4-fold elevation in ACTH, whereas 11.6% of patients had little to no increase in ACTH. Gaillard et al (32) showed that ACTH increases were greater in response to increasing single mifepristone doses (4.5 and 6 mg/kg) with no incremental increase at a dose of 2.2 mg/kg compared with control (no mifepristone). Our study showed that ACTH concentrations were directly related to dose and increased linearly across the dose range. A dose of 300 mg of mifepristone did not result in a statistically significant difference in ACTH compared with baseline. Significant changes in ACTH required dose changes ≥600 mg. The relatively weak association of dose and ACTH levels may in part be due to the pharmacokinetics of mifepristone (35) or the heterogeneity of the tumor's responsiveness to cortisol negative feedback and GR antagonism. Increasing doses of mifepristone lead to less than dose proportionality increases in total (bound and unbound) drug concentration (36, 37). The increases in ACTH during mifepristone treatment were correlated with increases in cortisol.
Radiographic studies of the adrenal glands after prolonged exposure to elevated ACTH levels were not obtained in this study. However, there were no reports of treatment-emergent events of increase in adrenal size, mass, or hyperplasia during the study. The relatively recent discovery of GR expression in human adrenal cortex (38–41) suggests that ACTH increases with commensurate increases in cortisol that occurs with GR antagonism would portend a different effect on adrenal cortical tissue than that seen in the absence of GR antagonism. That is, increased ACTH levels with GR antagonism may not result in the same trophic effects on adrenal cortical tissue as may occur in untreated CD (42). A long-term prospective imaging study would be required to evaluate the effects on the adrenal glands in CD patients treated with mifepristone.
Corticotroph tumor progression has been demonstrated after medical treatment (12, 20) and BLA (7, 16, 43). Macroadenomas may be at greater risk for growth than smaller tumors (16). An increase in ACTH has been suggested to predict progression after BLA (16). However, increases in ACTH may occur in patients without progression as well. Although ACTH elevations during mifepristone treatment were common in our study, the tumors of 32 patients did not progress. Progression of corticotroph tumor was confirmed in only 4 patients, 3 of whom had macroadenomas at baseline and 1 patient with normal MRI (nonvisible tumor) had a visible microadenoma at follow-up. Although the image of the progression at 6 months appears somewhat unremarkable (Figure 4B), the measurement exceeded the predefined limit of 2 mm; thus, this case was identified as a progression. Regression of corticotroph tumors was confirmed in 2 patients, one with a macroadenoma and one with a microadenoma. For their behavior, presentation, and outcome, ACTH-secreting macroadenomas represent a distinct profile compared with microadenomas, although they probably represent one end of a spectrum of tumor autonomy, with specific growth and biochemical characteristics (44). One patient with tumor progression had a large silent corticotrophic adenoma (Figure 4A) that became active before entering the study and the patient was withdrawn from therapy early due to increasing tumor size. The aggressive behavior of such atypical tumors has been previously described (45–47), particularly in tumors with high Ki67 levels; the previous behavior of this tumor suggests that mifepristone treatment may not have been responsible for this enlargement. Our study was not adequately powered to identify predictors of tumor progression; other than the suggestion of larger tumor burden at study start, there were no other distinguishing characteristics we could identify among the few patients with progression. Indeed, ACTH increases among the 4 progression cases were similar to those cases that did not progress. The clinical and metabolic parameters assessed during SEISMIC (21) and the LTE (global clinical response, weight change, body mass index, waist circumference, glucose control, blood pressure, total cholesterol, low-density lipoprotein, high-density lipoprotein, and triglycerides) for the 4 progression cases were also similar to those that did not progress (data not shown). We observed 2 cases of regression: a microadenoma not previously treated with radiation and a macroadenoma that had been previously treated with radiation. Given the design of the study and the lack of available literature on this topic, we cannot exclude the possibility that all of these findings represent the natural history of these tumors. Furthermore, it is important to recognize that GR antagonism with mifepristone is not equivalent to complete removal of cortisol or all adrenal steroids as is caused by a BLA. Mifepristone's interaction with the GR is not simply one of preventing cortisol binding to the cytosolic GR monomers and blocking all downstream events. Instead, it causes conformational changes in GR that modulate receptor dimerization, corepressor/activator interaction, DNA binding, and transcription (48–50) and modulates gene expression in distinct ways (51, 52). Other considerations include the emerging role that mineralocorticoid (53) and progesterone (53, 54) play in regulating the hypothalamic-pituitary-adrenal axis. The effect of mifepristone on hypothalamic and pituitary GR must be considered within the context of preserved or increased mineralocorticoid activity and antiprogestin activity during its treatment of CD.
Although the largest long-term prospective study of mifepristone on ACTH and pituitary tumor size in patients with CD, the relatively small sample size and duration of follow up could be considered a limitation of our research. Differences in image acquisition and resolution due to the lack of a uniform study-wide MRI scanning protocol between scans may have created the potential for interpretation errors. We believe that the careful retrospective central MRI analysis of an entire series of images on each individual patient, conducted by 2 well-skilled physicians using a standardized evaluation system (23, 24), limited potential errors, and we believe this analysis provides a fairly precise estimation of tumor size changes. Another limitation is that our study population included only CD patients from SEISMIC who were required to have type 2 diabetes/impaired glucose tolerance or a diagnosis of hypertension as part of the inclusion criteria. Although these 2 manifestations are distinguishing features of endogenous Cushing's syndrome, our findings may not be generalizable to CD patients without these cardiometabolic comorbidities.
An important finding of our study is that increases in ACTH during treatment with mifepristone or a history of previous radiation was not predictive of corticotroph tumor progression. There was a low overall frequency of increase in tumor size during long-term mifepristone treatment in CD. There were 3 progressions and 1 regression noted among 7 patients with macroadenomas. To be consistent with current clinical practice, post-treatment monitoring of pituitary MRI is recommended, especially in macroadenomas (3, 6). Depending on clinical circumstances, it may also be reasonable to monitor and assess tumor size in microadenomas on a yearly basis.
In conclusion, GR antagonism with mifepristone increases ACTH in the large majority of patients with CD refractory to surgery, independent of previous pituitary radiation exposure. These changes are observed within the first few weeks of treatment, are dose-dependent, and generally remain stable over time. Both corticotroph tumor progression and regression may occur over time, and patients may have significant increases in ACTH levels without evidence of tumor growth.
Acknowledgments
We acknowledge the SEISMIC investigators: Richard Auchus, University of Texas Southwestern Medical Center, Dallas, TX; Timothy Bailey, AMCR Institute Inc, Escondido, CA; Beverly M. K. Biller, Massachusetts General Hospital, Harvard Medical School, Boston, MA; Ty Carroll, Medical College of Wisconsin, Milwaukee, WI; Kathleen Colleran, University of New Mexico Health Sciences Center, Albuquerque, NM; Henry Fein, Sinai Hospital of Baltimore, Baltimore, MD; James W. Findling, Medical College of Wisconsin, Milwaukee, WI; Maria Fleseriu, Oregon Health & Science University, Portland, OR; Amir Hamrahian, Cleveland Clinic Foundation, Cleveland, OH; Laurence Katznelson, Stanford University Medical Center, Stanford, CA; Janice Kerr, University of Colorado Health Science Center at Fitzsimons, Aurora, CO; Mark Kipnes, Cetero Research/Diabetes and Glandular Disease Research, San Antonio, TX; Lawrence Kirschner, Ohio State University Medical Center, Columbus, OH; Christian Koch, University of Mississippi Medical Center, Jackson, MS; Sam Lerman, The Center for Diabetes and Endocrine Care, Hollywood, FL; Timothy Lyons, Oklahoma University Health Science Center, Oklahoma City, OK; Michael McPhaul, University of Texas Southwestern Medical Center, Dallas, TX; Mark E. Molitch, Northwestern University Feinberg Medical, Chicago, IL; David E. Schteingart, University of Michigan Medical Center, Ann Arbor, MI; T. Brooks Vaughan III, University of Alabama at Birmingham School of Medicine, Birmingham, AL; and Roy Weiss, The University of Chicago, Chicago, IL.
We thank Dat Nguyen of Corcept Therapeutics and Sarah Mizne and Courtney Breuel of MedVal Scientific Information Services, LLC, for editorial assistance.
This work was supported by Corcept Therapeutics. Funding to support editorial and graphics assistance was provided by Corcept Therapeutics. The authors did not receive any grants in support of writing this manuscript.
All drafts of the manuscript were written and reviewed by all the authors.
Disclosure Summary: M.F. has been a principal investigator in clinical trials sponsored by Corcept Therapeutics, Ipsen, and Novartis Pharmaceuticals, with research support provided to Oregon Health & Science University (OHSU). She also has been an ad hoc consultant for Genetech, Ipsen, Novartis Pharmaceuticals, and Pfizer. J.W.F. has served as a consultant and investigator for Corcept Therapeutics and Novartis. C.A.K. has served as a consultant for Novo Nordisk, Ipsen, and Bayer and as an investigator of the SEISMIC trial sponsored by Corcept Therapeutics and the TR321 trial sponsored by Ipsen, with research support provided to the University of Mississippi Medical Center. He also has been/is a speaker for Ipsen. S.-M.S. and M.B. have received honoraria from Corcept Therapeutics for reviewing magnetic resonance images as well as from Novartis, Pfizer, and Ipsen. C.G. is an employee of Corcept Therapeutics.
Footnotes
- BLA
- bilateral adrenalectomy
- CD
- Cushing's disease
- GR
- glucocorticoid receptor
- LTE
- long-term extension
- MRI
- magnetic resonance imaging
- RT
- radiation therapy
- SEISMIC
- Study of the Efficacy and Safety of Mifepristone in the Treatment of Endogenous Cushing's Syndrome
- UFC
- urinary free cortisol.
References
- 1. Clayton RN, Raskauskiene D, Reulen RC, Jones PW. Mortality and morbidity in Cushing's disease over 50 years in Stoke-on-Trent, UK: audit and meta-analysis of literature. J Clin Endocrinol Metab. 2011;96:632–642. [DOI] [PubMed] [Google Scholar]
- 2. Dekkers OM, Horváth-Puhó E, Jørgensen JO, et al. Multisystem morbidity and mortality in Cushing's syndrome: a cohort study. J Clin Endocrinol Metab. 2013;98:2277–2284. [DOI] [PubMed] [Google Scholar]
- 3. Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93:2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atkinson AB, Kennedy A, Wiggam MI, McCance DR, Sheridan B. Long-term remission rates after pituitary surgery for Cushing's disease: the need for long-term surveillance. Clin Endocrinol (Oxf). 2005;63:549–559. [DOI] [PubMed] [Google Scholar]
- 5. Loeffler JS, Shih HA. Radiation therapy in the management of pituitary adenomas. J Clin Endocrinol Metab. 2011;96:1992–2003. [DOI] [PubMed] [Google Scholar]
- 6. Fleseriu M, Molitch ME, Gross C, Schteingart DE, Vaughan TB, 3rd, Biller BM. A new therapeutic approach in the medical treatment of Cushing's syndrome: glucocorticoid receptor blockade with mifepristone. Endocr Pract. 2013;19:313–326. [DOI] [PubMed] [Google Scholar]
- 7. Ritzel K, Beuschlein F, Mickisch A, et al. Outcome of bilateral adrenalectomy in Cushing's syndrome: a systematic review. J Clin Endocrinol Metab. 2013;98:3939–3948. [DOI] [PubMed] [Google Scholar]
- 8. Tritos NA, Biller BM. Medical management of Cushing's disease. J Neurooncol. 2014;117:407–414. [DOI] [PubMed] [Google Scholar]
- 9. Fleseriu M, Petersenn S. Medical management of Cushing's disease: what is the future? Pituitary. 2012;15:330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carroll T, Findling JW. The use of mifepristone in the treatment of Cushing's syndrome. Drugs Today. 2012;48:509–518. [DOI] [PubMed] [Google Scholar]
- 11. Verhelst JA, Trainer PJ, Howlett TA, et al. Short and long-term responses to metyrapone in the medical management of 91 patients with Cushing's syndrome. Clin Endocrinol. 1991;35:169–178. [DOI] [PubMed] [Google Scholar]
- 12. Baudry C, Coste J, Bou Khalil R, et al. Efficiency and tolerance of mitotane in Cushing's disease in 76 patients from a single center. Eur J Endocrinol. 2012;167:473–481. [DOI] [PubMed] [Google Scholar]
- 13. Sonino N, Boscaro M, Paoletta A, Mantero F, Ziliotto D. Ketoconazole treatment in Cushing's syndrome: experience in 34 patients. Clin Endocrinol (Oxf). 1991;35:347–352. [DOI] [PubMed] [Google Scholar]
- 14. Feelders RA, Hofland LJ, de Herder WW. Medical treatment of Cushing's syndrome: adrenal-blocking drugs and ketaconazole. Neuroendocrinology. 2010;92(Suppl 1):111–115. [DOI] [PubMed] [Google Scholar]
- 15. Castinetti F, Fassnacht M, Johanssen S, et al. Merits and pitfalls of mifepristone in Cushing's syndrome. Eur J Endocrinol. 2009;160:1003–1010. [DOI] [PubMed] [Google Scholar]
- 16. Assié G, Bahurel H, Coste J, et al. Corticotroph tumor progression after adrenalectomy in Cushing's disease: a reappraisal of Nelson's syndrome. J Clin Endocrinol Metab. 2007;92:172–179. [DOI] [PubMed] [Google Scholar]
- 17. Bertagna X, Guignat L. Approach to the Cushing's disease patient with persistent/recurrent hypercortisolism after pituitary surgery. J Clin Endocrinol Metab. 2013;98:1307–1318. [DOI] [PubMed] [Google Scholar]
- 18. Barber TM, Adams E, Ansorge O, Byrne JV, Karavitaki N, Wass JA. Nelson's syndrome. Eur J Endocrinol. 2010;163:495–507. [DOI] [PubMed] [Google Scholar]
- 19. Mehta GU, Sheehan JP, Vance ML. Effect of stereotactic radiosurgery before bilateral adrenalectomy for Cushing's disease on the incidence of Nelson's syndrome. J Neurosurg. 2013;119:1493–1497. [DOI] [PubMed] [Google Scholar]
- 20. Castinetti F, Morange I, Jaquet P, Conte-Devolx B, Brue T. Ketoconazole revisited: a preoperative or postoperative treatment in Cushing's disease. Eur J Endocrinol. 2008;158:91–99. [DOI] [PubMed] [Google Scholar]
- 21. Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J Clin Endocrinol Metab. 2012;97:2039–2049. [DOI] [PubMed] [Google Scholar]
- 22. Katznelson L, Loriaux DL, Feldman D, Braunstein GD, Schteingart DE, Gross C. Global clinical response in Cushing's syndrome patients treated with mifepristone. Clin Endocrinol (Oxf). 2014;80:562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buchfelder M, Weigel D, Droste M, et al. Pituitary tumor size in acromegaly during pegvisomant treatment: experience from MR re-evaluations of the German Pegvisomant Observational Study. Eur J Endocrinol. 2009;161:27–35. [DOI] [PubMed] [Google Scholar]
- 24. van der Lely AJ, Biller BM, Brue T, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97:1589–1597. [DOI] [PubMed] [Google Scholar]
- 25. Bertagna X, Guignat L, Raux-Demay MC, Guilhaume B, Girard F. Cushing's disease. In: Melmed S, ed. The Pituitary. 3rd ed New York, NY: Elsevier; 2011:533–617. [Google Scholar]
- 26. Bourgeois S, Pfahl M, Baulieu EE. DNA binding properties of glucocorticosteroid receptors bound to the steroid antagonist RU-486. EMBO J. 1984;3:751–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heikinheimo O, Kontula K, Croxatto H, Spitz I, Luukkainen T, Lähteenmäki P. Plasma concentrations and receptor binding of RU 486 and its metabolites in humans. J Steroid Biochem. 1987;26:279–284. [DOI] [PubMed] [Google Scholar]
- 28. Chrousos GP, Laue L, Nieman LK, Udelsman R, Kawai S, Loriaux DL. Clinical applications of RU 486, a prototype glucocorticoid and progestin antagonist. In: Mantero F, Scoggins B, Takeda R, Biglieri E, Funder J, eds. The Adrenal Hypertension: From Cloning to Clinic. New York, NY: Raven Press; 1989:273–284. [Google Scholar]
- 29. Laue L, Chrousos GP, Loriaux DL, et al. The antiglucocorticoid and antiprogestin steroid RU 486 suppresses the adrenocorticotropin response to ovine corticotropin releasing hormone in man. J Clin Endocrinol Metab. 1988;66:290–293. [DOI] [PubMed] [Google Scholar]
- 30. Laue L, Lotze MT, Chrousos GP, Barnes K, Loriaux DL, Fleisher TA. Effect of chronic treatment with the glucocorticoid antagonist RU 486 in man: toxicity, immunological, and hormonal aspects. J Clin Endocrinol Metab. 1990;71:1474–1480. [DOI] [PubMed] [Google Scholar]
- 31. Garrel DR, Moussali R, De Oliveira A, Lesiège D, Larivière F. RU 486 prevents the acute effects of cortisol on glucose and leucine metabolism. J Clin Endocrinol Metab. 1995;80:379–385. [DOI] [PubMed] [Google Scholar]
- 32. Gaillard RC, Riondel A, Muller AF, Herrmann W, Baulieu EE. RU 486: a steroid with antiglucocorticosteroid activity that only disinhibits the human pituitary-adrenal system at a specific time of day. Proc Natl Acad Sci U S A. 1984;81:3879–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bertagna X, Bertagna C, Laudat MH, Husson JM, Girard F, Luton JP. Pituitary-adrenal response to the antiglucocorticoid action of RU 486 in Cushing's syndrome. J Clin Endocrinol Metab. 1986;63:639–643. [DOI] [PubMed] [Google Scholar]
- 34. Basina M, Liu H, Hoffman AR, Feldman D. Successful long-term treatment of Cushing disease with mifepristone (RU486). Endocr Pract. 2012;18:e114–e120. [DOI] [PubMed] [Google Scholar]
- 35. Heikinheimo O, Kekkonen R, Lähteenmäki P. The pharmacokinetics of mifepristone in humans reveal insights into differential mechanisms of antiprogestin action. Contraception. 2003;68:421–426. [DOI] [PubMed] [Google Scholar]
- 36. Shi YE, Ye ZH, He CH, et al. Pharmacokinetic study of RU 486 and its metabolites after oral administration of single doses to pregnant and non-pregnant women. Contraception. 1993;48:133–149. [DOI] [PubMed] [Google Scholar]
- 37. Heikinheimo O. Pharmacokinetics of the antiprogesterone RU 486 in women during multiple dose administration. J Steroid Biochem. 1989;32:21–25. [DOI] [PubMed] [Google Scholar]
- 38. Condon J, Gosden C, Gardener D, et al. Expression of type 2 11β-hydroxysteroid dehydrogenase and corticosteroid hormone receptors in early human fetal life. J Clin Endocrinol Metab. 1998;83:4490–4497. [DOI] [PubMed] [Google Scholar]
- 39. Paust HJ, Loeper S, Else T, et al. Expression of the glucocorticoid receptor in the human adrenal cortex. Exp Clin Endocrinol Diabetes. 2006;114:6–10. [DOI] [PubMed] [Google Scholar]
- 40. Tacon LJ, Soon PS, Gill AJ, et al. The glucocorticoid receptor is overexpressed in malignant adrenocortical tumors. J Clin Endocrinol Metab. 2009;94:4591–4599. [DOI] [PubMed] [Google Scholar]
- 41. Bourdeau I, Lacroix A, Schürch W, Caron P, Antakly T, Stratakis CA. Primary pigmented nodular adrenocortical disease: paradoxical responses of cortisol secretion to dexamethasone occur in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab. 2003;88:3931–3937. [DOI] [PubMed] [Google Scholar]
- 42. Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing's syndrome. Arq Bras Endocrinol Metabol. 2007;51:1319–1328. [DOI] [PubMed] [Google Scholar]
- 43. Blevins LS, Jr, Christy JH, Khajavi M, Tindall GT. Outcomes of therapy for Cushing's disease due to adrenocorticotropin-secreting pituitary macroadenomas. J Clin Endocrinol Metab. 1998;83:63–67. [DOI] [PubMed] [Google Scholar]
- 44. Woo YS, Isidori AM, Wat WZ, et al. Clinical and biochemical characteristics of adrenocorticotropin-secreting macroadenomas. J Clin Endocrinol Metab. 2005;90:4963–4969. [DOI] [PubMed] [Google Scholar]
- 45. Melcescu E, Gannon AW, Parent AD, et al. Silent or subclinical corticotroph pituitary macroadenoma transforming into Cushing disease: 11-year follow-up. Neurosurgery. 2013;72:E144–E146. [DOI] [PubMed] [Google Scholar]
- 46. Cornell RF, Kelly DF, Bordo G, et al. Chemotherapy-induced regression of an adrenocorticotropin-secreting pituitary carcinoma accompanied by secondary adrenal insufficiency. Case Rep Endocrinol. 2013;2013:675298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Phillips J, East HE, French SE, et al. What causes a prolactinoma to be aggressive or to become a pituitary carcinoma? Hormones. 2012;11:477–482. [DOI] [PubMed] [Google Scholar]
- 48. Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform β: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66:3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cadepond F, Ulmann A, Baulieu EE. RU486 (mifepristone): mechanisms of action and clinical uses. Annu Rev Med. 1997;48:129–156. [DOI] [PubMed] [Google Scholar]
- 50. Kauppi B, Jakob C, Färnegårdh M, et al. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem. 2003;278:22748–22754. [DOI] [PubMed] [Google Scholar]
- 51. Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor β binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27:2266–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoshikawa N, Nagasaki M, Sano M, et al. Ligand-based gene expression profiling reveals novel roles of glucocorticoid receptor in cardiac metabolism. Am J Physiol Endocrinol Metab. 2009;296:E1363–E1373. [DOI] [PubMed] [Google Scholar]
- 53. Handa RJ, Weiser MJ. Gonadal steroid hormones and the hypothalamo-pituitary-adrenal axis. Front Neuroendocrinol. 2014;35:197–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee EE, Nieman LK, Martinez PE, Harsh VL, Rubinow DR, Schmidt PJ. ACTH and cortisol response to Dex/CRH testing in women with and without premenstrual dysphoria during GnRH agonist-induced hypogonadism and ovarian steroid replacement. J Clin Endocrinol Metab. 2012;97:1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]