

Abstract
Wood decay mechanisms in Agaricomycotina have been traditionally separated in two categories termed white and brown rot. Recently the accuracy of such a dichotomy has been questioned. Here, we present the genome sequences of the white rot fungus Cylindrobasidium torrendii and the brown rot fungus Fistulina hepatica both members of Agaricales, combining comparative genomics and wood decay experiments. Cylindrobasidium torrendii is closely related to the white-rot root pathogen Armillaria mellea, while F. hepatica is related to Schizophyllum commune, which has been reported to cause white rot. Our results suggest that C. torrendii and S. commune are intermediate between white-rot and brown-rot fungi, but at the same time they show characteristics of decay that resembles soft rot. Both species cause weak wood decay and degrade all wood components but leave the middle lamella intact. Their gene content related to lignin degradation is reduced, similar to brown-rot fungi, but both have maintained a rich array of genes related to carbohydrate degradation, similar to white-rot fungi. These characteristics appear to have evolved from white-rot ancestors with stronger ligninolytic ability. Fistulina hepatica shows characteristics of brown rot both in terms of wood decay genes found in its genome and the decay that it causes. However, genes related to cellulose degradation are still present, which is a plesiomorphic characteristic shared with its white-rot ancestors. Four wood degradation-related genes, homologs of which are frequently lost in brown-rot fungi, show signs of pseudogenization in the genome of F. hepatica. These results suggest that transition towards a brown rot lifestyle could be an ongoing process in F. hepatica. Our results reinforce the idea that wood decay mechanisms are more diverse than initially thought and that the dichotomous separation of wood decay mechanisms in Agaricomycotina into white rot and brown rot should be revisited.
Keywords: Agaricales, wood decay, white rot, brown rot, reconciliation, pseudogenes, genome sequencing
1. Introduction
The plant cell wall (PCW) is a significant carbon pool in terrestrial ecosystems (Albersheim et al., 2011). Saprotrophic Agaricomycotina exploit this pool as a carbon and energy source, acting as wood or litter decomposers. Wood decomposers follow different strategies of decomposition termed white and brown rot. White-rot fungi cause the degradation of all wood components including the recalcitrant lignin and crystalline cellulose mainly through enzymatic processes (Kersten & Cullen, 2007; Baldrian & Valaskova, 2008). In contrast, brown-rot fungi cause complete degradation of polysaccharides, but only partial degradation of lignin (Blanchette, 1995; Worrall et al, 1997; Niemenmaa et al., 2007; Yelle et al., 2008).
Enzymes implicated in lignin degradation by white-rot fungi include Class II peroxidases (POD), dye-decolorizing peroxidases (DyP) and laccases sensu stricto (Cullen & Kersten, 2004; Martinez et al., 2005; Bourbonnais et al., 1995; Eggert et al., 1996; Eggert et al., 1997; Gronqvist et al., 2005; Liers et al., 2010). Enzymes involved in the degradation of crystalline cellulose by white-rot fungi include mainly cellobiohydrolases (glycoside hydrolases GH6 & GH7) and lytic polysaccharide monooxygenases (LPMO) (Harris et al., 2010). In addition to those enzymes, white-rot fungi employ diverse sets of other carbohydrate active enzymes (CAZY) involved in the degradation of the PCW (Kirk & Cullen, 1998). In brown-rot fungi, polysaccharide degradation takes place through non-enzymatic processes, at least during the initial stages of degradation. Hydroxyl radicals generated through the Fenton reaction have been suggested to be the major agent in non-enzymatic degradation of polysaccharides by brown-rot species (Kirk & Highley, 1973; Illman, 1991).
Recent genome investigations (Martinez et al., 2004; Martinez et al., 2009; Eastwood et al., 2011; Floudas et al., 2012) revealed that white-rot species are enriched in genes related to the degradation of lignin (POD, DyP, laccases s.s.), crystalline cellulose (GH6, GH7, LPMO) and other carbohydrates (GH43, GH74). Furthermore, white-rot species are rich in copies of the cellulose-binding module 1 (CBM1), which facilitates attachment of enzymes to crystalline cellulose (Boraston et al., 2004). In contrast, brown-rot fungi appear to have few or no gene copies in these families and CBM1. It has been suggested that the role of hydroxyl radicals in carbohydrate degradation renders extensive enzymatic lignin and carbohydrate degradation redundant (Worrall et al., 1997). Thus, gene losses accompanied the transitions from a white-rot to a brown-rot lifestyle. Less is known regarding such processes in litter decomposers, but it has been suggested that the latter group causes mostly white rot (Osono, 2007).
The separation of lignicolous Agaricomycotina into white-rot and brown-rot categories could be an oversimplification. Species that do not seem to follow typical brown-rot or white-rot strategies have been noted, for example in the Boletales. Even though the order includes saprotrophic brown-rot species, species of Coniophora and Serpula appear to be able to degrade cellulose in a similar manner to white-rot species (Redhead & Ginns 1985; Nilsson, 1974; Nilsson and Ginns, 1979). In addition, Schizophyllum commune (Agaricales) (Ohm et al., 2010), Jaapia argillacea (Jaapiales) and Botryobasidium botryosum (Cantharellales) (Riley et al., 2014) have reduced numbers of POD, DyP and laccases s.s., similar to brown-rot species, but they are enriched in genes related to the degradation of the PCW carbohydrates, including enzymes involved in the degradation of crystalline cellulose, similar to white-rot species. Schizophyllum commune and B. botryosum have been associated with white rot, but the former species appears to cause only weak wood degradation (Ginns & Lefebvre, 1993; Schmidt & Liese, 1980).
Most studies on wood decay mechanisms have focused on model species such as Rhodonia placenta (Postia placenta, Polyporales), Phanerochaete chrysosporium (Polyporales) and Gloeophyllum trabeum (Gloeophyllales). Less attention has been given to members of Agaricales, except for the genus Pleurotus, which has been mainly studied for its ligninolytic potential (Cerniglia, 1997; Pointing, 2001; Ruiz-Duenas et al., 2007; Faraco et al., 2007).
The Agaricales is a diverse order with more than 13,000 described species (Kirk et al., 2008) that manifest diverse lifestyles, including biotrophs and saprotrophs (Matheny et al., 2006). Saprotrophic Agaricales comprise litter decomposing, coprophilous, humicolous, and lignicolous species. The latter group is mostly associated with white rot (Kaarik, 1965; Worrall et al., 1997). Brown rot is a rare nutritional strategy in Agaricales, associated with the small genera Fistulina, Ossicaulis, and Hypsizygus (Redhead & Ginns, 1985). Ossicaulis and Hypsizygus are members of Lyophylleae and they seem to be closely related (Moncalvo et al., 2002), but Fistulina is an isolated brown-rot genus closely related to Schizophyllum, and the little-known Auriculariopsis and Porodisculus (Ginns, 1997; Binder et al., 2004). Until recently, sequenced genomes of Agaricales species related to PCW degradation included only the cacao pathogen Moniliophthora perniciosa (Mondego et al., 2008), the litter decomposer Coprinopsis cinerea (Stajich et al., 2010) and the lignicolous S. commune (Ohm et al., 2010). This picture has been changing with an increasing number of sequenced Agaricales genomes (Morin et al., 2012; Wawrzyn et al., 2012; Bao et al., 2013; Aylward et al., 2013; Collins et al., 2013; Hess et al., 2014).
Here, we report the newly sequenced draft genomes of the “beefsteak fungus” Fistulina hepatica and Cylindrobasidium torrendii. Both species are members of the Agaricales, but the former species causes brown rot on hardwood (Schwarze et al., 2000a), while the latter species is associated with white rot most frequently on hardwood (Ginns & Lefebvre, 1993). We compare the wood degradation strategies of each species with those of other wood-degrading fungi and we explore the evolution of plant cell-wall degradation strategies in Agaricales based on gene tree/species tree reconciliation analyses.
2. Materials and Methods
2.1 Strain info and nucleic acid extraction
We sequenced the single spore isolates of F. hepatica (ATCC 64428, isolated from a sporophore growing on a Castanea dentata rootstock, North Carolina) and C. torrendii (HHB-15055, ss-10, isolated from an Acer rubrum log, WI, USA, deposited at the Forest Products Laboratory culture collection).
RNA was isolated from F. hepatica by the incubation of liquid nitrogen-ground mycelia from YM agar plates in a CTAB-SDS extraction buffer at 65°C, with sequential LiCl and Na acetate precipitations, DNAase treatment, and a phenol-chloroform extraction. DNA from F. hepatica was isolated from similarly pulverized tissue pretreated with methanol+1% β-mercaptoethanol and lyophilized. The tissue was slurried in TES buffer, incubated with proteinase K, and then heated with a high salt - 0.9% CTAB buffer at 65 °C. The mixture was extracted with phenol/chloroform/isoamyl alcohol, and centrifuged to remove the organic soluble components and debris. Nucleic acids were precipitated with ammonium acetate and then, following an RNase A treatment, DNA was pelleted with isopropanol. High-quality genomic DNA was isolated by passage through Qiagen genomic DNA columns.
Culturing of C. torrendii was done in 0.25 l liquid media of malt extract (20 g/l) and yeast extract (0.5 g/l) at 30 C in darkness. Harvested mycelium was filtered, washed and immediately stored at −80 C until the time of DNA or RNA extraction. Genomic DNA extraction from liquid cultures of C. torrendii was done using Qiagen 500/G tips and following the lysis protocol for tissue in the Qiagen Blood & Cell Culture DNA Kit. RNA extractions were done using the Qiagen RNeasy Midi Kit. The protocol for animal tissue (Qiagen) was followed for isolation of total RNA including on-column DNase digestion.
2.2 Genome and transcriptome sequencing
General aspects of library construction and sequencing can be found at the JGI website http://www.jgi.doe.gov/. The genome of F. hepatica was sequenced using two constructed libraries. A 4 kb library was made from LFPE (ligation-free paired end) mate pair fragments generated using the 5500 SOLiD Mate-Paired Library Construction Kit (SOLiD®). 15μg of genomic DNA was sheared using the Covaris g-TUBE™ (Covaris), and gel size was selected for 4kb. The sheared DNA was end-repaired, and ligated with biotinylated internal linkers. The DNA was circularized using intra-molecular hybridization of the internal linkers. The circularized DNA was then treated with Plasmid-Safe (Epicentre) to remove non-circularized products, and nick-translated and treated with T7 exonuclease and S1 nuclease to yield fragments containing internal linkers with genomic tags on each end. The mate pair fragments were A-tailed and purified using Streptavidin bead selection (Invitrogen). The purified fragments were ligated with Illumina adaptors and amplified using 10 cycles of PCR with Illumina primers (Illumina) to generate the final library. qPCR was used to determine the concentration of the libraries and were sequenced on the Illumina Hiseq.
A 270 bp library was prepared by shearing 1 μg of DNA using the Covaris E210 (Covaris), and size-selected using SPRI beads (Beckman Coulter). The fragments were treated with end-repair, A- tailing, and ligation of Illumina-compatible adapters (IDT, Inc.) using the KAPA-Illumina library creation kit (KAPA biosystems). qPCR was used to determine the concentration of the libraries to be sequenced on the Illumina Hiseq.
The 4 kb and 270 bp libraries of F. hepatica genomic DNA were then sequenced using the Illumina HiSeq platform. An additional sequencing run using PacBio v2 chemistry, 3 kb, 42 SMRT cells provided an additional 1610382 post-filtered reads for H. hepatica.
The genome of C. torrendii was sequenced from a 270 bp fragments library following the same methodology used for the construction of the 270 bp library for F. hepatica.
The transcriptome libraries of both organisms were prepared by purifying 2 μg (5 μg for C. torrendii) of total RNA using Dynabeads® mRNA Purification Kit (Invitrogen) and chemically fragmented to 200–250 bp (Ambion). mRNA was reverse transcribed with SuperScript II using random hexamers. Second Strand cDNA was synthesized using dNTP/dUTP mix (Thermo Scientific), E. coli DNA Ligase, E. coli DNA polymerase I, and E coli RnaseH (Invitrogen). The fragmented cDNA was treated with end-pair, A-tailing, adapter ligation using the TruSeq Sample Preparation Kit (Illumina). Second strand cDNA was removed by AmpErase UNG (Applied Biosystems) to generate strandedness. qPCR was used to determine the concentration of the unamplified libraries. Libraries were sequenced on the Illumina Hiseq.
2.3 Genome assembly and annotation
The F. hepatica genome was assembled with AllPathsLG release version R42328 (Gnerre et al., 2011). PBJelly (English et al., 2012), was then used to fill and reduce gaps by aligning PacBio data to draft assemblies. This resulted in a 137.9 X coverage assembly with 588 scaffolds.
The C. torrendii genome was initially assembled with Velvet (Zerbino, 2010). The resulting assembly was used to create a long mate pair library with insert 3 kb +/− 300 bp, which was then assembled with the original Illumina reads with AllPathsLG release version R42328. This resulted in a 134.3 X coverage assembly with 1149 scaffolds. Additional statistics on both genome assemblies are given in Table S1.
Transcriptome reads for both organisms were assembled into contigs with Rnnotator (Martin et al., 2010) and mapped to genome contigs using BLAT (Kent, 2002). Table S2 summarizes the transcriptome data, and mapping to the genome, for each organism.
Both genomes were annotated using the JGI annotation pipeline (Grigoriev et al., 2006), which combines several gene prediction and functional annotation methods with transcriptome data and integrates the result in Mycocosm (Grigoriev et al., 2014), a web-based resource for fungal comparative genomics. Before gene prediction, assembly scaffolds were masked using RepeatMasker (http://www.repeatmasker.org), RepBase library (Jurka et al., 2005), and frequent (>150 times) repeats were recognized by RepeatScout (Price et el., 2005). The following combination of gene predictors was run on the masked assembly: ab initio Fgenesh (Salamov & Solovyev, 2000) and GeneMark (Ter-Hovhannisyan et al, 2008), homology-based Fgenesh+ (Salamov & Solovyev, 2000) and Genewise (Birney & Durbin, 2000) seeded by BLASTx (Altschul et al., 1990) alignments against NCBI NR database (http://www.ncbi.nlm.nih.gov), and, in the case of C. torrendii, transcriptome-based assemblies. Transcriptome data for F. hepatica, were not used for gene prediction. In addition to protein coding genes for both genomes, tRNAs were predicted using tRNAscan-SE (Lowe & Eddy, 1997). All predicted proteins were functionally annotated using SignalP (Nielsen et al., 1997) for signal sequences; TMHMM (Melen et al., 2003) for transmembrane domains; InterProScan (Quevillon et al., 2005) for integrated collection of functional and structure protein domains; and protein alignments to the NCBI nr, SwissProt (http://www.expasy.org/sprot/), KEGG (Kanehisa et al., 2006), and KOG (Koonin et al., 2004) databases. Interpro and SwissProt hits were used to map gene ontology (GO) terms (Ashburner et al., 2000). For each genomic locus, the best representative gene model was selected based on a combination of protein homology and (in the case of C. torrendii) EST support, which resulted in the final sets of genes analyzed in this work. Table S3 summarizes, for both organisms, the predicted gene sets and support metrics.
2.4 Clustering
For comparative purposes we clustered the predicted protein sequences from F. hepatica and C. torrendii with the predicted proteins from eleven additional genomes of saprotrophic Agaricales and one species of Amylocorticiales (Plicaturopsis crispa, Binder et al., 2010), which served as an outgroup. The clustering was done using the MCL clustering algorithm (Enright et al., 2002) and an inflation parameter of 2.0. Genome sampling included the genomes of Agaricus bisporus var. bisporus (H97) v 2.0 (Agabi), Amanita thiersii Skay4041 v 1.0 (Amath), Armillaria mellea (Armme), C. cinerea (Copci), Galerina marginata v 1.0 (Galma), Gymnopus luxurians v 1.0 (Gymlu), Hypholoma sublateritium v 1.0 (Hypsu), Omphalotus olearius (Ompol), Pleurotus ostreatus PC15 v 2.0 (Pleos), P. crispa v 1.0 (Plicr), S. commune v 2.0 (Schco), and Volvariella volvacea (Volvo) (Ohm et al., 2010; Stajich et al., 2010; Morin et al., 2012; Wawrzyn et al., 2012; Bao et al., 2013; Collins et al., 2013; Riley et al. 2014; Hess et al., 2014; Kohler et al., unpublished data). The 390,268 protein sequences from these organisms were grouped into 32,532 clusters. The results can be browsed at http://genome.jgi.doe.gov/clustering/pages/cluster/clusters.jsf?runId=2610.
2.5 Data assembly of single copy genes and wood-degrading enzymes
We selected twenty-six single-copy genes from a subset of a larger dataset of 71 genes that we have previously used (Floudas et al., 2012) for organismal phylogenetics. We assembled each of the 26 gene datasets by identifying its cluster in the cluster run mentioned in section 2.3 (Table S4). Four of the identified clusters included distantly related paralogs that we separated based on phylogenetic analyses. We also removed the paralogs from potential recent gene duplications in four genes. Two genes were not present in the gene catalog and we retrieved them by performing blastp searches on all predicted models. We replaced fragmented models by complete ones, when this was possible (Table S4).
We also assembled datasets for 33 gene families thought to be involved in various ways in PCW degradation across the 14 genomes, using the same cluster run (Table 1). The oxidative enzymes dataset consists of six gene families (Table 1), four of which are related to degradation of lignin or lignin-like compounds by white-rot fungi (Cullen & Kersten, 2004; Martinez et al., 2005; Bourbonnais et al., 1995; Eggert et al., 1996; Eggert et al., 1997; Gronqvist et al., 2005; Liers et al., 2010; Hofrichter & Ullrich, 2006, Gutierrez et al., 2011). The other two families include copper radical oxidases (CRO) and cellobiose dehydrogenases (CDH), which are involved in production of hydrogen peroxide and hydroxyl radicals respectively (Cullen & Kersten, 2004; Henriksson et al., 2000). Hydrogen peroxide or hydroxyl radical production is accomplished through various pathways in Agaricomycetes (Cullen & Kersten, 2004; Daniel et al., 1994; Guillen et al., 1994; Volc et al, 1996; Daniel et al., 2007, Arantes et al., 2012). However, we included here only datasets for CRO and CDH, which appear to be differentially maintained between white-rot and brown-rot fungi (Floudas et al., 2012). The other twenty-seven gene families are separated into bulk carbohydrate active enzymes (CAZY) and accessory CAZY (De Vries et al., 2010) and show diverse catalytic activity on carbohydrates (Table 1).
Table 1.
Gene families sampled in the study and their proposed functions in wood degradation.
| Gene family | Abbreviation | Activity related to PCW degradation | literature |
|---|---|---|---|
| Class II peroxidases | POD | lignin degradation | Cullen & Kersten, 2004; Martinez et al., 2005 |
| Dye decolorizing peroxidases | DyP | lignin degradation | Liers et al., 2010 |
| Heme-thiolate peroxidases | HTP | potential lignin degradation | Hofrichter & Ullrich, 2006 |
| Multicopper oxidases | MCO | lignin degradation | Kües & Rühl, 2011 |
| Copper radical oxidases | CRO | hydrogen peroxide generation | Cullen & Kersten, 2004 |
| Cellobiose dehydrogenases | CDH | hydroxyl radical generation and iron reduction | Henriksson et al., 2000 |
| Bulk carbohydrate CAZY | GH5-5 | endoglucanase | De Vries et al., 2010 |
| GH5-7 | endomannanase | De Vries et al., 2010 | |
| GH6 | cellobiohydrolase | De Vries et al., 2010 | |
| GH7 | cellobiohydrolase | De Vries et al., 2010 | |
| LPMO (GH61) | monoxygenase activity on cellulose | Harris et al., 2010 | |
| GH10 | endoxylanase | De Vries et al., 2010 | |
| GH11 | endoxylanase | De Vries et al., 2010 | |
| GH12 | endoglucanase | De Vries et al., 2010 | |
| GH28 | pectinase activity | Marcovic & Janecek, 2001 | |
| GH45 | endoglucanase | De Vries et al., 2010 | |
| GH74 | xyloglucanase | De Vries et al., 2010 | |
| Accessory CAZY | GH1 | β-mannosidase/β-glucosidase | De Vries et al., 2010 |
| GH2 | β-mannosidase | De Vries et al., 2010 | |
| GH3 | β-glucosidase/β-xylosidase | De Vries et al., 2010 | |
| GH27 | α-galactosidase | De Vries et al., 2010 | |
| GH29 | α-fucosidase | De Vries et al., 2010 | |
| GH35 | β-galactosidase | De Vries et al., 2010 | |
| GH43 | α-arainofuranosidase/β-xylosidase | De Vries et al., 2010 | |
| GH51 | α-arabinofuranosidase | De Vries et al., 2010 | |
| GH95 | α-fucosidase | De Vries et al., 2010 | |
| GH115 | α-glucuronidase | De Vries et al., 2010 | |
| CE1 | acetyl-xylan-esterase, ferruloyl esterase, cinnamoyl esterase | Crepin et al., 2003; Kroon et al., 2000 | |
| CE5 | cutinase | Rubio et al., 2008 | |
| CE8 | pectin methylesterase | Marcovic & Janecek, 2004 | |
| CE12 | acetylesterase | Molgaard et al., 2000 | |
| CE15 | 4-O-methyl-glucuronoyl methylesterase | Li et al., 2007 | |
| CE16 | acetyl-xylan-esterase, ferruloyl esterase, cinnamoyl esterase | Li et al., 2008 |
Each dataset was assembled using the JGI cluster run mentioned above, and the use of InterPro and PFAM domains (Table S5). We also identified the CBM1-containing gene copies by searching for the corresponding PFAM domain PF00734 (Table 2). We used proteins annotated by CAZYbase (Lombard et al., 2014, Kohler et al., unpublished data) to identify gene families without a specific PFAM or InterPro domain, and to verify the recovered gene numbers for all annotated genomes. For a subset of 15 gene families and the CBM1, we obtained data from 18 additional Agaricomycotina genomes from previous studies (Floudas et al., 2012; Riley et al., 2014). We replaced low quality models by improved ones found on the genome browser of each genome or otherwise we excluded them from the datasets (Table S6). We subclassified MCO and CRO based on characterized sequences and preliminary phylogenetic analyses (Table S7). We also subclassified POD into manganese peroxidases (MnP), versatile peroxidases (VP), lignin peroxidases (LiP), generic peroxidases (GP) and also the atypical MnP and VP (Table S7), based on the completeness of the manganese binding site and the presence of the long range electron transfer tryptophan, as we have previously done (Floudas et al., 2012). Finally, we separated the MnP into the short and long/extra long types based on preliminary phylogenetic analyses (data not shown) with other previously characterized sequences (Floudas et al., 2012).
Table 2.
Gene copy numbers across 33 gene families related to wood degradation, and CBM1 copies found across 13 Agaricales genomes and P. crispa. Absence of gene copies for a gene family is highlighted in grey. For an explanation of the species acronyms, see the materials and methods.
| Fishe | Schco | Cylto | Gymlu | Ompol | Armme | Galma | Hypsu | Pleos | Plicr | Copci | Agabi | Amath | Volvo | total copies | average number of copies | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POD | 0 | 0 | 0 | 5 | 5 | 10 | 23 | 14 | 9 | 7 | 1 | 2 | 0 | 7 | 83 | 6 |
| DyP | 0 | 0 | 0 | 12 | 1 | 4 | 5 | 2 | 4 | 0 | 4 | 0 | 1 | 3 | 36 | 3 |
| HTP | 3 | 3 | 5 | 19 | 8 | 6 | 24 | 13 | 4 | 3 | 8 | 24 | 4 | 3 | 127 | 9 |
| Lac | 3 | 2 | 3 | 16 | 6 | 23 | 8 | 12 | 11 | 5 | 17 | 12 | 15 | 11 | 144 | 10 |
| GLX | 0 | 0 | 0 | 5 | 2 | 0 | 4 | 3 | 4 | 0 | 0 | 3 | 2 | 0 | 23 | 2 |
| CDH | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 16 | 1 |
| total oxidative enzymes per species | 7 | 6 | 10 | 58 | 23 | 45 | 65 | 45 | 33 | 16 | 31 | 42 | 23 | 25 | 429 | 31 |
| GH6 | 0 | 1 | 3 | 1 | 1 | 2 | 3 | 1 | 3 | 2 | 5 | 1 | 1 | 5 | 29 | 2 |
| GH7 | 4 | 2 | 5 | 7 | 4 | 4 | 8 | 4 | 16 | 1 | 6 | 1 | 1 | 11 | 74 | 5 |
| LPMO | 10 | 22 | 26 | 13 | 7 | 19 | 19 | 14 | 29 | 9 | 35 | 11 | 16 | 28 | 258 | 18 |
| GH10 | 2 | 5 | 3 | 5 | 4 | 6 | 8 | 7 | 3 | 2 | 5 | 2 | 4 | 18 | 74 | 5 |
| GH11 | 0 | 1 | 1 | 4 | 0 | 2 | 8 | 2 | 2 | 0 | 6 | 2 | 0 | 0 | 28 | 2 |
| GH12 | 2 | 1 | 4 | 3 | 1 | 3 | 4 | 1 | 2 | 2 | 1 | 2 | 3 | 2 | 31 | 2 |
| GH5_5 | 1 | 2 | 3 | 4 | 4 | 1 | 7 | 6 | 4 | 2 | 1 | 3 | 3 | 1 | 42 | 3 |
| GH5_7 | 0 | 1 | 1 | 5 | 2 | 3 | 7 | 2 | 4 | 2 | 3 | 1 | 1 | 1 | 33 | 2 |
| GH28 | 6 | 3 | 9 | 19 | 7 | 21 | 19 | 7 | 6 | 10 | 3 | 5 | 5 | 3 | 123 | 9 |
| GH74 | 0 | 1 | 2 | 1 | 1 | 2 | 2 | 1 | 3 | 1 | 1 | 1 | 1 | 1 | 18 | 1 |
| GH45 | 1 | 1 | 2 | 2 | 2 | 3 | 2 | 1 | 2 | 1 | 0 | 1 | 2 | 1 | 21 | 2 |
| total bulk CAZY per species | 26 | 40 | 59 | 64 | 33 | 66 | 87 | 46 | 74 | 32 | 66 | 30 | 37 | 71 | 731 | 52 |
| GH1 | 1 | 3 | 1 | 3 | 3 | 8 | 5 | 3 | 3 | 3 | 2 | 1 | 3 | 3 | 42 | 3 |
| GH2 | 1 | 4 | 3 | 4 | 4 | 2 | 3 | 3 | 3 | 5 | 2 | 2 | 2 | 2 | 40 | 3 |
| GH3 | 14 | 12 | 7 | 17 | 10 | 13 | 11 | 9 | 11 | 10 | 7 | 7 | 10 | 9 | 147 | 11 |
| GH27 | 4 | 1 | 9 | 6 | 3 | 6 | 8 | 6 | 7 | 2 | 0 | 4 | 4 | 2 | 62 | 4 |
| GH29 | 1 | 2 | 0 | 1 | 1 | 3 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 12 | 1 |
| GH35 | 3 | 4 | 2 | 5 | 5 | 8 | 10 | 4 | 5 | 2 | 0 | 1 | 4 | 4 | 57 | 4 |
| GH43 | 2 | 19 | 19 | 11 | 3 | 7 | 6 | 3 | 8 | 3 | 4 | 3 | 6 | 14 | 108 | 8 |
| GH51 | 1 | 2 | 2 | 5 | 2 | 3 | 5 | 2 | 3 | 2 | 1 | 1 | 1 | 3 | 33 | 2 |
| GH95 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 17 | 1 |
| GH115 | 4 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 3 | 3 | 23 | 2 |
| CE1 | 0 | 11 | 6 | 5 | 3 | 1 | 2 | 3 | 2 | 4 | 3 | 1 | 1 | 4 | 46 | 3 |
| CE5 | 0 | 2 | 2 | 3 | 0 | 0 | 6 | 2 | 0 | 0 | 6 | 6 | 2 | 1 | 30 | 2 |
| CE8 | 2 | 2 | 3 | 6 | 3 | 8 | 3 | 4 | 2 | 3 | 0 | 2 | 2 | 3 | 43 | 3 |
| CE12 | 0 | 2 | 5 | 3 | 1 | 1 | 4 | 3 | 2 | 0 | 1 | 2 | 3 | 1 | 28 | 2 |
| CE15 | 0 | 2 | 1 | 2 | 1 | 4 | 1 | 2 | 1 | 1 | 8 | 1 | 1 | 1 | 26 | 2 |
| CE16 | 7 | 11 | 12 | 13 | 4 | 11 | 10 | 7 | 10 | 11 | 5 | 11 | 4 | 7 | 123 | 9 |
| total accessory CAZY per species | 41 | 81 | 75 | 86 | 45 | 77 | 78 | 53 | 59 | 50 | 40 | 46 | 48 | 58 | 837 | 60 |
| toal copies per species | 74 | 127 | 144 | 208 | 101 | 188 | 230 | 144 | 166 | 98 | 137 | 118 | 108 | 154 | 1997 | 143 |
| CBM1 | 0 | 5 | 0 | 32 | 15 | 10 | 51 | 28 | 31 | 15 | 44 | 13 | 10 | 51 | 305 | 22 |
Only complete CDH (containing both the GMC and cytochrome domains) genes are reported.
2.6 Alignments and phylogenetics
We aligned all the datasets using the online version of PRANK (http://www.ebi.ac.uk/goldman-srv/webprank/) with the default settings (Löytynoja & Goldman, 2010). We removed poorly aligned areas of the alignments for each of the 26 datasets for the organismal phylogeny using Gblocks v. 0.91b (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) with less stringent settings (Castresana, 2000). We manually examined and removed poorly aligned areas of the alignments of wood-degrading enzymes datasets using MacClade v.4.08 (Maddison and Maddison 2002). Maximum likelihood (ML) analyses were performed for each alignment using RAxML v. 7.6.6 (Stamatakis et al., 2008) under the GTR model with CAT distributed rate heterogeneity and the WAG substitution matrix with 500 rapid bootstrap replicates (200 replicates for ML analyses of wood degradation enzymes datasets). Bayesian analyses were performed using MrBayes 3.2.2 (Ronquist et al., 2012) for seven million generations, with four chains and sampling every 1000 generations. The burn-in proportion was set to 0.25, which was found to be adequate after examining the likelihood scores using Tracer v1.5 (http://tree.bio.ed.ac.uk/software/tracer/). All phylogenetic analyses were performed at Cipres (Miller et al., 2010; http://www.phylo.org/index.php/portal/).
2.7 Species tree/Gene tree reconciliation
We estimated the number of gene copies for each gene family related to wood degradation at the ancestral nodes of the organismal phylogeny using Notung (Durand et al., 2006). Midpoint rooting was used to root gene trees prior to reconciliation. Reconciliation analyses were performed using the default cost of duplications and losses and the edge weight threshold was set to 90.
2.8 Wood decay experiments
Studies used to determine wood decay mechanisms by Cylindrobasidium torrendii, Fistulina hepatica or Schizophyllum commune were set up using 10 × 10 × 1 mm wood wafers of aspen (Populus sp.). Fifteen wafers were used for each isolate and each time point. Following determination of oven dry weight, wafers were hydrated to 80–100% and sterilized in an autoclave for 60 minutes at 120°C. Wafers were then placed on mycelium growing on 2% malt yeast extract agar (15 g malt extract, 2 g yeast extract, 15 g agar, 1000 ml water). After 45 and 90 days, 12 wafers were removed and dried to determine mass loss and 3 wafers were frozen at −20° C for microscopy. Micromorphological characteristics were described using scanning electron microscopy methods as previously described (Blanchette et al. 2010). Samples were examined and photographed using a Hitachi S3500 N (Hitachi, Tokyo, Japan) scanning electron microscope.
3. Results
3.1 Gene copies of wood-degrading enzymes in Agaricales and P. crispa
We collected in total 1997 protein models from 14 genomes, which can be separated into 429 oxidative enzymes, 731 bulk CAZYs, and 837 accessory CAZYs (Table 2). Fistulina hepatica has 74 copies across only 22 gene families, which is the smallest number of copies seen in the Agaricales, while C. torrendii has 144 copies across 29 gene families, which is close to the average number of copies across the 14 genomes. Fistulina hepatica and C. torrendii are the only species of the 14 genomes dataset that lack a CBM1.
The POD, MCO and CRO were subclassified recognizing 6 categories of genes for POD and 5 categories for both MCO and CRO (Table S7). PODs represent mostly different types of manganese peroxidases (MnP), while 7 genes were fragments and could not be assigned to any type. MCO are dominated by laccases s.s. (LAC s.s.), while other types of MCOs such as laccase-like genes (LAC-like), L-ascorbate oxidases, and melanin synthesis related genes (MS) are found only in marasmioid species. Glyoxal oxidase (GLX) is the only category of CRO that has scattered representation in our dataset. We reconciled laccases s.s and GLX within the MCO and CRO respectively. The reason for that choice was based on the role of these subsets of enzymes during wood degradation (Kersten & Kirk, 1987; Kües & Rühl, 2011). Laccase-like enzymes appear to play a similar role to laccases s.s. for some species (Rodriguez-Rincon et al., 2010) but they were excluded from reconciliation because of their scarce presence in the dataset.
3.2 Organismal phylogeny and reconciliation results using Notung
We performed gene tree/species tree reconciliation analyses using the species tree we generated based on the dataset of 26 single copy genes (Table S4). The final concatenated alignment included 21167 amino acid characters after exclusion of the poorly aligned regions. The resulting phylogenetic trees from ML and Bayesian analyses have identical, fully resolved topology (Fig. 1). Our results largely agree with a previous study in Agaricales based on five genes (Matheny et al., 2006). Fistulina hepatica is closely related to S. commune, as has been shown before (Matheny et al., 2006; Binder et al., 2004), while C. torrendii appears to be related to the white-rot A. mellea. Both F. hepatica and C. torrendii belong in the Marasmioid clade, which includes here six species.
Figure 1.
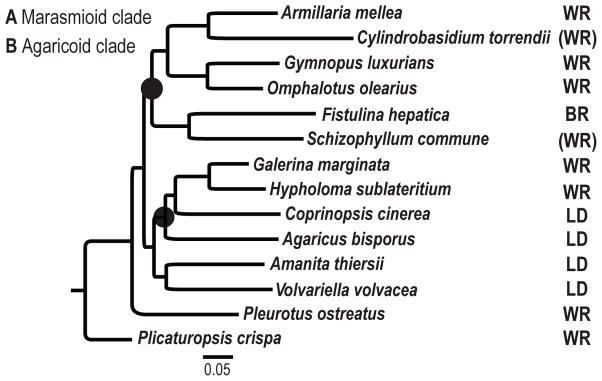
Species phylogeny of 13 Agaricales species and P. crispa (Amylocorticiales) as outgroup. Both ML and Bayesian analyses of 21167 amino acid characters from 26 single copy genes resulted in identical topologies and received maximum bootstrap and posterior probability support at all nodes. The ML tree is shown here. WR, white rot; BR, brown rot; LD, litter decomposer; (WR), reported as white rot, but wood decay strategy is uncertain.
The common ancestor of Agaricales is estimated to have had 21 copies of oxidative enzymes (Fig. 2a). Seven of these copies represent PODs (Fig. S1). The ancestor is also suggested to have had a rich repertoire of CAZY (61 bulk and 73 accessory enzymes, Fig. 2b and 2c). Among the largest CAZY families suggested to have been present are the LPMO, GH28, and GH43 (Fig. S2a and S3a), while 3 and 2 copies of GH6 and GH7 were present respectively (Fig. S2a). In comparison to the ancestor of Agaricales, both F. hepatica and C. torrendii have reductions for oxidative enzymes (Fig. 2). These reductions are mainly related to POD and DyP (Fig. S1). Fistulina hepatica has additional reductions for bulk and accessory CAZY (Figs. 2b, 2c) especially related to LPMOs and GH43 genes (Figs. S2, S3a, S5, S6), while C. torrendii has maintained CAZY copy numbers similar to the Agaricales ancestor (Fig. 2b and 2c).
Figure 2.

Species tree/gene tree reconciliation results. Summed reconciliation results of oxidative enzymes related to lignin degradation (a), bulk carbohydrate CAZY (b), and accessory CAZY (c). Numbers at the tips represent the summed number of copies for the corresponding category of gene families in the genome of each species. Numbers at internal nodes represent the predicted summed number of copies for the corresponding category of gene families for each ancestral species. The size of the circles is proportional to these numbers (shaded in dark green for the common ancestor of Agaricales). Nutritional strategies are coded as in Figure 1.
3.3 Comparison of Agaricales with brown-rot and white-rot species from other orders
To place the PCW degradation machineries of the 13 Agaricales genomes in a broader context we compared them with 18 genomes from 11 orders across Agaricomycotina. Eight of the genomes belong to white-rot species, while eight genomes represent brown-rot species from 4 independently evolved brown-rot lineages. We focused on the CBM1 and a subset of 15 families of the 33 gene-families dataset (Fig. 3). White-rot species possess 46 to 118 gene copies in eleven to fifteen gene families. At the same time, brown-rot species possess only 10 to 50 copies in four to twelve gene families (Fig. 3). The litter decomposers A. bisporus and A. thiersii are intermediate between white-rot and brown-rot species, while V. volvacea and C. cinerea have gene repertoires similar to typical white-rot species.
Figure 3.

Copy numbers for fifteen gene families and CBM1 across 32 Agaricomycotina genomes. The columns on the right side of the table represent the summed number of genes for the fifteen gene families. Species on the table have been grouped in three categories; brown-rot (brown), litter decomposers (grey), white-rot (yellow) or uncertain type of rot (orange). Within each category, the species have been arranged based on the total number of gene copies they have. White-rot and uncertain type of rot have been grouped together for this purpose. Light blue indicates copy number below or equal to the average number of copies for the gene family, while dark blue indicates copy number above the average number of copies for the gene family. * One potential pseudogene is found for each of these gene families on the genome of F. hepatica. Data from: **Floudas et al., 2012, *** Kohler et al., unpublished data.
3.4 Pseudogenization of genes related to wood degradation in F. hepatica
Four decay-related pseudogenes were detected in F. hepatica. Three are represented in the gene catalog by protein models Fishe 57906 (DyP), Fishe 73885 (GH74 xyloglucanase), and Fishe 71082 (GH5-7 endomannanase). The fourth pseudogene was identified during a manual search of all the predicted models for cellobiohydrolase GH6 genes using the identifier PF01341. One to two predicted protein models represent each one of the four loci (except for the GH6 locus, which is represented by 5 predicted protein models). All predicted models are either fused with an adjacent gene, which is a gene prediction artifact due to the incomplete reading frame of the gene, or represent fragments (Fig S4a–b). Additionally, the automated functional annotation for DyP and GH5-7 failed to recognize the expected domains IPR006314 and IPR001547, respectively. Phylogenetic analyses of all the predicted models from the four putative pseudogenes with homologs from other genomes that the F. hepatica genes are on long branches, suggesting accumulation of many amino acid changes on the predicted proteins (Figs. 4, S7a–S7c).
Figure 4.

Phylogenetic relationships of the five predicted models of the potential GH6 pseudogene from F. hepatica with homologs from the 14 genomes showing the resulting long branch (in red color) and comparison with similar analyses of the adjacent genes. Numbers on the branches represent branch length. The scaffold graph shows the orientation of each potential pseudogene with its adjacent genes. Red dots for GH6 models of F. hepatica indicate models interrupted by stop codons. The protein models that represent the product of the adjacent genes are shown in blue on their corresponding phylogeny.
To assess whether the inferred pseudogenes could be artifacts resulting from poor assembly quality, we inspected alignments of the Illumina read data to the assembled consensus produced by AllPathsLG R42328 and called variant bases using SAMtools 1.19. Although the scaffolds harboring the pseudogenes contained varying numbers of SNPs (scaffold_92 336 SNPs; scaffold_142 159 SNPs; scaffold_272 99 SNPs; scaffold_437 8 SNPs), none of these SNPs lay within the boundaries of any of the four proposed pseudogenes, suggesting that the genome assembly is of high quality in the relevant regions.
Additionally, we examined genes upstream and downstream of each of the four loci. We generated phylogenetic trees from the flanking genes and their homologs in the 13 other genomes. Seven of the 8 genes adjacent to the four potential pseudogenes on the genome of F. hepatica do not result in long branches (Figs. 4, S7a–S7c), suggesting good quality sequencing at these areas of the genome. Model Fishe1 43738 (upstream of xyloglucanase Fishe1 73885) is the only gene placed on a longer branch and it is coupled with model Schco2 1215620, which is also on a long branch (Fig. S7a). The fragmented predicted models of the four loci, combined with the good quality of the assembly of the genome in these areas and the good quality of the predicted models for adjacent genes suggest that the four loci represent pseudogenes. However, additional experimental data are needed to verify that the four loci represent pseudogenes.
3.5 Wood decay by F. hepatica, C. torrendii and S. commune
Wood colonized in the laboratory by Cylindrobasidium torrendii, Fistulina hepatica or Schizophyllum commune was examined using scanning electron microscopy. After 45 days of colonization, all three fungi did not cause appreciable decay alteration of the wood cell walls, but after 90 days evidence of cell wall attack was observed. The wood substrate had relatively small amounts of biomass lost corresponding to 17.8% 2.3% and 7.2% for C. torrendii, F. hepatica and S. commune, respectively, after 90 days.
Transverse sections of wood decayed by C. torrendii after 90 days, showed a pattern of cell wall attack that was typical for white rot fungi that cause a simultaneous degradation of all cell wall components (Figs. 5A, 5B). In localized areas of the wood, fibers and vessels had eroded secondary cell walls. As the fungus removed the secondary wall, the middle lamella became weak, cells separated and voids in the wood cells were formed. This attack, however, was limited and occurred in some cells, while adjacent cells remained unaltered. Degradation by S. commune after 90 days, presented for comparison with C. torrendii, also appeared to be a white rot type of cell wall degradation (Figs. 5E, 5F). The secondary walls were eroded and thinned leaving the middle lamella intact in most cells. Some breakage of the residual middle lamella was evident in a few cells causing voids to be seen in the wood but in most cell walls the middle lamella remained in areas that were degraded (Fig. 5F).
Figure 5.

Scanning electron micrographs of transverse sections of aspen (Populus) wood decay by Cylindrobasidium torrendii (A and B), Fistulina hepatica (C and D) and Schizophyllum commune (E and F). A and B. Localized degradation of all cell wall components with erosion of the wall taking place from the cell lumen towards the middle lamella. Small voids occurred in the wood cells where all cell wall layers were degraded. C and D. A diffuse attack on wood cells resulted in cells with altered walls. No cell wall erosion took place but walls were slightly swollen and cells were partially collapsed and appeared convoluted. E and F. Thinning and eroded secondary wall layers were evident in wood cells. In some cells, the secondary wall was completely degraded but the middle lamella between cells remained. The thinned cell wall broke and detached in some areas resulting in small voids. Bar = 100 μm in A, 20 μm in B and 50 μm in C, D, E, F.
Decay by F. hepatica was evident in wood cells near the surface of the wood wafers after 90 days. Decay observed had an appearance of a typical brown rot with cell walls that displayed a diffuse attack resulting in slightly swollen secondary walls and a loss of cell wall integrity (Figs. 5C, 5D). The weakened fiber cell walls lost rigidity and assumed convoluted shapes.
4. Discussion
4.1 The common ancestor of Agaricales had similar types of wood decay genes with those seen in extant white-rot Agaricales species
The ancestor of Agaricales is estimated to have had genes from all 6 oxidative gene families examined here, including 7 POD and 3 DyP (Fig. S1), which is similar to white-rot species of Agaricales, even though the overall number of oxidative enzymes is lower than that of some extant white-rot Agaricales such as G. marginata or G. luxurians (Fig. 2). Additionally, the reconstructed 19 LPMOs, GH6 and GH7 cellobiohydrolases (Fig. S2a) and CDH (Fig. S1) suggests the presence of a rich system for utilization of crystalline cellulose and cellobiose. Taken together these results suggest that gene networks related to white-rot wood decay are plesiomorphic in Agaricales, as in the Agaricomycotina as a whole (Floudas et al., 2012).
4.2 Plant cell-wall decomposition similarities between litter decomposers and white-rot species
Litter decomposers in Agaricales (A. bisporus, A. thiersii, C. cinerea and V. volvacea) have maintained the plesiomorphic enzymatic degradation of cellulose and other large carbohydrates. This is shown by the presence of complete enzymatic systems for cellulose degradation (GH6, GH7, LPMO) and the diverse set of CAZYs involved in hemicellulose degradation (Table 1) similar to the Agaricales ancestor (Fig. 2b and 2c) and to white-rot species from other orders (Fig. 3).
The picture of lignin degradation is more complex among litter decomposers in Agaricales. Volvariella volvacea has a complete system of lignin-degrading enzymes including POD, DyP and laccases s.s. However, A. thiersii, A. bisporus and C. cinerea lack or have reduced numbers of POD or DyP, suggesting weaker ability for lignin degradation. The numbers of copies of shared oxidative gene families among litter decomposers show variation. Volvariella volvacea has 5 or 6 ligninolytic PODs (Table S7), but has only 3 HTPs, while A. bisporus has 24 HTPs, but only two PODs. Laccases s.s. are represented by abundant copies in all litter decomposers, suggesting an important role in their lifestyle (Theuerl & Buscot, 2010).
Lignin concentration increases from the upper towards the lower layers of the soil, but in addition its structure changes as result of decomposition (Osono, 2007; Osono et al., 2008). The differences in types and copy numbers of lignin degrading enzymes present in litter decomposers could be connected to the diverse microenvironments found in the soil that provide different forms and amounts of recalcitrant carbon.
The shared gene content for the enzymatic degradation of lignin, cellulose and other carbohydrates between litter decomposers and white-rot Agaricales suggests that transitions between the two nutritional strategies are possible across Agaricales. Volvariella volvacea is the litter decomposer in the dataset closest to white-rot species regarding its wood-degrading apparatus. In agreement with this observation, it has been suggested that the transition from a litter decomposing towards a lignicolous white-rot lifestyle has happened twice in the genus (Justo et al., 2010). Additionally, G. luxurians appears to be one of the richest in PCW-degrading enzymes of the white-rot species in this dataset and is nested within a clade that contains both white-rot species and litter decomposers (Mata et al., 2004; Arnolds, 1995).
4.4 Cylindrobasidium torrendii and S. commune do not fit in the white-rot/brown-rot dichotomy
The wood-degrading apparatus of C. torrendii shows similarities to that of S. commune. Both species carry a complete set of enzymes for the enzymatic degradation of crystalline cellulose (GH6, GH7, LPMO), including large number of LPMO copies (Table 2, Fig. 3) and they have rich repertoires of other CAZY enzymes (Table 2, Fig. 3, Ohm et al., 2010). These characteristics may be plesiomorphic and indicate similarities of S. commune and C. torrendii with white-rot fungi and the common ancestor of Agaricales (Fig. 2). In spite of the rich CAZY content seen for both species, CBM1 copies are absent (C. torrendii) or very few are present (S. commune). In addition, both species have reduced ligninolytic gene content (Table 2). The reduced ligninolytic gene content for the two species shows similarities to brown-rot fungi (Fig. 3) and appears to be an apomorphic characteristic that has independently evolved in the two lineages from ancestors with more diverse repertoire of ligninolytic enzymes (Figs. 2, S1). The gene content of both species related to wood degradation places them in an intermediate position between white rot and brown rot species.
Microscopy, especially scanning electron microscopy, can provide a precise characterization for the type of decay present (Eriksson et al., 1990). The decay caused by C. torrendii and S. commune, appeared to be a simultaneous white rot causing degradation of all cell wall components. The secondary wall was attacked and the erosion of the wall progressed from the lumen toward the middle lamella. In some cells, the secondary wall had been completely degraded, but the middle lamella remained. The middle lamella between cells was detached or degraded in some areas. This may be due to a very localized attack that destroyed this region of the middle lamella or from the weakened condition of the thinned cell wall that remained. This caused small voids in the wood as cells separated. There appeared to be limited effect on the middle lamellae as compared to results from other studies of degradation patterns produced by different species of white rot fungi. The overall pattern of decay appeared more similar to a Type II form of soft rot where in advanced stages of degradation entire secondary walls are completely degraded but the middle lamella is not (Eriksson et al. 1990). As has been found with other white rot fungi, the type and amount of lignin within cell walls can influence how white rot fungi can attack certain types of cells (Blanchette et al. 1988).
The ability of C. torrendii to decay wood has not been studied previously. More information is available for Cylindrobasidium laeve (syn. Corticium laeve), a closely related species to C. torrendii. Both C. laeve and S. commune have been grouped with brown-rot fungi in oxidative enzymes tests (Kaarik, 1965). However, both species of Cylindrobasidium and S. commune have been associated with white rot (Ginns & Lefebvre, 1993). Schizophyllum commune does not seem to cause extensive wood degradation (Schmidt & Liese, 1980), and it has been shown to have a preference for degrading ray parenchyma cells with other cells such as fibers and fiber tracheids being more resistant to attack (Padhiar and Albert 2011). Additionally, wood decay by Cylindrobasidium laeve (syn. Corticium laeve), was shown to resemble soft rot showing similarities to wood degradation caused by Fusarium (Henningsson, 1967). The idea of soft rot caused by basidiomycetes has been suggested more recently as well (Schwarze et al., 2000b).
The decay mechanisms of C. torrendii and S. commune resemble those of J. argillacea (Jaapiales) and B. botryosum (Cantharellales), which are described by Riley et al. (2014). All four species cause weak and localized wood decay that resembles white rot. At the same time they share the reduced ligninolytic gene content, typical of brown rot species, but have enriched CAZY gene content related to carbohydrate degradation, which is usually characteristic of white rot fungi.
The phylogenetic placement of the four species and the reconciliation results for Agaricales suggest that this mode of decay has evolved multiple times across Agaricomycotina from white-rot ancestors through losses of their lignin decay related genes. In agreement with their intermediate wood decay characteristics, S. commune and J. argillacea are placed in areas where transitions from white-rot to brown-rot could have taken place such as the lineage leading towards F. hepatica and the lineage leading towards the Gloeophyllales respectively (Fig. 1; Riley et al., 2014).
The reasons behind these intermediate characteristics and how they are related to the species biology are largely unknown. A possible explanation could be that some of these species act along with other wood degraders or they take advantage of the presence of efficient wood decayers at the same substrate. Fruitbodies of S. commune frequently appear with fruitbodies of other basidiomycetes on wood (Essig 1922, personal observations) and the species can act as destructive mycoparasite on other fungi (over 50 species) of different phyla (Tzean and Estey 1978). Alternatively, some of these species may act as plant parasites that rely selectively on living tissues of the plant stem such as the sap or the bark of living trees (Takemoto et al., 2010). Our results suggest that wood degradation strategies in Agaricomycotina as traditionally viewed should be revisited, as the potential exists that such strategies could be more diverse than previously thought and highlight the need for more functional studies of wood degradation strategies (Ohm et al., 2014).
4.5 Fistulina hepatica and brown-rot Boletales still possesses complete or partial systems for the enzymatic degradation of crystalline cellulose
Our results confirm the placement of F. hepatica as an isolated brown-rot lineage in the Marasmioid clade (Fig. 1) related to S. commune (Binder et al., 2004; Binder et al., 2010). The wood-degrading apparatus of F. hepatica is reduced compared to those of other PCW decomposing Agaricales (Table 2). Fistulina hepatica has the smallest sets of oxidative enzymes and bulk CAZYs and among the smallest sets of accessory CAZYs. The types of enzymes missing largely agree with what has been shown for other brown-rot fungi (Fig. 3, Floudas et al., 2012; Martinez et al., 2009). The major similarities include the reduced enzymatic content related to lignin (POD, DyP, GLX) and bulk carbohydrates degradation such as crystalline cellulose (GH6, LPMO, CBM1).
Despite the overall similarity of the gene content related to wood degradation among brown-rot fungi, differences exist. Sequenced species in Polyporales, Gloeophyllales and Dacrymycetales lack GH6 and GH7 cellobiohydrolases and they have few copies of LPMOs. This suggests that they largely lack the ability to enzymatically degrade crystalline cellulose, even though GH5 processive endoglucanases could degrade crystalline cellulose in some of those species (Cohen et al., 2005; Yoon et al., 2008). Therefore, these species represent typical brown rotters. However, F. hepatica and saprotrophic members of the Boletales harbor complete (H. pinastri and C. puteana) or partial (F. hepatica and S. lacrymans) sets of cellobiohydrolases, CDH, intermediate numbers of LPMO genes (except for S. lacrymans), and in the case of H. pinastri increased CBM1 copies. These results suggest that F. hepatica and members of the Boletales still possess genes related to the degradation of cellulose, similar to the white-rot fungi from which brown-rot fungi have been suggested to have evolved (Floudas et al., 2012).
The ability of members of Boletales to degrade cellulose has been shown before (Nilsson, 1974; Nilsson and Ginns, 1979; Schmidhalter & Canevascini, 1993), while some Boletales have been shown to produce weaker iron-reducing potential on wood in comparison with brown-rot species from other lineages, similarly to white-rot species (Goodell et al., 2006). Less is known about the wood decay strategy of F. hepatica. In agreement with its reduced ligninolytic gene content, F. hepatica caused brown rot in the wood decay experiments. However, wood decay was limited with 2.3% loss observed after 90 days. The limited weight loss indicates that any degradation observed would be restricted to localized areas of the wood. Small number of decayed cells were observed. Previous investigations with F. hepatica indicate that this fungus can readily colonize wood and impart a brownish stain but biomass loss is minimal (Schwarze et al. 2000a). In a study of wood artificially inoculated in the laboratory, only 1.2% weight loss was observed after 6 months and 4.1% loss after 18 months (Schwarze et al. 2000a). This reduced capacity for decaying wood as compared to other brown rot fungi is the likely reason that no appreciable loss of strength is associated with decay by Fistulina in wood affected in natural environments (Schwarze et al. 2000a). The limited amount of decay and its localization within wood caused by F. hepatica suggests that this type of brown rot is different from that produced by other brown rot fungi with only small zones of cells being attacked while adjacent cells remain unaltered. Additionally, it raises the question whether F. hepatica makes any use of the cellulose degradation related genes and under what conditions.
4.6 Gene losses and pseudogenization of GH5-7, GH6, GH74, and DyP genes in F. hepatica could be associated with transition to brown rot
The smaller content of wood-degrading enzymes seen for F. hepatica is suggested to be the result of gene losses. These losses are associated with transition from a potential white-rot towards a brown-rot lifestyle, as has been suggested for other brown-rot lineages (Martinez et al., 2009; Floudas et al., 2012). In addition, our results suggest that this transition could have taken place in two stages. The first stage consists of a shared reduction of oxidative enzymes in the common ancestor of S. commune and F. hepatica. The last common ancestor of the Marasmioid clade is suggested to have had 23 copies of enzymes from the six gene families similar to extant white-rot species (Fig. 2). In contrast, the common ancestor of S. commune and F. hepatica is suggested to have lost 16 members of those gene families, including all PODs and DyPs (Fig. S1). This ancestor appears to be more similar to S. commune in its overall wood-degrading enzymes diversity (Fig. 2). The second stage might have taken place in the lineage leading to F. hepatica and included mainly losses of CAZYs. During this second step 16 and 28 gene losses might have taken place for the bulk and accessory CAZYs (Figs. 2b–c), which represent 38% and 40% of the CAZYs present at the ancestral species respectively. The most extensive gene losses were inferred for GH61, GH43, and CE1 (Figs. S2a, S3a, S5, S6), similar to other brown-rot lineages (Floudas et al., 2012).
Our results support the presence of four wood decay-related genes with signs of pseudogenization in the genome of F. hepatica, but it is known if these genes are still functional. Low quality predicted genes or potential pseudogenes are not rare especially for the draft version of sequenced genomes (Table S6). However, the original genes appear to have been members of the DyP, GH6, GH74 and GH5-7 families. The first 3 gene families are frequently absent or are represented in low copies in brown-rot species (Fig. 3). At the same time F. hepatica seems to belong in the second category of brown-rot fungi having GH7 cellobiohydrolases and an intermediate number of LPMO copies. Taken together, these results suggest that the partial maintenance of enzymatic cellulolysis, along with the potential pseudogenization events in wood-degrading gene families, could be part of an ongoing transition of F. hepatica towards the brown-rot lifestyle.
5. Conclusions
The wood decay gene networks of F. hepatica and C. torrendii deviate from typical brown-rot and white-rot species respectively. Fistulina hepatica has undergone extensive gene losses related to the enzymatic degradation of lignocellulose, but we found few remaining genes related to the degradation crystalline cellulose. Furthermore, we found four potential pseudogenes of genes that are frequently lost in brown-rot fungi, suggesting that transition towards a brown-rot lifestyle could be an ongoing process for F. hepatica. The genome of C. torrendii is enriched in CAZYs similar to white-rot species, but lacks most of the genes related to the degradation of lignin similar to brown-rot species. Therefore, it takes an intermediate placement between white-rot and brown-rot fungi, sharing this characteristic with S. commune, J. argillacea and B. botryosum, which have been suggested to belong in a grey zone of rot types (Riley et al., 2014). Our results suggest that such transitions could have taken place multiple times across Agaricomycotina. Wood decay experiments largely support our results. Fistulina hepatica causes brown rot, while C. torrendii causes a simultaneous white-rot. However, both species do not show complete brown-rot and white-rot characteristics, since they are weak wood decayers and decay wood only locally, while decay by C. torrendii has soft-rot characteristics as well. The reasons behind the limited wood decay are not completely understood and need to be further studied, but they could be related to alternative strategies these species follow to gain nutrients in addition to the weak wood decay they cause.
Supplementary Material
Figure S1. Estimated number of copies for the six oxidative gene families at the ancestral nodes of the 14 genomes phylogeny based on genetree/species tree reconciliation with the edge weight threshold set to 90. The estimated number of copies per family for the common ancestor of Agaricales is shown in red. Uncertainty on the estimated number of copies present at an ancestral node is indicated with question mark next to the number. F. hepatica, C. torrendii and S. commune are the only species in the dataset that lack both POD and DyP.
Figure S2 a & b. Estimated number of copies for bulk CAZY at the ancestral nodes of the 14 genomes phylogeny as for Fig. S1. F. hepatica has the largest number of families with zero copies and it is the only species that has completely lost GH6.
Figure S3a–S3c. Estimated number of copies for accessory CAZY at the ancestral nodes of the 14 genomes phylogeny as for Fig. S1. F. hepatica has the largest number of families without any members, while is the only species that has lost all members of the GH6 family. F. hepatica has a higher number of losses for carbohydrate esterase gene families and very reduced GH43 content.
Figures S4a & S4b. Alignments of GH6, DyP-clade A (a) and GH74, GH5-7 (b) after manual removal of poorly aligned regions, showing the fragmentation of all the F. hepatica models from each loci. Colored columns represent constant amino acid positions. All the predicted models of F. hepatica for these loci represent fragments of the complete protein, having gaps even in areas of very conserved amino acids. Numbers on the grey bar above each alignment indicate the length of the alignment.
Figures S5 & S6. ML phylogenetic analysis of GH43 and LMPO (former GH61) respectively. Sequences of species in the Marasmioid clade have been coded with green, yellow and brown (see inset species tree). Stars indicate areas where S. commune, but not F. hepatica, has maintained genes copies, suggesting gene losses for the later species. Bootstrap values shown ≥ 70.
Figure S7a–S7c. Phylogenetic relationships of the predicted models of the potential F. hepatica pseudogenized loci in GH74 (a), DyP (b), and GH5-7 (c) with homologs from the 14 genomes showing the resulting long branches (highlighted in red) and comparison with similar analyses of the adjacent genes. The placement of the LPMO model Fishe1_24835 can be seen in Figure S6. Numbers on the branches represent branch length. The scaffold graphs show the orientation of each potential pseudogene with its adjacent genes. Filled black circles next to a protein ID indicate the placement of the protein product the adjacent gene on the phylogeny.
Highlights.
We sequenced the genomes of Cylindrobasidium torrendii and Fistulina hepatica.
We examined the evolution of wood decay mechanisms in Agaricales.
We performed wood decay experiments for both species and Schizophyllum commune.
Both species do not have typical white or brown rot characteristics.
Atypical wood decayers are more frequent than initially thought.
Acknowledgments
This work was supported by the PolyPEET project, Taxonomy and Evolution of the Polyporales (Basidiomycota, Fungi) under the NSF award DEB-0933081 (DSH) and the Open Tree of Life project under the NSF award DEB-1208719 (DSH). It was also partially supported by the National Institutes of Health (7R15 GM069493 REM), National Science Foundation (MCB 0919743 REM), and the Center for Membrane Biosciences, IUPUI. The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under contract DE-AC02-05CH11231. We thank Francis Martin for kindly providing permission to use data of the unpublished genomes of G. luxurians, H. sublateritium, and P. crispa. The alignments of sequence data for the organismal phylogeny have been deposited at TreeBASE (###). Assemblies and annotations of the reported genomes of F. hepatica and C. torrendii are available from the JGI fungal portal MycoCosm (http://jgi.doe.gov/fungi) and from DDBJ/EMBL/GenBank under the following accessions: ###, ###.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Benjamin W. Held, Email: bheld@umn.edu.
Robert Riley, Email: RWRiley@lbl.gov.
Laszlo G. Nagy, Email: LNagy@clarku.edu.
Gage Koehler, Email: kgk@iupui.edu.
Anthony S. Ransdell, Email: aransdel@iumail.iu.edu.
Hina Younus, Email: hinayounus@rediffmail.com.
Julianna Chow, Email: JChow@lbl.gov.
Jennifer Chiniquy, Email: JLChiniquy@lbl.gov.
Anna Lipzen, Email: ALipzen@lbl.gov.
Andrew Tritt, Email: AJTritt@lbl.gov.
Hui Sun, Email: HSun@lbl.gov.
Sajeet Haridas, Email: SHaridas@lbl.gov.
Kurt LaButti, Email: klabutti@lbl.gov.
Robin A. Ohm, Email: raohm@lbl.gov.
Ursula Kües, Email: ukuees@gwdg.de.
Robert A. Blanchette, Email: robertb@umn.edu.
Igor V. Grigoriev, Email: IVGrigoriev@lbl.gov.
Robert E. Minto, Email: rminto@iupui.edu.
David S. Hibbett, Email: dhibbett@clarku.edu.
Literature cited
- Albersheim P, Darvill A, Roberts K, Sederoff R, Staehelin A. Plant cell walls. Garland Science, Taylor & Francis Group, LLC; New York, NY: 2011. Plant cell walls: A renewable material source; pp. 365–409. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arantes V, Jellison J, Goodell B. Peculiarities of brown-rot fungi and biochemical Fenton reaction with regard to their potential as a model for bioprocessing biomass. Appl Microbiol Biotechnol. 2012;94(2):323–338. doi: 10.1007/s00253-012-3954-y. [DOI] [PubMed] [Google Scholar]
- Arnolds E. Collybia. In: Bas C, Kuyper ThW, Noordeloos ME, Vellinga EC, editors. Flora Agaricina Neerlandica. Vol. 3. A.A. Balkema; Rotterdam: 1995. pp. 106–123. [Google Scholar]
- Arnolds E. Marasmiellus. In: Bas C, Kuyper ThW, Noordeloos ME, Vellinga EC, editors. Flora Agaricina Neerlandica. Vol. 3. A.A. Balkema; Rotterdam: 1995. pp. 123–129. [Google Scholar]
- Arnolds E. Micromphale. In: Bas C, Kuyper ThW, Noordeloos ME, Vellinga EC, editors. Flora Agaricina Neerlandica. Vol. 3. A.A. Balkema; Rotterdam: 1995. pp. 129–132. [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward FO, Burnum-Johnson E, Tringe SG, Teiling C, Tremmel DM, Moeller JA, Scott JJ, Barry KW, Piehowski PD, Nicora CD, Malfatti SA, Monroe ME, Purvine SO, Goodwin LA, Smith RD, Weinstock GM, Gerardo NM, Suen G, Lipton MS, Currie CR. Leucoagaricus gongylophorus produces diverse enzymes for the degradation of recalcitrant plant polymers in leaf-cutter ant fungus gardens. App Environ Microbiol. 2013;79 (12):3770–3778. doi: 10.1128/AEM.03833-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldrian P, Valaskova V. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol Rev. 2008;32:501–521. doi: 10.1111/j.1574-6976.2008.00106.x. [DOI] [PubMed] [Google Scholar]
- Bao D, Gong M, Zheng H, Chen M, Zhang L, Wang H, Jiang J, Wu L, Zhu Y, Zhou Y, Li C, Wang S, Zhao Y, Zhao G, Tan Q. Sequencing and comparative analysis of the straw mushroom (Volvariella volvacea) genome. PLoS ONE. 2013;8 (3):e58294. doi: 10.1371/journal.pone.0058294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder M, Hibbett DS, Wang Z, Farnham WF. Evolutionary relationships of Mycaureola dilseae (Agaricales), a basidiomycete pathogen of a subtidal rhodophyte. Am J Bot. 2004;93(4):547–556. doi: 10.3732/ajb.93.4.547. [DOI] [PubMed] [Google Scholar]
- Binder M, Larsson KH, Matheny PB, Hibbett DS. Amylocorticiales ord. nov. and Jaapiales ord. nov.: Early diverging clades of Agaricomycetidae dominatedby corticioid forms. Mycologia. 2010;102 (4):865–880. doi: 10.3852/09-288. [DOI] [PubMed] [Google Scholar]
- Birney E, Durbin R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000;10:547–548. doi: 10.1101/gr.10.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette RA, Obst JR, Hedges JI, Weliky K. Resistance of hardwood vessels to degradation by white rot Basidiomycetes. Can J Bot. 1988;66:1841–1847. [Google Scholar]
- Blanchette R. Degradation of the lignocellulose complex in wood. Can J Bot. 1995;73(Suppl 1):999–1010. [Google Scholar]
- Blanchette RA, Held BW, Arenz BE, Jurgens JA, Baltes NJ, Duncan SM, Farrell RL. An Antarctic hot spot for fungi at Shackleton’s historic hut on cape Royds. Microl Ecol. 2010;60:29–38. doi: 10.1007/s00248-010-9664-z. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Bolam BN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbonnais R, Paice MG, Reid ID, Lanthier P, Yaguchi M. Lignin oxidation by laccase isozymes from Trametes versicolor and role of the mediator 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonate) in kraft lignin depolymerization. Appl Environ Microbiol. 1995;61 (5):1876–1880. doi: 10.1128/aem.61.5.1876-1880.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Cerniglia CE. Fungal metabolism of polycyclic aromatic hydrocarbons: past, present and future applications in bioremediation. J Ind Microbiol Biot. 1997;19:324–333. doi: 10.1038/sj.jim.2900459. [DOI] [PubMed] [Google Scholar]
- Cohen R, Suzuki MR, Hammel KE. Processive endoglucanase active in crystalline cellulose hydrolysis by the brown rot basidiomycete Gloeophyllum trabeum. Appl Environ Microbiol. 2005;71(5):2412–2417. doi: 10.1128/AEM.71.5.2412-2417.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C, Keane TM, Turner DJ, O’Keeffe G, Fitzpatrick DA, Doyle S. Genomic and proteomic dissection of the ubiquitous plant pathogen, Armillaria mellea: Towards anew infection model system. J Proteome Res. 2013;12:2552–2570. doi: 10.1021/pr301131t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creppin VF, Faulds CB, Connerton IF. A non-modular type B feruloyl esterase from Neurospora crassa exhibits concentration-dependent substrate inhibition. Biochem J. 2003;370:417–427. doi: 10.1042/BJ20020917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen D, Kersten PJ. Enzymology and molecular biology of lignin degradation. In: Brambl R, Marzluf GA, editors. The Mycota III; Biochemistry and Molecular Biology. 2. Springer-Verlag; Berlin-Heidelberg: 2004. pp. 249–273. [Google Scholar]
- Daniel G, Volc J, Kubatova E. Pyranose oxidase, a major source of H2O2 during wood degradation by Phanerochaete chrysosporium, Trametes versicolor, and Oudemansiella mucida. Appl Environ Microbiol. 1994;60 (7):2524–2532. doi: 10.1128/aem.60.7.2524-2532.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel G, Volc J, Filonova L, Plihal O, Kubatova E, Halada P. Characteristics of Gloeophyllum trabeum alcohol oxidase, an extracellular source of H2O2 in brown rot decay of wood. Appl Environ Microbiol. 2007;73 (19):6241–6253. doi: 10.1128/AEM.00977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries RP, Battaglia E, Coutinho PM, Henrissat B, Visser J. (Hemi-)cellulose degrading enzymes and their encoding genes from Aspergillus and Trichoderma. In: Hofrichter M, editor. The Mycota X. Industrial Applications. 2. Springer-Verlag; Berlin Heidelberg: 2010. pp. 341–355. [Google Scholar]
- Durand D, Halldorsson BV, Vernot B. A hybrid micro-macroevolutionary approach to gene tree reconstruction. J Comput Biol. 2006;13:320–335. doi: 10.1089/cmb.2006.13.320. [DOI] [PubMed] [Google Scholar]
- Eastwood D, Floudas D, Binder B, Majcherczyk A, Schneider P, Aerts A, Asiegbu FO, Baker SE, Barry K, Bendiksby M, Blumentritt M, Coutinho PM, Cullen D, de Vries RP, Gathman A, Goodell B, Henrissat B, Ihrmark K, Kauserud H, Kohler A, LaButti K, Lapidus L, Lavin JL, Lee YH, Lindquist E, Lilly W, Lucas S, Morin M, Murat C, Oguiza JA, Park J, Pisabarro AG, Riley R, Rosling A, Salamov A, Schmidt O, Schmutz J, Skrede I, Stenlid J, Wiebenga A, Xie X, Kües U, Hibbett DS, Hoffmeister D, Högberg N, Martin F, Grigoriev IV, Watkinson SC. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science. 2011;333:762–765. doi: 10.1126/science.1205411. [DOI] [PubMed] [Google Scholar]
- Eggert C, Temp U, Dean JFD, Eriksson KEL. A fungal metabolite mediates degradation of non-phenolic lignin structure and synthetic lignin by laccase. FEBS Lett. 1996;391:144–148. doi: 10.1016/0014-5793(96)00719-3. [DOI] [PubMed] [Google Scholar]
- Eggert C, Temp U, Eriksson KEL. Laccase is essential for lignin degradation by the white-rot fungus Pycnoporus cinnabarinus. EBS Lett. 1997;407:89–92. doi: 10.1016/s0014-5793(97)00301-3. [DOI] [PubMed] [Google Scholar]
- English AC, Richards S, Han YM, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, Gibbs RA. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One. 2012;7(11):e47768. doi: 10.1371/journal.pone.0047768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KEL, Blanchette RA, Ander P. Microbial and enzymatic degradation of wood and wood components. Springer-Verlag; Berlin, New York: 1990. [Google Scholar]
- Essig FM. The morphology, development, and economic aspects of Schizophyllum commune Fries. University of California Publications in Botany. 1922;7(14):447–498. plates 51–61. [Google Scholar]
- Faraco V, Piscitelli A, Sannia G, Giardina P. Identification of a new member of the dye-decolorizing peroxidase family from Pleurotus ostreatus. World J Microbiol Biotechnol. 2007;23:889–893. [Google Scholar]
- Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martínez AT, Otillar R, Spatafora JW, Yadav JS, Aerts A, Benoit I, Boyd A, Carlson A, Copeland A, Coutinho PM, de Vries RP, Ferreira P, Findley K, Foster B, Gaskell J, Glotzer D, Górecki P, Heitman J, Hesse C, Hori C, Igarashi K, Jurgens JA, Kallen N, Kersten P, Kohler A, Kües U, Kumar TKA, Kuo A, LaButti K, Larrondo LF, Lindquist E, Ling A, Lombard V, Lucas S, Lundell T, Martin R, McLaughlin DJ, Morgenstern I, Morin E, Murat C, Nagy LG, Nolan M, Ohm RA, Patyshakuliyeva A, Rokas A, Ruiz-Dueñas FJ, Sabat G, Salamov A, Samejima M, Schmutz J, Slot JC, St John F, Stenlid J, Sun H, Sun S, Syed K, Tsang A, Wiebenga A, Young D, Pisabarro A, Eastwood DC, Martin F, Cullen D, Grigoriev IV, Hibbett DS. The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- Ginns J, Lefebvre MNL. Lignicolous corticioid fungi (Basidiomycota) of North America. Systematics, distribution, and ecology. Mycol Mem. 1993:19. [Google Scholar]
- Ginns J. Porodisculus pendulus: systematic, cultural characters, and Canadian records. Can J Bot. 1997;75:220–227. [Google Scholar]
- Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, Berlin AM, Aird D, Costello M, Daza R, Williams L, Nicol R, Gnirke A, Nusbaum C, Lander ES, Jaffe DB. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011;108(4):1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell B, Daniel G, Jellison J, Qian Y. Iron reducing capacity of low-molecular-weight compounds produced in wood by fungi. Holzforschung. 2006;60:630–636. [Google Scholar]
- Grigoriev IV, Martinez DA, Salamov AA. Fungal genomic annotation. Appl Mycol Biotechnol. 2006;6:123–142. [Google Scholar]
- Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Slamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(1):D699–704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronqvist S, Viikari L, Niku-Paavola ML, Orlandi M, Canevali C, Buchert J. Oxidation of milled wood lignin with laccase, tyrosinase and horseradish peroxidase. Appl Microbiol Biotechnol. 2005;67:489–494. doi: 10.1007/s00253-004-1800-6. [DOI] [PubMed] [Google Scholar]
- Guillen F, Martinez AT, Martinez MJ, Evans CS. Hydrogen-peroxide-producing system of Pleurotus eryngii involving the extracellular enzyme aryl-alcohol oxidase. Appl Microbiol Biotechnol. 1994;41:465–470. [Google Scholar]
- Gutierrez A, Babot ED, Ullrich R, Hofrichter M, Martinez AT, del Rio JC. Regioselective oxygenation of fatty acids, fatty alcohols and other aliphatic compounds by a basidiomycete heme-thiolate peroxidase. Arch Biochem Biophys. 2011;514:33–43. doi: 10.1016/j.abb.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Harris PV, Welner D, McFarland KC, Re E, Navarro Poulsen JC, Brown K, Salbo R, Ding H, Vlasenko E, Merino S, Xu F, Cherry J, Larsen S, Leggio LL. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: Structure and function of a large, enigmatic family. Biochemistry. 2010;49:3305–3316. doi: 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]
- Henningsson B. Changes in impact bending strength, weight and alkali solubility following fungal attack on birch wood. Studia Forestalia Suecica. 1967;41:1–20. [Google Scholar]
- Henriksson G, Johansson G, Pettersson G. A critical review of cellobiose dehydrogenases. J Biotechnol. 2000;78:93–113. doi: 10.1016/s0168-1656(00)00206-6. [DOI] [PubMed] [Google Scholar]
- Hess J, Skrede I, Wolfe BE, LaButti K, Ohm RA, Grigoriev IV, Pringle A. Transposable element dynamics among asymbiotic and ectomycorrhizal Amanita fungi. Genome Biol Evol. 2014;6(7):1564–1578. doi: 10.1093/gbe/evu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofrichter M, Ullrich R. Heme-thiolate haloperoxidases: versatile biocatalysts with biotechnological and environmental significance. Appl Microbiol Biotechnol. 2006;71:276–288. doi: 10.1007/s00253-006-0417-3. [DOI] [PubMed] [Google Scholar]
- Illman BL. Oxidative degradation of wood by brown-rot fungi. In: Pell E, Steffen K, editors. Active oxygen/oxidative stress and plant metabolism. American Society of Plant Physiologists; Rockville, MD: 1991. pp. 97–196. [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110 (1–4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Justo A, Vizzini A, Minnis AM, Menolli N, Jr, Capelari M, Rodriguez O, Malysheva E, Contu M, Ghignone S, Hibbett DS. Phylogeny of the Pluteaceae (Agaricales, Basidiomycota): taxonomy and character evolution. Fungal Biol. 2010;115 (1):1–20. doi: 10.1016/j.funbio.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Kaarik A. The identification of the mycelia of wood-decay fungi by their oxidation reactions with phenolic compounds. Studia Forestalia Suecica. 1965;31:3–81. [Google Scholar]
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT-The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten P, Kirk TK. Involvement of a new enzyme, glyoxal oxidase, in extracellular H2O2 production by Phanerochaete chrysosporium. J Bacteriol. 1987;169(5):2195–2201. doi: 10.1128/jb.169.5.2195-2201.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten P, Cullen D. Extracellular oxidative systems of the lignin-degrading Basidiomycete Phanerochaete chrysosporium. Fungal Genet Biol. 2007;44:77–87. doi: 10.1016/j.fgb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Kirk PM, Cannon PF, Minter DW, Stalpers JA. Dictionary of the fungi. 10. CAB INTERNATIONAL; Wallingford, UK: 2008. [Google Scholar]
- Kirk TK, Highley TL. Quantitive changes in structural components of conifer woods during decay by white- and brown-rot fungi. Phytopathology. 1973;63:1338–1342. [Google Scholar]
- Kirk TK, Cullen D. Enzymology and molecular genetics of wood degradation by white-rot fungi. In: Young RA, Akhtar M, editors. Environmentally friendly technologies for the pulp and paper industry. John Wiley & Sons, Inc; New York, NY: 1998. pp. 273–307. [Google Scholar]
- Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN*, Rao BS, Rogozin IB, Smirnov S*, Sorokin AV, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5(2) doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon PA, Williamson G, Fish NM, Archer DB, Belshaw NJ. A modular esterase from Penicillium funiculosum which releases ferulic acid from plant cell walls and binds crystalline cellulose contains a carbohydrate binding module. Eur J Biochem. 2000;267:6740–6752. doi: 10.1046/j.1432-1033.2000.01742.x. [DOI] [PubMed] [Google Scholar]
- Kües U, Rühl M. Mutliple multi-copper oxidase gene families in Basidiomycetes – What for? Curr Genomics. 2011;12:72–94. doi: 10.2174/138920211795564377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-L, Spanikova S, de Vries RP, Biely P. Identification of genes encoding microbial glucuronoyl esterases. FEBS Lett. 2007;581:4029–4035. doi: 10.1016/j.febslet.2007.07.041. [DOI] [PubMed] [Google Scholar]
- Li X-L, Skory CD, Cotta MA, Puchart V, Biely P. Novel family of carbohydrate esterases, based on identification of the Hypocrea jecorina acetyl esterase gene. Appl Environ Microbiol. 2008;74 (24):7482–7489. doi: 10.1128/AEM.00807-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liers C, Bobeth C, Pecyna M, Ullrich R, Hofrichter M. DyP-like peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl Microbiol Biotechnol. 2010;85:1869–1879. doi: 10.1007/s00253-009-2173-7. [DOI] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The Carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics. 2010;11:579. doi: 10.1186/1471-2105-11-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison DR, Maddison WP. MacClade4: Analysis of phylogeny and character evolution. Sunderland, MA: Sinauer Associates; 2002. [Google Scholar]
- Marcovic O, Janecek S. Pectin degrading glycoside hydrolases of family 28: sequence-structural features, specificity and evolution. Protein Eng. 2001;14(9):615–631. doi: 10.1093/protein/14.9.615. [DOI] [PubMed] [Google Scholar]
- Marcovic O, Janecek S. Pectin methylesterases: sequence-structural features and phylogenetic relationships. Carbohydr Res. 2004;339:2281–2295. doi: 10.1016/j.carres.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Martin J, Bruno VM, Fang Z, Meng X, Blow M, Zhang T, Sherlock G, Snyder M, Wang Z. Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genomics. 2010;11:663–670. doi: 10.1186/1471-2164-11-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez AT, Speranza M, Ruiz-Duenas FJ, Ferreira P, Camarero S, Guillen F, Martinez MJ, Gutierrez MJ, del Rio JC. Biodegradation of lignocellulosics: microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int Microbiol. 2005;8:195–204. [PubMed] [Google Scholar]
- Martinez D, Larrondo LF, Putnam N, Gelpke MDS, Huang K, Chapman J, Helfenbein KG, Ramaiya P, Detter JC, Larimer F, Coutinho PM, Henrissat B, Berka R, Cullen D, Rokhsar D. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium RP78. Nat Biotechnol. 2004;22 (6):695–700. doi: 10.1038/nbt967. [DOI] [PubMed] [Google Scholar]
- Martinez D, Challacombe J, Morgenstern I, Hibbett D, Schmoll M, Kubicek C, Ferreira P, Ruiz-Duenas FJ, Martinez AT, Kersten P, Hammel KE, Vanden Wymelenberg A, Gaskell J, Lindquist E, Sabat G, Splinter BonDurant S, Larrondo LF, Canessa P, Vicuna R, Yadav J, Doddapaneni H, Subramanian V, Pisabarro AG, Lavin JL, Oquiza JA, Master E, Henrissat B, Coutinho PM, Harris P, Magnuson JK, Baker SE, Bruno K, Kenealy W, Hoegger PJ, Kües U, Ramaiya P, Lucas S, Salamov A, Shapiro H, Tu H, Chee CL, Misra M, Xie G, Teter S, Yaver D, James T, Mokreis M, Posposek M, Grigoriev IV, Brettin T, Rokhsar D, Berka R, Cullen D. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci USA. 2009;106 (6):1954–1959. doi: 10.1073/pnas.0809575106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata JL, Hughes KW, Petersen RH. Phylogenetic placement of Marasmiellus juniperinus. Mycoscience. 2004;45:214–221. [Google Scholar]
- Matheny PB, Curtis JM, Hofstetter V, Aime MC, Moncalvo JM, Ge ZW, Yang ZL, Slot JC, Ammirati JF, Baroni TJ, Bougher NL, Hughes KW, Lodge DJ, Seidl MT, Aanen DK, DeNitis M, Daniele GM, Desjardin DE, Kropp BR, Norvell LL, Parker A, Vellinga EC, Vilgalys R, Hibbett DS. Major clades of Agaricales: a multilocus phylogenetic overview. Mycologia. 2006;98(6):982–995. doi: 10.3852/mycologia.98.6.982. [DOI] [PubMed] [Google Scholar]
- Melen K, Krogh A, von Heijne G. Reliability measures for membrane protein topology prediction algorithms. J Mol Biol. 2003;327:735–744. doi: 10.1016/S0022-2836(03)00182-7. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceeding of the Gateway Computing Environments Workshop (GCE); 14 Nov. 2010; New Orleans, LA. 2010. pp. 1–8. [Google Scholar]
- Moncalvo JM, Vilgalys R, Redhead SA, Johnson JE, James TY, Aime CA, Hofstetter V, Verduin SJW, Larsson E, Baroni TJ, Thorn RG, Jacobsson S, Clemencon H, Miller OK., Jr One hundred and seventeen clades of euagarics. Mol Phylogenet Evol. 2002;23:357–400. doi: 10.1016/S1055-7903(02)00027-1. [DOI] [PubMed] [Google Scholar]
- Mondego JMC, Carazzolle MF, Costa GGL, Formighieri EF, Parizzi LP, Rincones J, Cottomaci C, Carraro DM, Cunha AF, Carrer H, Vidal RO, Estrela RC, Garcia O, Thomazella DPT, de Oliveira BV, Pires ABL, Rio MCS, Araujo MRR, de Moraes MH, Castro LAB, Gramacho KP, Goncalves MS, Moura Neto JP, Goes Neto A, Barbosa LV, Guiltinan MJ, Bailey BA, Meinhardt LW, Cascardo JCM, Pereira GAG. A genome survey of Moniliophthora perniciosa gives new insights into Witches’ Broom disease of cacao. BMC Genomics. 2008;9:548. doi: 10.1186/1471-2164-9-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgaard A, Kauppinen S, Larsen S. Rhamnogalacturonan acetylesterase elucidates the structure and function of a new family of hydrolases. Structure. 2000;8:373–383. doi: 10.1016/s0969-2126(00)00118-0. [DOI] [PubMed] [Google Scholar]
- Morin M, Kohler A, Baker AR, Foulongne-Oriol M, Lombard V, Nagy LG, Ohm RA, Patyshakuliyeva A, Brun A, Aerts AL, Bailey AM, Billette C, Coutinho PM, Deakin G, Doddapaneni H, Floudas D, Grimwood J, Hildén K, Kües U, LaButti KM, Lapidus A, Lindquist EA, Lucas SM, Murat C, Riley RW, Salamov AA, Schmutz J, Subramanian V, Wösten HAB, Xu J, Eastwood DC, Foster GD, Sonnenberg ASM, Cullen D, de Vries RP, Lundell T, Hibbett DS, Henrissat B, Burton KS, Kerrigan RW, Challen MP, Grigoriev IV, Martin F. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc Natl Acad Sci USA. 2012;109:17501–17506. doi: 10.1073/pnas.1206847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10(1):1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Niemenmaa O, Uusi-Rauva A, Hatakka A. Demethoxylation of [O(14)CH (3)]-labelled lignin model compounds by the brown-rot fungi Gloeophyllum trabeum and Poria (Postia) placenta. Biodegradation. 2007;19:555–565. doi: 10.1007/s10532-007-9161-3. [DOI] [PubMed] [Google Scholar]
- Nilsson T. Comparative study on the cellulolytic activity of white-rot and brown-rot fungi. Mater Organismen. 1974;9:173–198. [Google Scholar]
- Nilsson T, Ginns J. Cellulolytic activity and the taxonomic position of selected brown-rot fungi. Mycologia. 1979;71:170–177. [Google Scholar]
- Ohm RA, de Jong JF, Lugones LG, Aerts A, Kothe E, Stajich JE, de Vries RP, Record E, Levasseur A, Baker SE, Bartholomew KA, Coutinho PM, Erdmann S, Fowler TJ, Gahman AC, Lombard V, Henrissat B, Knabe N, Kües U, Lilly WW, Lindquist E, Lucas S, Magnuson JK, Piumi F, Raudaskoski M, Salamov A, Schmutz J, Schwarze FWMR, vanKuyk PA, Horton JS, Grigoriev IV, Wösten HAB. Genome sequence of the model mushroom Schizophyllum commune. Nat Biotechnol. 2010;28:957–963. doi: 10.1038/nbt.1643. [DOI] [PubMed] [Google Scholar]
- Ohm RA, Riley R, Salamov A, Min B, Choi I-G, Grigoriev IV. Genomics of wood-degrading fungi. Fungal Genet Biol. 2014 doi: 10.1016/j.fgb.2014.05.001. http://dx.doi.org/10.1016/j.fgb.2014.05.001. [DOI] [PubMed]
- Osono T. Ecology of ligninolytic fungi associated with leaf litter decomposition. Ecol Res. 2007;22:955–974. [Google Scholar]
- Osono T, Takeda H, Azuma J. Carbon isotope dynamics during leaf litter decomposition with reference to lignin fractions. Ecol Res. 2008;23:51–55. [Google Scholar]
- Padhiar A, Albert S. Anatomical changes in Syzygium cumuini Linn. wood decayed by two white rot fungi Schizophyllum commune Fries. and Flavodon flavus (Klotzsch) Ryvarden. J Indian Acad Wood Sci. 2011;8:11–20. [Google Scholar]
- Pointing SB. Feasibility of bioremediation by white-rot fungi. Appl Microbiol Biotechnol. 2001;57:20–33. doi: 10.1007/s002530100745. [DOI] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(suppl 1):i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redhead SA, Ginns JH. A reappraisal of agaric genera associated with brown rots of wood. Trans Mycol Soc Japan. 1985;26:349–381. [Google Scholar]
- Rodriguez-Rincon F, Suarez A, Lucas M, Larrondo LF, de la Rubia T, Polaina J, Martinez J. Molecular and structural modeling of the Phanerochaete flavido-alba extracellular laccase reveals its ferroxidase structure. Arch Microbiol. 2010;192:883–892. doi: 10.1007/s00203-010-0616-2. [DOI] [PubMed] [Google Scholar]
- Riley R, Salamov AA, Brown DW, Nagy LG, Floudas D, Held BW, Levasseur A, Lombard V, Morin E, Otillar R, Lindquist EA, Sun H, LaButti KM, Schmutz J, Jabbour D, Luo, Baker SE, Pisabarro AG, Walton JD, Blanchette RA, Henrissat B, Martin F, Cullen D, Hibbett DS, Grigoriev IV. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci USA. 2014 doi: 10.1073/pnas.1400592111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres D, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61 (3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MB, Cardoza RE, Hermosa R, Gutiérrez S, Monte E. Cloning and characterization of the Thcut1 gene encoding a cutinase of Trichoderma harzianum T34. Curr Genet. 2008;54:301–312. doi: 10.1007/s00294-008-0218-6. [DOI] [PubMed] [Google Scholar]
- Ruiz-Duenas FJ, Morales M, Perez-Boada M, Choinowski T, Martinez MJ, Piontek K, Martinez AT. Manganese oxidation site in Pleurotus eryngii versatile peroxidase: A site-directed mutagenesis, kinetic, and crystallographic study. Biochemistry. 2007;46:66–77. doi: 10.1021/bi061542h. [DOI] [PubMed] [Google Scholar]
- Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidhalter DR, Canevascini G. Characterization of the cellulolytic system from the brown-rot fungus Coniophora puteana. App Microbiol Biotechnol. 1993;37:431–436. [Google Scholar]
- Schmidt O, Liese W. Variability of wood degrading enzymes of Schizophyllum commune. Holzforschung. 1980;34:67–72. [Google Scholar]
- Schwarze FWMR, Baum S, Fink S. Dual modes of degradation by Fistulina hepatica in xylem cell walls of Quercus robur. Mycol Res. 2000a;104:846–852. [Google Scholar]
- Schwarze FWMR, Engels J, Mattheck C. Fungal strategies of wood decay in trees. Springer-Verlag; Berlin-Heidelberg: 2000b. pp. 76–81. [Google Scholar]
- Stajich JE, Wilke SK, Ahren D, Au CH, Birren BW, Borodovsky M, Burns C, Canbäck B, Casselton LA, Cheng CK, Deng J, Dietrich FS, Fargo DC, Farman ML, Gathman AC, Goldberg J, Guigo R, Hoegger PJ, Hooker JB, Huggins A, James TY, Kamada T, Kilaru S, Kodira C, Kües U, Kupfer D, Kwan HS, Lomsadze A, Li W, Lilly WW, Ma LJ, Mackey AJ, Manning G, Roe BA, Shenoy N, Stanke M, Ter-Hovhannisyan V, Tunlid A, Velagapudi R, Vision TJ, Zeng Q, Zolan ME, Pukkila PJ. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus) Proc Natl Acad Sci USA. 2010;107 (26):11889–11894. doi: 10.1073/pnas.1003391107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Takemoto S, Nakamura H, Imamura EY, Shimane T. Schizophyllum commune as a ubiquitous plant parasite. Jpn Agr Res Q. 2010;44(4):357–364. [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theuerl S, Buscot F. Laccases: toward disentangling their diversity and functions in relation to soil organic matter cycling. Biol Fertil Soils. 2010;46:215–225. [Google Scholar]
- Tzean SS, Estey RH. Schizophyllum commune Fr. as a destructive mycoparasite. Can J Microbiol. 1978;24(7):780–784. doi: 10.1139/m78-131. [DOI] [PubMed] [Google Scholar]
- Volc J, Kubatova E, Daniel G, Pikrilova V. Only C-2 specific glucose oxidase activity is expressed in ligninolytic cultures of the white rot fungus Phanerochaete chrysosporium. Arch Microbiol. 1996;165:421–424. doi: 10.1007/s002030050348. [DOI] [PubMed] [Google Scholar]
- Wawrzyn GT, Quin MB, Choudhary S, Lopez-Gallego F, Schmidt-Dannert C. Draft genome of Omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in Basidiomycota. Chem Biol. 2012;19 (6):772–783. doi: 10.1016/j.chembiol.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrall JJ, Anagnost SE, Zabel RA. Comparison of wood decay among diverse lignicolous fungi. Mycologia. 1997;89 (2):199–219. [Google Scholar]
- Yelle DJ, Ralph J, Lu F, Hammel KE. Evidence for cleavage of lignin by a brown rot basidiomycete. Environ Microbiol. 2008;10(7):1844–1849. doi: 10.1111/j.1462-2920.2008.01605.x. [DOI] [PubMed] [Google Scholar]
- Yoon JJ, Cha CJ, Kim YS, Kim W. Degradation of cellulose by the major endoglucanase produced from the brown-rot fungus Fomitopsis pinicola. Biotechnol Lett. 2008;30:1373–1378. doi: 10.1007/s10529-008-9715-4. [DOI] [PubMed] [Google Scholar]
- Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics. 2010 doi: 10.1002/0471250953.bi1105s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Estimated number of copies for the six oxidative gene families at the ancestral nodes of the 14 genomes phylogeny based on genetree/species tree reconciliation with the edge weight threshold set to 90. The estimated number of copies per family for the common ancestor of Agaricales is shown in red. Uncertainty on the estimated number of copies present at an ancestral node is indicated with question mark next to the number. F. hepatica, C. torrendii and S. commune are the only species in the dataset that lack both POD and DyP.
Figure S2 a & b. Estimated number of copies for bulk CAZY at the ancestral nodes of the 14 genomes phylogeny as for Fig. S1. F. hepatica has the largest number of families with zero copies and it is the only species that has completely lost GH6.
Figure S3a–S3c. Estimated number of copies for accessory CAZY at the ancestral nodes of the 14 genomes phylogeny as for Fig. S1. F. hepatica has the largest number of families without any members, while is the only species that has lost all members of the GH6 family. F. hepatica has a higher number of losses for carbohydrate esterase gene families and very reduced GH43 content.
Figures S4a & S4b. Alignments of GH6, DyP-clade A (a) and GH74, GH5-7 (b) after manual removal of poorly aligned regions, showing the fragmentation of all the F. hepatica models from each loci. Colored columns represent constant amino acid positions. All the predicted models of F. hepatica for these loci represent fragments of the complete protein, having gaps even in areas of very conserved amino acids. Numbers on the grey bar above each alignment indicate the length of the alignment.
Figures S5 & S6. ML phylogenetic analysis of GH43 and LMPO (former GH61) respectively. Sequences of species in the Marasmioid clade have been coded with green, yellow and brown (see inset species tree). Stars indicate areas where S. commune, but not F. hepatica, has maintained genes copies, suggesting gene losses for the later species. Bootstrap values shown ≥ 70.
Figure S7a–S7c. Phylogenetic relationships of the predicted models of the potential F. hepatica pseudogenized loci in GH74 (a), DyP (b), and GH5-7 (c) with homologs from the 14 genomes showing the resulting long branches (highlighted in red) and comparison with similar analyses of the adjacent genes. The placement of the LPMO model Fishe1_24835 can be seen in Figure S6. Numbers on the branches represent branch length. The scaffold graphs show the orientation of each potential pseudogene with its adjacent genes. Filled black circles next to a protein ID indicate the placement of the protein product the adjacent gene on the phylogeny.