

Abstract
Selective blockade of the Orexin-1 receptor has been suggested as a potential approach to drug addiction therapy because of its role in modulating the brain's reward system. We have recently reported a series of tetrahydroisoquinoline-based OX1 selective antagonists. Aimed at elucidating SAR requirements in other regions of the molecule and further enhancing OX1 potency and selectivity, we have designed and synthesized a series of analogs bearing a variety of substituents at the 1-position of the tetrahydroisoquinoline. The results show that an optimally substituted benzyl group is required for activity at the OX1 receptor. Several compounds with improved potency and/or selectivity have been identified. When combined with structural modifications that were previously found to improve selectivity, we have identified compound 73 (RTIOX-251) with an apparent dissociation constant (Ke) of 16.1 nM at the OX1 receptor and >620-fold selectivity over the OX2 receptor. In vivo, compound 73 was shown to block the development of locomotor sensitization to cocaine in rats.
Keywords: Orexin, antagonist, selective, tetrahydroisoquinoline
Introduction
Orexins (hypocretins), including orexin A and orexin B, are neuropeptides exclusively produced in hypothalamic neurons arising in the dorsomedial hypothalamus (DMH), perifornical area (PFA), and lateral hypothalamus (LH).1, 2 The orexin-producing neurons in the hypothalamus project widely to key areas of the central nervous system (CNS) that are commonly thought to control sleep–wake states, modulation of food intake, panic, anxiety, reward and addictive behaviors, suggesting diverse roles for these peptides.3-5 Orexin A and B bind and activate two G protein-coupled receptors (GPCRs), orexin-1 (OX1) and orexin-2 (OX2), with OX1 signaling via Gq proteins and OX2 signaling via Gq or Gi/o proteins.6, 7 The OX1 receptor has 10-fold higher affinity for orexin A than for orexin B; whereas OX2 has equal affinity for both peptides. Interestingly, these receptors are differentially distributed throughout the brain, suggesting different physiological roles for each receptor.1, 8, 9 Originally known for regulation of metabolic, circadian and stress systems, the orexin system has recently been associated with drug addiction.10-13 The fact that orexin neurons project to the ventral tegmental area (VTA) and other brain regions involved in reward processing supports this notion. Selective blockade of the OX1 receptor has been shown to attenuate stress- and cue-induced reinstatement of previously extinguished cocaine-, morphine-, and alcohol-seeking behavior.14-18 Together, these findings suggest that OX1 antagonists may have therapeutic utility for the treatment of drug addiction.
In order to elucidate the physiological role of the orexin receptors and explore the potential of orexin receptor antagonists as therapeutics, a number of groups, mostly from the pharmaceutical industry, have developed orexin receptor antagonists.19-23 In these endeavors, the majority of research has focused on dual orexin receptor antagonists for new sleep medications development.24, 25 Several dual antagonists, including almorexant (1) and SB-649828 (2), have been advanced into clinical trials, but their development was later halted because of tolerability and toxicity concerns, respectively. The most success was seen with suvorexant (3), a dual orexin antagonist developed by Merck. Recently, suvorexant was approved at a lower dose (20 mg) than initially proposed for the treatment of insomnia, and is currently marketed under the trade name Belsomra.26 Several OX1 receptor selective antagonists have also been developed to probe the importance of this receptor.27 Among these, the OX1 antagonist SB-334867 (4) was the first OX1 selective antagonist reported and has been extensively studied because it has favorable preclinical pharmacokinetics.28 Its affinity for OX1 is ~50-fold higher than for OX2, but some in vivo studies using high doses should be viewed cautiously because those doses may block both receptors. Additionally, Rottapharm Madaus has reported a series of azaspiro compounds as selective OX1 antagonists, and identified the spiro moiety as a key structural feature for OX1 receptor selectivity.29, 30 Several other OX1 selective antagonists have also been reported, including GSK-1059865 and its analogs, as well as ACT-335827, although these compounds mostly retained a significant amount of OX2 activity.31-33
Our group has been developing OX1 selective antagonists for the potential treatment of drug addiction and related disorders.34-36 We recently described our progress toward selective OX antagonists based on the tetrahydroisoquinoline scaffold, which is found both in 1 and the OX2 receptor selective antagonist TCS-OX2-29 (5).37 The structural modifications focused on the 7-position of the tetrahydroisoquinoline ring and the acetamide positions, resulting in several potent and selective OX1 antagonists. In particular, RTIOX-276 (6) showed excellent OX1 potency and selectivity, and attenuated cocaine-induced conditioned place preference (CPP) in rats.34 However, the structure-activity relationship (SAR) requirements in other regions of the structure have yet to be explored. Interestingly, at the 1-position of the tetrahydroisoquinoline, the dual orexin antagonist 1 has a 4-trifluoromethylphenylethyl group, whereas the OX2 selective antagonist 5 does not bear any substitution, suggesting that this position may play an important role in receptor subtype selectivity. Therefore, we have examined a series of analogs bearing a variety of modifications at the 1-position of the tetrahydroisoquinoline. Herein, we report our effort in the design, synthesis, and in vitro and in vivo characterization of these 1-substituted analogs.
Results and Discussion
Chemistry
The overall approach to the synthesis followed methods detailed in our earlier work (Scheme 1).34, 36 Briefly, commercially available 3,4-dimethoxyphenethylamine (7) and phenylacetic acid 8a were coupled using HBTU or BOP to give the amide 9. Cyclization of 9 via the Bischler-Napieralski reaction using phosphorus oxychloride in toluene afforded the dihydroisoquinoline, which was readily reduced to the tetrahydroisoquinoline 10 with sodium borohydride. The nitrogen was alkylated using N-benzyl bromoacetamide with diisopropylethylamine as base to give final product 11. Similarly, compound 12 was synthesized from 8b (R = H). Elaboration of the phenol 12 was achieved by alkylation using the appropriate alkyl bromide in the presence of K2CO3, esterification via BOP-mediated coupling, or by sulfonylation using the sulfonyl chloride with triethylamine as base, to afford target compounds 13-23.
Scheme 1.

Synthesis of 1-substituted tetrahydroisoquinolines 11-23a
Aniline derivatives were synthesized following a similar route (Scheme 2). Amide coupling of 24 with the amine 7 followed by alkylation of the hydroxyl group afforded intermediate 25, which was converted to 27 via Bischler-Napieralski reaction, alkylation of the nitrogen and reduction of the nitro group. The free aniline in 27 was then modified by reductive amination using sodium triacetoxyborohydride or sodium cyanoborohydride, alkylation via an alkyl bromide or by amide coupling using BOP in DMF to provide final compounds 28-35. Similarly, the benzoxazole 38 was prepared from 24 via the 4-hydroxy-3-amino intermediate 37 by condensation with ethyl acetimidate. In addition to the 3,4-disubstituted, several monosubstituted analogs were prepared in analogous fashion (Scheme 3), starting from acid 39, via the tetrahydroisoquinoline 40 with further elaboration at phenol or aniline. Urea 50 was prepared by reaction with n-hexyl isocyanate in toluene.
Scheme 2.

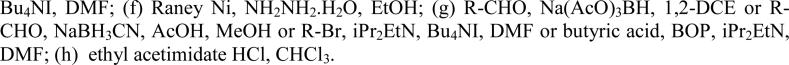
Synthesis of 1-aminobenzyl substituted tetrahydroisoquinolines 28-38a
Scheme 3.


Synthesis of 1-substituted tetrahydroisoquinolines 41-56a
Compounds with substituents other than the benzyl group at the 1-position were made via Pictet-Spengler condensation between 7 and the corresponding aldehydes in toluene and trifluoroacetic acid at 140 °C for 30 minutes in the microwave, followed by N-alkylation as described above (Scheme 4). The olefin 61 was reduced by hydrogenation on Pd/C in ethanol to give the saturated analog 62. Non-commercial aldehydes in the Pictet-Spengler reaction were prepared via pyridinium chlorochromate oxidation of the appropriate alcohols.
Scheme 4.

Synthesis of 1-substituted tetrahydroisoquinolines 59-69a
The 7-propoxy derivative 73 was prepared in a similar fashion as described above (Scheme 5), starting from 4-hydroxy-3-methoxy-phenethylamine (70). Amide coupling between 70 and 8b, followed by alkylation of the two hydroxyl groups afforded intermediate 71. Cyclization followed by reduction provided amine 72 and then a final N-alkylation with the bromoacetamide gave 73.
Scheme 5.

Synthesis of 7-propoxy tetrahydroisoquinoline 73.a
Biological Evaluation
Activity of the target compounds at the human OX1 and OX2 receptors was evaluated using Fluorescent Imaging Plate Reader technology (FLIPRTM, Molecular Devices), which measures intracellular calcium mobilization in live cells. The apparent dissociation constant Ke was calculated from compound-mediated inhibition of orexin A activity as previously described.34-36 In these assays, the EC50 for orexin A at OX1 and OX2 is 0.13 ± 0.02 nM and 4.2 ± 0.2 nM, respectively. All the compounds that had OX1 Ke values < 1 μM were also tested for agonist activity at 10 μM; none of them were active.
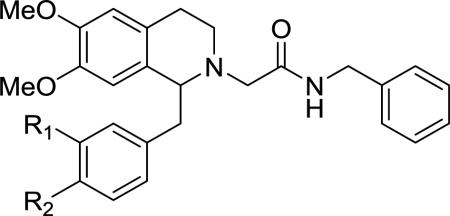
Our studies aimed at mapping out the SAR requirements at the 1-positon based on both the steric and electronic considerations. An examination of several reported orexin antagonists based on the tetrahydroisoquinoline scaffold revealed the importance of the 1-postion. The dual orexin antagonist 1 has a 4-trifluoromethylphenylethyl group at the 1-position. Conversely, the OX2 selective antagonist 5 does not bear any substitution at the 1-position. Compound 11, the hit identified in a high-throughput screening campaign by Actelion, has a 3,4-dimethoxybenzyl group at the 1-position of the tetrahydroisoquinoline, and showed some selectivity for the OX1 receptor (Table 1).34, 38 The OX1 receptor selective antagonist 6 developed in our lab also has the 3,4-dimethoxy substitution. Taken together, these results suggest that the 1-position substituents may have differential effects on the activity of ligands at the two orexin receptors.
Table 1.
Effect of benzyl substitution on OX antagonism
| |||||
|---|---|---|---|---|---|
| Number | R1 | R2 | Ke (OX1, nM)b | Ke (OX2, nM)c | OX2/OX1 |
| 11 | OMe | OMe | 199 ± 47 | >10,000 | >50.3 |
| 12 | OMe | OH | 419 ± 64 | >10,000 | >24 |
| 13 | OMe | O-n-Butyl | 48 ± 27 | 2000 ± 860 | 42 |
| 14 | OMe | O-n-Hexyl | 120 ± 20 | >10,000 | >83 |
| 15 | OMe | O-(CH2)2-piperidinyl | >10,000d | a | |
| 16 | OMe | OCO(CH2)2CH3 | 43.5 ± 3.7 | 2080 ± 600 | 48 |
| 17 | OMe | OSO2Me | 480 ± 180 | a | |
| 18 | OMe | OBenzyl | 399 ± 22 | a | |
| 19 | OMe | O-(CH2)4-OPh | 385 ± 96 | a | |
| 20 | OMe | OSO2(4-Me)Ph | > 10,000 | a | |
| 21 | OMe | OCH2-2-Pyridyl | 153 ± 43 | a | |
| 22 | OMe | OCH2-3-Pyridyl | 250 ± 120 | a | |
| 23 | OMe | OSO2Ph | 1820 ± 680 | a | |
| 26 | NO2 | OMe | 1500 ± 230 | a | |
| 28 | NMe2 | OMe | 12.7 ± 2.8 | 970 ± 350 | 76.7 |
| 29 | NHEt | OMe | 309 ± 39 | >10,000 | >32 |
| 30 | NEt2 | OMe | 208 ± 38 | >10,000 | >48 |
| 31 | NH-n-Propyl | OMe | 920 ± 140 | >10,000 | >11 |
| 32 | N(n-Propyl)2 | OMe | 857 ± 380d | a | |
| 33 | NH-Benzyl | OMe | >10,000d | a | |
| 34 | NHAc | OMe | >10,000d | a | |
| 35 | NHCO(CH2)2CH3 | OMe | 320± 30 | a | |
| 36 | NO2 | OH | >10,000d | a | |
| 37 | NH2 | OH | >10,000d | a | |
| 38 | 2-Methyl-5-benzoxazole | 2620± 870d | a | ||
< 35% inhibition at 10 μM
Values are the mean ± SEM of at least three independent experiments performed in duplicate
Values are the mean ± SEM of at least two independent experiments performed in duplicate; for compounds with Ke < 100 nM at OX1 at least three independent experiments in duplicate were performed.
Values are the mean ± SEM of two independent experiments performed in duplicate.
The small set of analogs of 11 reported by Actelion suggests that the substitution pattern on the 1-benzyl group may have a significant effect on the activity at the orexin receptors.38 For instance, the corresponding phenyl analog which lacked the dimethoxy groups at the 3 and 4-positons, showed little activity at either receptor. Removing one of the methoxy groups also resulted in significant loss of activity at both receptors. Therefore, we first evaluated a series of 3,4-disubstituted benzyl analogs that retained the 3-methyoxy group but had different alkyl-substituted groups at the 4-position of the benzyl group (Table 1). All of the target compounds were prepared racemically, although the 1-position stereochemistry is undoubtedly important, as evidenced by both 1 and our own findings.34, 39 While removing the methyl group at the 4-methoxy position gave a slight drop in potency in phenol 12, the potency was regained and even increased by substituting with a larger alkyl group such as butyl 13 or hexyl 14, with 13 having slightly higher potency. Further increasing the size to a piperidinylethyl group, which may provide improved drug-like properties due to the basic nitrogen, resulted in a total loss of activity, suggesting limited size tolerance at this site. Interestingly, the butyric ester 16 was equally potent to the butyl analog 13, suggesting polar groups can be tolerated at this site. Analog 17 with a mesyl group, which has the size between a methyl (11) and butyl groups (13) but a high electron deficiency, had a Ke = 485 nM, only slightly less potent than 11. These findings indicate that steric bulk may play a more prominent role at this position than electronic effects. A series of aromatic substituents at the 4-alkoxy position were then examined (18-22). The O-benzyl analog 18 was 2-fold less potent than 11. Extending the phenyl group away from the ring system had no effect on potency (19). The corresponding sulfonyl analog (20) showed no potency at the OX1 receptor. The two pyridylmethyl derivatives 21 and 22 showed similar potency to compound 11. Finally, the phenylsulfonyl analog (23) had a ~9-fold decrease in potency.
We next synthesized a series of compounds that had modifications at the 3-position of the 1-benzyl group, while retaining the 4-methoxy group (Table 1). Replacement of the oxygen moiety with a series of nitrogen-containing groups, with the aim of reducing logP and improving the drug-likeness, gave some interesting results. The 3-nitro analog (26) showed ~7-fold reduced potency, whereas dimethylamino analog 28 gave a significant improvement of potency at the OX1 receptor (Ke = 13 nM) but also increased the OX2 receptor potency. However, larger N-alkyl groups were not as well tolerated and the potency decreased with the increase of the size of the alkyl groups (29-33), with the benzyl analog (33) having no activity at concentrations up to 10 μM. Interestingly, while the acetyl derivatives 34 had a significant drop in potency, 35, which had a larger acyl group, regained most of the potency. Finally, the corresponding 4-hydroxyl substituent had a significant potency reduction compared to its 4-methoxy analog (36 vs. 26). The benzoxazole 38 had a Ke in the micromolar range.
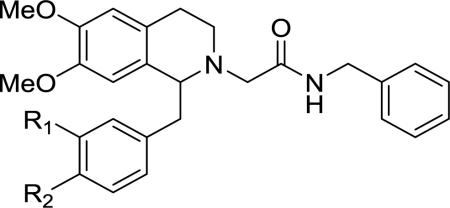
All of the 1-benzyl analogs discussed thus far have a 3,4-substitution pattern. The relative importance of each of those positions was then examined by preparing a series of 3-substituted and 4-substituted analogs (Table 2). As previously reported, analogs singly substituted with a methoxyl group at either the 3- or 4-position had diminished activity at both OX1 and OX2 receptors.38 Therefore, we have examined a series of analogs that bear substituents other than a methoxyl group at these positions. The 3-isopropoxyl analog (41) had a ~7-fold drop in OX1 potency. In the 3-nitrogen containing analogs, the 3-nitro 42 showed activity only at OX2 (1200 nM), whereas the aniline 43 showed modest potency at the OX1 receptor and no activity at OX2. Potency at the OX1 receptor was further increased by dimethylation (44), which was the most potent mono-substituted compound in the series. However, 44 also showed significant potency at the OX2 receptor (Ke = 660 nM). It appears that the 4-position may contribute more to the OX1 potency as the 4-substituted mono isopropoxy derivative (46) showed increased potency compared to the corresponding 3-substituted derivative (41). The differences in OX1 potency between the n-propyl 45 and the isopropyl 46 were modest and both were slightly less potent than 11. In the 4-nitrogen series, the 4-aniline 47 showed no OX1 activity and again this was restored with the 4-dimethylamino analog 48, which had similar potency to compound 11 (253 v 199 nM). As with the disubstituted analogs, acylation as the acetamide 49 or the urea 50 caused a significant drop in potency. Finally, the isopropyl analog 51 had good potency, with a Ke of 85 nM, further reinforcing the idea that steric factors are the overriding factor in potency. The OX2 selectivity of the nitro 42 indicates some preference for electron-withdrawing substituents for OX2 potency and indeed a trifluoromethyl substituent may contribute to the OX2 potency of 1.
Table 2.
Mono-substituted benzylic substituents at the 1-position and their effect on OX antagonism
| |||||
|---|---|---|---|---|---|
| Number | R1 | R2 | Ke (OX1, nM)b | Ke (OX2, nM)c | OX2/OX1 |
| 11 | OMe | OMe | 199± 47 | >10,000 | >50.3 |
| 41 | O-Isopropyl | H | 1470± 70 | >10,000 | >6.8 |
| 42 | NO2 | H | >10,000 | 1200± 160 | <0.12 |
| 43 | NH2 | H | 1310± 90 | a | |
| 44 | NMe2 | H | 75.3± 1.3 | 660±160 | 8.8 |
| 45 | H | O-n-Propyl | 370± 50 | >10,000 | >27 |
| 46 | H | O-Isopropyl | 489± 68 | >10,000 | >20 |
| 47 | H | NH2 | >10,000d | a | |
| 48 | H | NMe2 | 253± 85 | >10,000 | >40 |
| 49 | H | NHAc | >10,000d | a | |
| 50 | H | NHCONH-n-hexyl | >10,000d | a | |
| 51 | H | Isopropyl | 85± 21 | >10,000 | >118 |
< 35% inhibition at 10 μM
Values are the mean ± SEM of at least three independent experiments performed in duplicate
Values are the mean ± SEM of at least two independent experiments in performed duplicate; for compounds with Ke < 100 nM at OX1 at least three independent experiments in duplicate were performed.
Values are the mean ± SEM of two independent experiments performed in duplicate.
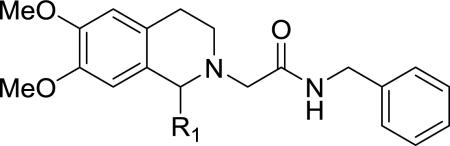
We then investigated a series of alternate substituents, including differentially substituted benzylic and non-aromatic systems to further examine the SAR requirements at this position (Table 3). Surprisingly, the 3,4,5-trimethoxy analog 52 was mostly inactive at both receptors. This clearly shows that the 1-benzyl substituent is highly sensitive to substitutions, confirming the earlier observations. The 3,4-dimethylbenzyl analog 53 showed higher potency than 11, further illustrating the importance of steric effects on potency. The naphthyl (54) and quinoline (55) analogs showed a modest reduction in potency compared to 11. Interestingly, removing the methylene group resulted in a total loss of activity (59 vs. 11). Elongation of the methylene group (60, 62, 63) also led to significant loss of activity at the OX1 receptor. This is somewhat surprising as the dual orexin antagonist 1 has a phenethyl group and has high potency at both receptors. Introduction of rigidity (61) led to diminished activity. The requirement for an aromatic system was then examined with the replacement of the 1-benzyl with nonaromatic systems (Table 3). A series of alkyl analogs were prepared and their OX potency evaluated; however, none of them (64-69) showed detectable antagonism in our assay at concentrations of 10 μM. Taken together, these findings clearly confirm that an optimally substituted benzyl group is required for OX1 potency.
Table 3.
Other substituents at the 1-position and their effect on OX antagonism
| ||||
|---|---|---|---|---|
| Number | R1 | Ke (OX1, nM)b | Ke (OX2, nM)c | OX2/OX1 |
| 11 |
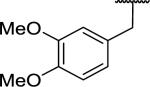
|
199 ± 47 | >10,000 | >50.3 |
| 52 |
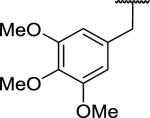
|
>10,000d | >10,000 | |
| 53 |
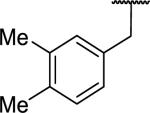
|
94 ± 28 | >10,000 | >32 |
| 54 |
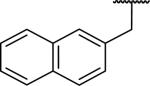
|
245 ± 40 | 1620 ± 230 | 6.6 |
| 55 |
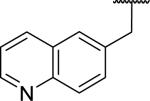
|
432 ± 22 | >10,000 | >23 |
| 56 |
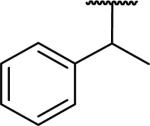
|
>10,000d | >10,000 | |
| 59 |
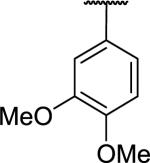
|
>10,00d | a | |
| 60 |
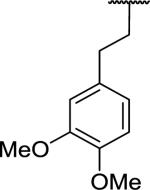
|
>10,00d | >10,000 | |
| 61 |
|
>10,00d | a | |
| 62 |
|
>10,000d | a | |
| 63 |
|
>10,000d | a | |
| 64 |
|
>10,000d | a | |
| 65 |
|
>10,000d | a | |
| 66 |
|
>10,000d | a | |
| 67 |
|
>10,000d | a | |
| 68 |
|
>10,000d | a | |
| 69 |
|
>10,000d | a | |
< 35% inhibition at 10 μM
Values are the mean ± SEM of at least three independent experiments performed in duplicate
Values are the mean ± SEM of at least two independent experiments in performed duplicate; for compounds with Ke < 100 nM at OX1 at least three independent experiments in duplicate were performed.
Values are the mean ± SEM of two independent experiments performed in duplicate.
The structural modifications at the 1-position have led to the identification of several compounds that have improved OX1 potency compared to the 3,4-dimethoxy analog 11. Several compounds also showed improved OX1 selectivity, greater than 50-fold (e.g. 16, 28, 51), even though this improvement is only modest. As previously reported by our group, introducing larger alkyl groups such as a 7-propoxy at the 7-methoxy position led to improved potency and selectivity. Therefore, we synthesized a dipropoxy analog which has a propoxy group at the 7-position of the tetrahydroisoquinoline and the 4-position of the 1-benzyl group, respectively (73, RTIOX-251, Figure 2). Indeed, these modifications resulted in an analog that showed improvement in both OX1 potency and selectivity (73). Compound 73 had a Ke of 16.1 nM (vs. 48 nM for the close analog 13), but it was selectivity where the greatest improvement was seen in 73, which increased to approximately >620-fold.
Figure 2.
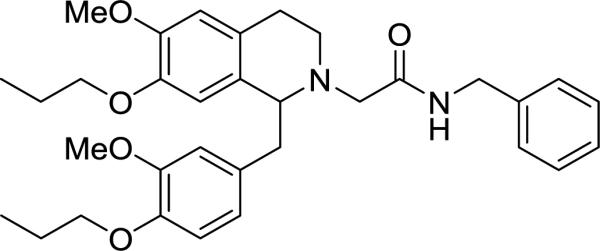
7-Propoxy-1-(4-propoxy)benzyl tetrahydroisoquinoline derivative 73.
Cocaine-induced Behavioral Sensitization
OX1 receptor selective antagonist SB334867 has been reported to block the development of locomotor sensitization to cocaine when administrated i.p.40 We next moved on to test one of the compounds that showed the highest potency and selectivity in this series of compounds, compound 73, on the development of behavioral sensitization to cocaine in rats. As expected, acute cocaine treatment induced a dose-dependent hyperactivity (open circles, left panel, Fig. 3). Daily treatment with 15 mg/kg cocaine for 7 days induced a significant locomotor sensitization, as demonstrated by a leftward shift of the cocaine dose-effect curve on day 15 as compared to day 1 (compare gray circles with open circles, right panel, Fig. 3). Two way ANOVA revealed significant main effects of repeated treatment (F [2, 14] = 24.7, P < 0.0001) and repeated treatment × cocaine dose interactions (F [2, 14] = 35.2, P < 0.0001). Post hoc analysis indicated that on day 15, the effects of 3.2 and 10 mg/kg cocaine were significantly increased while the effect of 32 mg/kg cocaine was significantly decreased. Although 10 mg/kg compound 73, a dose that alone did not significantly alter the spontaneous activity in rats (data not shown), did not alter acute cocaine-induced hyperactivity when given acutely (left panel, Fig. 3), repeated treatment with compound 73 significantly blocked the development of cocaine sensitization (compare open circles with open squares, right panel, Fig. 3). Two way ANOVA revealed significant main effects of repeated treatment (F [2, 14] = 7.2, P < 0.01) and repeated treatment × cocaine dose interactions (F [2, 14] = 10.5, P < 0.01). Post hoc analysis indicated that on day 15, the effects of 10 mg/kg cocaine were significantly lower while the effect of 32 mg/kg cocaine was significantly higher in the compound 73 treated group as compared to control group, suggesting a significant blockade of cocaine sensitization.
Figure 3.

Compound 73 attenuated the development of cocaine-induced behavioral sensitization. Left: compound 73 did not significantly alter acute cocaine-induced hyperactivity (n=8/group). Right: compound 73 significantly reduced the development of cocaine sensitization during the challenge test (* P < 0.05 as compared with control group). Dashed line represents the replotted data of control group in Day 1 for comparison in Day 15. Data represent the mean ± SEM. The absence of error bars indicates that the variability is contained within the data point. V, vehicle.
Conclusions
Recently, the orexin system has been indicated to play an important role in the reward pathway and OX1 receptor selective antagonists have been suggested to hold value for the treatment of addiction to a number of illicit drugs. While several OX1 antagonists have been reported so far, they tend to retain some activity at the OX2 receptor. In our continued effort to investigate the SAR on the tetrahydroisoquinoline scaffold and develop highly potent and selective OX1 antagonists, we have designed and synthesized a series of compounds with a range of substituents at the 1-position. These compounds were then evaluated for the potency at the OX1 and OX2 receptors in FLPR-based calcium mobilization assays. The SAR results indicate that an optimally substituted benzyl group is required for activity at the OX1 receptor. Shortening or elongation of the methylene unit in the benzyl group both led to dramatic decreases in OX1 potency. Other nonaromatic systems including straight chain or cyclic alkyl groups were also not well tolerated. A number of analogs (e.g. 13, 16 and 51) showed improvement on potency at the OX1 receptor compared with the dimethoxy substitution (11). In particular, the 3-dimethylamino-4-methyoxy substitution pattern (28) provided the best OX1 potency (Ke = 12.7 nM) and reasonable selectivity (76.7-fold), although some activity at the OX2 potency remains (Ke = 970 nM). When structural modifications at the 7-position of the tetrahydroisoquinoline that have been shown to improve potency were introduced, OX1 potency and selectivity were further enhanced. Compound 73 (RTIOX-251) is both a potent and highly selective OX1 receptor antagonist. At 10 mg/kg 73 did not alter acute cocaine-induced hyperactivity when given acutely, but blocked the development of locomotor sensitization to cocaine in rats when repeatedly administrated.
Experimental Procedures
General
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all were purchased from commercial vendors and used as received. Flash column chromatography was done on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used were hexane, ethyl acetate (EtOAc), dichloromethane (DCM), methanol and chloroform:methanol:ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds was established by a combination of high pressure liquid chromatography (HPLC), thin layer chromatography (TLC), mass spectrometry (MS) and nuclear magnetic resonance (NMR) analysis. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in chloroform-d or methanol-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in Hz. TLC was done on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. Low resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). High resolution mass spectra were obtained using an Agilent 6230 time-of-flight mass spectrometer. Melting points were determined using a Mel Temp II melting point apparatus and are uncorrected. All test compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm x 150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.1% CF3COOH and (B) MeCN, with a flow rate of 1.0 mL/min.
General procedures
N-[2-(3,4-Dimethoxyphenyl)ethyl]-2-(3-hydroxy-4-methoxyphenyl)acetamide (9)
3-Hydroxy-4-methoxyphenylacetic acid (1.0 g, 5.49 mmol), 3,4-dimethoxyphenethylamine (1.0 g, 0.93 mL, 5.49 mmol) and HBTU (2.08 g, 5.49 mmol) were combined in dry DMF (55 mL) at RT under N2. Diisopropylethylamine (1.77 g, 2.4 mL, 13.72 mmol) was added and the reaction stirred at RT overnight. The reaction was diluted with EtOAc, washed with 2N HCl, NaHCO3 solution and brine, dried over MgSO4 and the solvent removed under reduced pressure to give the desired amide as a yellow oil which solidified upon standing (1.50 g, 79%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.39 - 7.50 (m, 1H), 6.81 - 6.88 (m, 1H), 6.71 (d, J = 8.19 Hz, 1H), 6.57 - 6.67 (m, 3H), 6.50 (dd, J = 1.98, 8.10 Hz, 1H), 5.52 (br. s., 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.82 (s, 3H), 3.47 (s, 2H), 3.43 (t, J = 6.12 Hz, 2H), 2.67 (t, J = 6.83 Hz, 2H).
4-[(6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl)methyl]-2-methoxyphenol (10)
Amide 9 (1.51 g, 4.37 mmol) was suspended in anhydrous toluene (22 mL) and phosphorous oxychloride (4.02 g, 2.45 mL, 26.23 mmol) added slowly. The reaction was heated to 90 °C for 2 hr, during which the solid went into solution, then a red oil separated. The reaction was cooled, then quenched by slow addition of the reaction mixture to water and heated until a solution formed. The toluene layer was removed, 2N sodium hydroxide solution was added until pH was 8-9, then the solution was extracted 3 times with DCM. The combined extracts were dried over MgSO4 and the solvent removed under reduced pressure.
The crude dihydroisoquinoline was dissolved in methanol (25 mL) and cooled in an ice bath under N2. Sodium borohydride (0.83 g, 21.99 mmol) was added portionwise and the reaction allowed to warm slowly to RT overnight. The reaction was quenched with water then the methanol removed under reduced pressure. The aqueous solution was extracted 3 times with DCM and the combined extracts were dried over MgSO4 and the solvent removed under reduced pressure to give the desired tetrahydroisoquinoline as an off-white foam (0.73 g, 91%). 1H NMR (300 MHz, CHLOROFORM-d) δ 6.86 (d, J = 7.82 Hz, 1H), 6.70 - 6.79 (m, 2H), 6.66 (s, 1H), 6.60 (s, 1H), 4.13 (dd, J = 4.29, 9.18 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H), 3.11 - 3.27 (m, 2H), 2.83 - 2.98 (m, 4H), 2.63 - 2.79 (m, 2H).
N-Benzyl-2-{1-[(4-hydroxy-3-methoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (12)
Amine 10 (0.20 g, 0.61 mmol), N-benzyl-2-bromoacetamide (0.152 g, 0.67 mmol) and tetrabutylammonium iodide (0.045 g, 0.12 mmol) were combined in dry DMF (6 mL) and diisopropylethylamine (0.196 g, 0.26 mL, 1.52 mmol) was added. The reaction was stirred at RT overnight under N2. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine (x2), then dried over MgSO4 and the solvent removed under reduced pressure. The crude was purified by chromatography on silica (0-60% EtOAc in hexane) to obtain the desired product as a pale yellow oil (0.097 g, 33%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.21 - 7.36 (m, 3H), 7.14 (d, J = 6.69 Hz, 2H), 6.93 - 7.04 (m, 1H), 6.76 - 6.84 (m, 1H), 6.66 - 6.73 (m, 1H), 6.64 (d, J = 1.51 Hz, 1H), 6.59 (s, 1H), 6.45 (s, 1H), 5.50 (s, 1H), 4.48 (dd, J = 8.01, 14.69 Hz, 1H), 3.87 (s, 3H), 3.82 (s, 3H), 3.76 (s, 3H), 3.56 - 3.72 (m, 2H), 3.35 - 3.49 (m, 1H), 3.09 - 3.34 (m, 2H), 2.79 - 3.01 (m, 4H), 2.42 - 2.56 (m, 1H). m/z 477 (M+H). HRMS (ESI, CH3OH) m/z calcd for C28H33N2O5 [M+H]+ 477.2384, m/z found 477.2411.
N-Benzyl-2-{1-[(4-butoxy-3-methoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (13)
Phenol 12 (50 mg, 0.105 mmol), potassium carbonate (29 mg, 0.210 mmol) and tetrabutylammonium iodide (8 mg, 0.021 mmol) were combined in DMF (1 mL) and 1-bromobutane (16 mg, 12 μL, 0.115 mmol) was added. The reaction was heated at 50 °C overnight. It was diluted with EtOAc, washed with water and brine, dried over MgSO4 and the solvent removed under reduced pressure. The crude was purified by chromatography on silica (0-75% EtOAc in hexane) to give the desired product as a yellow glassy solid (47 mg, 84%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 3H), 7.11 (d, J = 6.50 Hz, 2H), 6.96 - 7.06 (m, 1H), 6.63 - 6.72 (m, 3H), 6.58 (s, 1H), 6.44 (s, 1H), 4.49 (dd, J = 8.05, 14.93 Hz, 1H), 3.83 - 3.91 (m, 5H), 3.81 (s, 3H), 3.79 (s, 3H), 3.58 - 3.71 (m, 2H), 3.34 - 3.51 (m, 1H), 3.10 - 3.34 (m, 2H), 2.78 - 3.00 (m, 4H), 2.41 - 2.56 (m, 1H), 1.72 - 1.87 (m, 2H), 1.39 - 1.53 (m, 2H), 0.97 (t, J = 7.35 Hz, 3H). m/z 533 (M+H). HRMS (ESI, CH3OH) m/z calcd for C32H41N2O5 [M+H]+ 533.3010, m/z found 533.3057.
N-Benzyl-2-(1-{[4-(hexyloxy)-3-methoxyphenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (14)
This was made by the general procedure from phenol 12 in 73% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.34 (m, 3H), 7.08 - 7.16 (m, 2H), 7.01 (dd, J = 5.23, 7.49 Hz, 1H), 6.63 - 6.73 (m, 3H), 6.59 (s, 1H), 6.44 (s, 1H), 4.49 (dd, J = 8.01, 14.88 Hz, 1H), 3.84 - 3.90 (m, 5H), 3.81 (s, 3H), 3.79 (s, 3H), 3.58 - 3.71 (m, 2H), 3.34 - 3.49 (m, 1H), 3.10 - 3.34 (m, 2H), 2.79 - 2.99 (m, 4H), 2.41 - 2.56 (m, 1H), 1.74 - 1.88 (m, 2H), 1.29 - 1.51 (m, 6H), 0.91 (t, J = 6.78 Hz, 3H). m/z 561 (M+H). HRMS (ESI, CH3OH) m/z calcd for C34H45N2O5 [M+H]+ 561.3323, m/z found 561.3362.
N-Benzyl-2-[6,7-dimethoxy-1-({3-methoxy-4-[2-(piperidin-1-yl)ethoxy]phenyl}methyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (15)
This was prepared from 12 using the general procedure in 27% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.35 (m, 3H), 7.12 (d, J = 6.69 Hz, 2H), 7.03 (dd, J = 5.09, 7.44 Hz, 1H), 6.66 - 6.73 (m, 3H), 6.59 (s, 1H), 6.43 (s, 1H), 4.48 (dd, J = 7.96, 14.93 Hz, 1H), 4.02 (t, J = 6.31 Hz, 2H), 3.87 (s, 3H), 3.81 (s, 3H), 3.78 (s, 3H), 3.59 - 3.72 (m, 2H), 3.34 - 3.48 (m, 1H), 3.11 - 3.34 (m, 2H), 2.83 - 2.99 (m, 4H), 2.76 - 2.82 (m, 2H), 2.42 - 2.56 (m, 5H), 1.61 (quin, J = 5.49 Hz, 4H), 1.40 - 1.51 (m, 2H). m/z 588 (M+H). HRMS (ESI, CH3OH) m/z calcd for C35H46N3O5 [M+H]+ 588.3432, m/z found 588.3489.
4-({2-[(Benzylcarbamoyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl}methyl)-2-methoxyphenyl butanoate (16)
To a mixture of phenol 12 (50 mg, 0.105 mmol), butyric acid (9 mg, 10 μL, 0.105 mmol) and BOP (46 mg, 0.105 mmol) in DCM (1 mL) was added diisopropylethylamine (34 mg, 46 μL, 0.262 mmol) and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with 2N HCl, NaHCO3 solution and brine, then dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-75% EtOAc in hexane) to give the desired ester as a yellow oil (54 mg, 95%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.23 - 7.35 (m, 3H), 7.14 - 7.21 (m, 2H), 7.07 (t, J = 6.22 Hz, 1H), 6.78 - 6.85 (m, 1H), 6.66 - 6.74 (m, 2H), 6.58 (s, 1H), 6.36 (s, 1H), 4.40 - 4.50 (m, 1H), 3.86 (s, 3H), 3.79 (s, 3H), 3.70 (s, 3H), 3.63 - 3.95 (m, 2H), 3.11 - 3.47 (m, 3H), 2.78 - 3.04 (m, 4H), 2.42 - 2.63 (m, 3H), 1.73 - 1.89 (m, 2H), 1.07 (t, J = 7.39 Hz, 3H). m/z 547 (M+H). HRMS (ESI, CH3OH) m/z calcd for C32H39N2O6 [M+H]+ 547.2803, m/z found 547.2845.
4-({2-[(Benzylcarbamoyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl}methyl)-2-methoxyphenyl methanesulfonate (17)
To a solution of phenol 12 (30 mg, 0.063 mmol) in DCM (0.5 mL) cooled in ice was added methanesulfonyl chloride (14 mg, 10 μL, 0.126 mmol) and triethylamine (16 mg, 22 μL, 0.157 mmol). The reaction was allowed to warm to RT overnight. The reaction mixture was applied directly to silica for chromatography (0-100% EtOAc in hexane) to give the desired sulfonate as a yellow oil (21 mg, 60%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.22 - 7.37 (m, 3H), 7.17 (d, J = 6.88 Hz, 2H), 7.09 (d, J = 8.29 Hz, 2H), 6.68 - 6.77 (m, 2H), 6.60 (s, 1H), 6.32 (s, 1H), 4.44 (dd, J = 7.25, 14.88 Hz, 1H), 3.95 (dd, J = 5.09, 14.98 Hz, 1H), 3.87 (s, 3H), 3.77 (s, 6H), 3.66 - 3.74 (m, 1H), 3.28 - 3.42 (m, 2H), 3.22 (br. s., 1H), 3.14 (s, 3H), 2.81 - 3.06 (m, 4H), 2.53 (d, J = 16.20 Hz, 1H). m/z 555 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H35N2O7S [M+H]+ 555.2160, m/z found 555.2212.
N-Benzyl-2-(1-{[4-(benzyloxy)-3-methoxyphenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (18)
This was prepared from phenol 12 by the general procedure in 15% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.45 (m, 8H), 7.12 (d, J = 6.78 Hz, 2H), 6.98 - 7.07 (m, 1H), 6.61 - 6.74 (m, 3H), 6.58 (s, 1H), 6.41 (s, 1H), 5.00 (s, 2H), 4.45 (dd, J = 8.01, 14.98 Hz, 1H), 3.86 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 3.55 - 3.76 (m, 2H), 3.34 - 3.48 (m, 1H), 3.10 - 3.33 (m, 2H), 2.77 - 2.99 (m, 4H), 2.41 - 2.55 (m, 1H). m/z 567 (M+H). HRMS (ESI, CH3OH) m/z calcd for C35H39N2O5 [M+H]+ 567.2854, m/z found 567.2917.
N-Benzyl-2-(6,7-dimethoxy-1-{[3-methoxy-4-(4-phenoxybutoxy)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (19)
This was prepared from 12 using the general procedure in 71% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.35 (m, 5H), 7.11 (d, J = 6.88 Hz, 2H), 7.02 (dd, J = 5.04, 7.39 Hz, 1H), 6.87 - 6.98 (m, 3H), 6.65 - 6.73 (m, 3H), 6.60 (s, 1H), 6.45 (s, 1H), 4.50 (dd, J = 8.01, 14.98 Hz, 1H), 4.04 (t, J = 5.89 Hz, 2H), 3.90 - 3.97 (m, 2H), 3.87 (s, 3H), 3.82 (s, 3H), 3.79 (s, 3H), 3.60 - 3.73 (m, 2H), 3.36 - 3.51 (m, 1H), 3.11 - 3.35 (m, 2H), 2.80 - 3.01 (m, 4H), 2.44 - 2.57 (m, 1H), 1.91 - 2.04 (m, 4H). m/z 625 (M+H). HRMS (ESI, CH3OH) m/z calcd for C38H45N2O6 [M+H]+ 625.3272, m/z found 625.3335.
4-({2-[(Benzylcarbamoyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl}methyl)-2-methoxyphenyl 4-methylbenzene-1-sulfonate (20)
This was prepared from 12 as sulfonate 17 above. Yield 80%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.76 (d, J = 8.29 Hz, 2H), 7.21 - 7.36 (m, 5H), 7.17 (d, J = 6.97 Hz, 2H), 7.05 (br. s., 1H), 6.91 (d, J = 8.10 Hz, 1H), 6.54 - 6.67 (m, 3H), 6.31 (s, 1H), 4.44 (dd, J = 7.30, 14.74 Hz, 1H), 3.92 (d, J = 4.90 Hz, 1H), 3.86 (s, 3H), 3.76 (s, 3H), 3.62 - 3.71 (m, 1H), 3.49 (s, 3H), 3.10 - 3.39 (m, 3H), 2.75 - 3.03 (m, 4H), 2.49 - 2.57 (m, 1H), 2.46 (s, 3H). m/z 631 (M+H). HRMS (ESI, CH3OH) m/z calcd for C35H39N2O7S [M+H]+ 631.2473, m/z found 631.2536.
N-Benzyl-2-(6,7-dimethoxy-1-{[3-methoxy-4-(pyridin-2-ylmethoxy)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (21)
This was prepared from 12 using the general procedure in 92% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.59 (dd, J = 0.85, 4.05 Hz, 1H), 7.61 - 7.71 (m, 1H), 7.53 (d, J = 7.82 Hz, 1H), 7.17 - 7.32 (m, 4H), 7.10 (d, J = 6.59 Hz, 2H), 6.96 - 7.05 (m, 1H), 6.69 - 6.75 (m, 2H), 6.62 - 6.68 (m, 1H), 6.59 (s, 1H), 6.42 (s, 1H), 5.15 (s, 2H), 4.46 (dd, J = 7.86, 14.84 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.58 - 3.74 (m, 2H), 3.34 - 3.48 (m, 1H), 3.11 - 3.33 (m, 2H), 2.78 - 3.00 (m, 4H), 2.50 (d, J = 15.82 Hz, 1H). m/z 568 (M+H). HRMS (ESI, CH3OH) m/z calcd for C34H38N3O5 [M+H]+ 568.2806, m/z found 568.2869.
N-Benzyl-2-(6,7-dimethoxy-1-{[3-methoxy-4-(pyridin-3-ylmethoxy)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (22)
This was prepared from 12 using the general procedure in 42% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.65 (d, J = 1.60 Hz, 1H), 8.57 (dd, J = 1.27, 4.76 Hz, 1H), 7.76 (d, J = 7.82 Hz, 1H), 7.18 - 7.34 (m, 4H), 7.12 (d, J = 6.59 Hz, 2H), 7.04 (br. s., 1H), 6.64 - 6.74 (m, 3H), 6.60 (s, 1H), 6.42 (s, 1H), 4.95 (s, 2H), 4.49 (dd, J = 7.91, 15.16 Hz, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.80 (s, 3H), 3.62 - 3.77 (m, 2H), 3.13 - 3.50 (m, 3H), 2.80 - 3.02 (m, 4H), 2.52 (d, J = 15.73 Hz, 1H). m/z 568 (M+H). HRMS (ESI, CH3OH) m/z calcd for C34H38N3O5 [M+H]+ 568.2806, m/z found 568.2869.
4-({2-[(Benzylcarbamoyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl}methyl)-2-methoxyphenyl benzenesulfonate (23)
This was prepared from 12 as sulfonate 17 above. Yield 69%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.85 - 7.92 (m, 2H), 7.62 - 7.71 (m, 1H), 7.48 - 7.57 (m, 2H), 7.21 - 7.36 (m, 3H), 7.17 (d, J = 6.78 Hz, 2H), 7.04 (t, J = 5.93 Hz, 1H), 6.93 (d, J = 8.10 Hz, 1H), 6.64 (d, J = 8.19 Hz, 1H), 6.55 - 6.60 (m, 2H), 6.31 (s, 1H), 4.44 (dd, J = 7.39, 14.74 Hz, 1H), 3.86 (s, 3H), 3.84 - 3.96 (m, 1H), 3.76 (s, 3H), 3.63 - 3.72 (m, 1H), 3.45 (s, 3H), 3.11 - 3.38 (m, 3H), 2.76 - 3.03 (m, 4H), 2.41 - 2.55 (m, 1H). m/z 617 (M+H). HRMS (ESI, CH3OH) m/z calcd for C34H37N2O7S [M+H]+ 617.2316, m/z found 617.2377.
N-Benzyl-2-{6,7-dimethoxy-1-[(4-methoxy-3-nitrophenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (26)
Phenol 36 (40 mg, 0.08 mmol) and potassium carbonate (17 mg, 0.12 mmol) were combined in DMF (2 mL) and iodomethane (14 mg, 6 μL, 0.097 mmol) was added. The reaction was heated to 50 °C for 2 hr. The reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the desired product (30 mg, 75%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.99 (s, 1H), 7.68 (d, J = 2.26 Hz, 1H), 7.28 - 7.39 (m, 4H), 7.02 - 7.14 (m, 1H), 6.75 (d, J = 8.29 Hz, 1H), 6.60 (s, 1H), 6.44 (s, 1H), 4.37 - 4.54 (m, 1H), 3.90 - 3.96 (m, 1H), 3.85 - 3.88 (m, 3H), 3.82 (s, 3H), 3.76 (s, 3H), 3.65 (dd, J = 5.46, 9.23 Hz, 1H), 3.38 - 3.47 (m, 2H), 3.11 - 3.22 (m, 1H), 2.90 - 3.00 (m, 4H), 2.46 - 2.58 (m, 1H). m/z 506 (M+H). HRMS (ESI, CH3OH) m/z calcd for C28H32N3O6 [M+H]+ 506.2286, m/z found 506.2327.
2-{1-[(3-Amino-4-methoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-benzylacetamide (27)
To the nitro derivative 26 (200 mg, 0.4 mmol) in ethanol (20 mL) was added hydrazine monohydrate (198 mg, 0.19 mL, 4.0 mmol) and then heated to 50 °C for 15 min. Raney nickel (2800 type as a slurry in water, 232 mg, 4.0 mmol) was added and heating continued for 1 hr. The reaction was filtered through Celite, rinsed with ethanol then the solvent was removed under reduced pressure to give the desired amine as a clear oil (100 mg, 53%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.68 (s, 2H), 7.20 - 7.35 (m, 5H), 7.09 (d, J = 6.78 Hz, 2H), 6.85 (br. s., 1H), 6.75 (d, J = 8.48 Hz, 1H), 6.59 (s, 1H), 6.43 (s, 1H), 4.47 (dd, J = 7.44, 15.16 Hz, 1H), 3.87 (s, 3H), 3.85 - 3.98 (m, 1H), 3.82 (s, 3H), 3.77 (s, 3H), 3.60 - 3.71 (m, 1H), 3.11 - 3.48 (m, 3H), 2.83 - 3.05 (m, 4H), 2.51 (d, J = 16.77 Hz, 1H). m/z 476 (M+H).
N-Benzyl-2-(1-{[3-(dimethylamino)-4-methoxyphenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (28)
To a solution of amine 27 (50 mg, 0.10 mmol) in methanol (1 mL) was added formaldehyde (37% solution in water, 1 mL) and glacial acetic acid (21 mg, 20 μL, 0.35 mmol). To this was then added sodium cyanoborohydride (31 mg, 0.5 mmol) and the reaction stirred at RT for 2 hr. 1N HCl (0.1 mL) was added then the reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-50% EtOAc in hexane) to give the desired dimethylamine (26 mg, 52%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 3H), 7.01 - 7.14 (m, 3H), 6.70 - 6.78 (m, 2H), 6.55 - 6.65 (m, 2H), 6.41 (s, 1H), 4.48 (dd, J = 7.82, 14.98 Hz, 1H), 3.86 (s, 3H), 3.80 (s, 3H), 3.75 (s, 3H), 3.59 - 3.72 (m, 2H), 3.37 - 3.51 (m, 1H), 3.13 - 3.35 (m, 2H), 2.79 - 3.00 (m, 4H), 2.75 (s, 6H), 2.44 - 2.56 (m, 1H). m/z 504 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H38N3O4 [M+H]+ 504.2857, m/z found 504.2906.
N-Benzyl-2-(1-{[3-(ethylamino)-4-methoxyphenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (29)
To amine 27 (30 mg, 0.06 mmol) in DMF (3 mL) was added 1-iodoethane (20 mg, 10 μL, 0.13 mmol) then diisopropylethylamine (20 mg, 26 μL, 0.16 mmol) and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-15% methanol in DCM) to give the desired amine as a white solid (15 mg, 47%): mp 122-125 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.32 (m, 3H), 7.01 - 7.11 (m, 3H), 6.41 - 6.61 (m, 5H), 4.50 (dd, J = 8.29, 14.88 Hz, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.71 (s, 3H), 3.39 - 3.66 (m, 3H), 3.02 - 3.32 (m, 4H), 2.78 - 2.99 (m, 4H), 2.42 - 2.54 (m, 1H), 1.26 (t, J = 7.16 Hz, 3H). m/z 504 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H38N3O4 [M+H]+ 504.2857, m/z found 504.2914.
N-Benzyl-2-(1-{[3-(diethylamino)-4-methoxyphenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (30)
This was prepared as per 29 using 1-iodoethane (3 eq). Yield 76%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.16 - 7.37 (m, 3H), 7.08 (d, J = 7.54 Hz, 2H), 6.27 - 6.75 (m, 5H), 4.50 (dd, J = 8.38, 14.98 Hz, 1H), 3.85 (d, J = 6.03 Hz, 6H), 3.71 (s, 3H), 3.56 - 3.66 (m, 2H), 3.38 - 3.56 (m, 2H), 3.21 - 3.35 (m, 1H), 3.02 - 3.19 (m, 3H), 2.77 - 3.00 (m, 7H), 0.91 - 1.34 (m, 6H). m/z 532 (M+H). HRMS (ESI, CH3OH) m/z calcd for C32H42N3O4 [M+H]+ 532.3170, m/z found 532.3223.
N-Benzyl-2-(6,7-dimethoxy-1-{[4-methoxy-3-(propylamino)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (31)
This was prepared as per 29 using 1-iodopropane (1 eq). Yield 15%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.16 - 7.34 (m, 3H), 6.99 - 7.12 (m, 3H), 6.40 - 6.64 (m, 5H), 4.51 (dd, J = 8.29, 15.07 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.70 (s, 3H), 3.38 - 3.67 (m, 4H), 3.09 - 3.32 (m, 2H), 2.98 - 3.08 (m, 1H), 2.77 - 2.93 (m, 4H), 2.42 - 2.57 (m, 1H), 1.60 - 1.72 (m, 2H), 1.01 (t, J = 7.44 Hz, 3H). m/z 518 (M+H). HRMS (ESI, CH3OH) m/z calcd for C31H40N3O4 [M+H]+ 518.3013, m/z found 518.3055.
N-Benzyl-2-(1-{[3-(dipropylamino)-4-methoxyphenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (32)
This was prepared as per 29 using 1-iodopropane. Yield 51%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.40 (m, 2H), 6.91 - 7.15 (m, 3H), 6.67 - 6.78 (m, 1H), 6.34 - 6.65 (m, 4H), 4.37 - 4.58 (m, 1H), 3.74 - 3.93 (m, 6H), 3.70 (s, 3H), 3.56 - 3.66 (m, 1H), 3.36 - 3.55 (m, 1H), 3.09 - 3.35 (m, 2H), 2.76 - 3.08 (m, 9H), 2.50 (dd, J = 4.05, 15.92 Hz, 1H), 1.29 - 1.71 (m, 4H), 0.67 - 1.15 (m, 6H). m/z 560 (M+H). HRMS (ESI, CH3OH) m/z calcd for C34H46N3O4 [M+H]+ 560.3483, m/z found 560.3551.
N-Benzyl-2-(1-{[3-(benzylamino)-4-methoxyphenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (33)
This was prepared as per 29 using benzyl bromide. Yield 82%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.06 - 7.34 (m, 10H), 6.93 (dd, J = 4.90, 7.91 Hz, 1H), 6.53 - 6.64 (m, 3H), 6.44 - 6.52 (m, 2H), 6.41 (d, J = 1.70 Hz, 1H), 4.51 (dd, J = 8.29, 15.07 Hz, 1H), 3.85 (s, 3H), 3.83 (s, 3H), 3.76 (d, J = 0.75 Hz, 2H), 3.73 (s, 3H), 3.47 - 3.62 (m, 2H), 3.25 - 3.40 (m, 1H), 3.01 - 3.24 (m, 2H), 2.58 - 2.96 (m, 4H), 2.42 (dd, J = 4.62, 16.86 Hz, 1H). m/z 566 (M+H). HRMS (ESI, CH3OH) m/z calcd for C35H40N3O4 [M+H]+ 566.3013, m/z found 566.3068.
N-Benzyl-2-{1-[(3-acetamido-4-methoxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (34)
To a solution of amine 27 (50 mg, 0.10 mmol) and diisopropylethlamine (32 mg, 41 μL, 0.25 mmol) in DCM (3 mL) under N2 cooled in an ice bath was added acetyl bromide (12 mg, 8 μL, 0.10 mmol). The reaction was stirred in ice for 10 min, then at RT for 3 hr. The reaction was diluted with NaHCO3 solution and extracted 3 times with EtOAc. The combined extracts were washed with brine, dried over MgSO4 and the solvents were removed under reduced pressure. The crude was purified by chromatography on silica (0-75% EtOAc in hexane) to give the desired amide as a yellow oil (32 mg, 62%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.34 (d, J = 1.51 Hz, 1H), 7.52 (s, 1H), 7.17 - 7.31 (m, 3H), 6.97 - 7.05 (m, 3H), 6.87 (dd, J = 1.70, 8.29 Hz, 1H), 6.66 (d, J = 8.29 Hz, 1H), 6.57 (d, J = 5.65 Hz, 2H), 4.42 (dd, J = 8.01, 15.35 Hz, 1H), 3.87 (s, 6H), 3.73 (s, 3H), 3.44 - 3.71 (m, 3H), 3.05 - 3.34 (m, 2H), 2.84 - 3.00 (m, 4H), 2.44 - 2.56 (m, 1H), 2.13 (s, 3H). m/z 518 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H36N3O5 [M+H]+ 518.2650, m/z found 518.271.
N-[5-({2-[(Benzylcarbamoyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl}methyl)-2-methoxyphenyl]butanamide (35)
To amine 27 (50 mg, 0.1 mmol) and BOP (44 mg, 0.1 mmol) in DMF (3 mL) was added butyric acid (9 mg, 9 μL, 0.1 mmol) then diisopropylethylamine (32 mg, 41 μL, 0.25 mmol) and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with 1N HCl, 1N NaOH solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-15% methanol in DCM) to give the desired amide as a yellow oil (22 mg, 40%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.40 (d, J = 1.88 Hz, 1H), 7.58 (s, 1H), 7.17 - 7.33 (m, 3H), 7.01 (d, J = 6.40 Hz, 3H), 6.86 (dd, J = 2.07, 8.29 Hz, 1H), 6.64 (d, J = 8.29 Hz, 1H), 6.55 - 6.60 (m, 2H), 4.43 (dd, J = 8.10, 15.26 Hz, 1H), 3.86 (s, 6H), 3.72 (s, 4H), 3.41 - 3.69 (m, 2H), 3.05 - 3.34 (m, 2H), 2.77 - 3.02 (m, 4H), 2.42 - 2.55 (m, 1H), 2.32 (t, J = 7.44 Hz, 2H), 1.74 (qd, J = 7.43, 14.81 Hz, 2H), 1.01 (t, J = 7.35 Hz, 3H). m/z 546 (M+H). HRMS (ESI, CH3OH) m/z calcd for C32H40N3O5 [M+H]+ 546.2963, m/z found 546.3021.
N-Benzyl-2-{1-[(4-hydroxy-3-nitrophenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (36)
This was made by the general procedure starting from 4-hydroxy-3-nitrophenylacetic acid in 4 steps in 15% overall yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.85 (s, 1H), 7.21 - 7.40 (m, 4H), 7.05 (d, J = 7.06 Hz, 1H), 6.93 (d, J = 8.57 Hz, 1H), 6.85 (br. s., 1H), 6.60 (s, 1H), 6.44 (s, 1H), 4.37 (dd, J = 7.06, 15.07 Hz, 1H), 3.97 (dd, J = 5.18, 14.98 Hz, 1H), 3.85 (d, J = 11.68 Hz, 6H), 3.57 - 3.71 (m, 1H), 3.26 - 3.49 (m, 3H), 3.10 - 3.24 (m, 1H), 2.80 - 3.03 (m, 4H), 2.52 (d, J = 16.39 Hz, 1H). m/z 492 (M+H). HRMS (ESI, CH3OH) m/z calcd for C27H30N3O6 [M+H]+ 492.2129, m/z found 492.2182.
2-{1-[(3-Amino-4-hydroxyphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-benzylacetamide (37)
To the nitro derivative 36 (100 mg, 0.2 mmol) in ethanol (10 mL) was added hydrazine monohydrate (100 mg, 0.1 mL, 2 mmol) and the reaction warmed to 50 °C. Raney nickel (2800 type as a slurry in water, 20 mg) was added and the reaction stirred at 50 °C for 1 hr. The reaction was cooled, filtered through Celite and washed with ethanol. The solvent was removed under reduced pressure and the crude purified by chromatography on silica (0-10% methanol in DCM) to give the aminophenol (56 mg, 57%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.36 (m, 3H), 7.11 - 7.18 (m, 2H), 7.04 - 7.11 (m, 1H), 6.55 - 6.60 (m, 2H), 6.49 - 6.54 (m, 1H), 6.42 - 6.49 (m, 2H), 5.49 (br. s., 1H), 4.48 (dd, J = 8.01, 14.98 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.72 (dd, J = 4.80, 14.98 Hz, 1H), 3.35 - 3.63 (m, 4H), 3.07 - 3.33 (m, 2H), 2.73 - 2.98 (m, 4H), 2.41 - 2.53 (m, 1H). m/z 462 (M+H). HRMS (ESI, CH3OH) m/z calcd for C27H32N3O4 [M+H]+ 462.2387, m/z found 462.2384.
N-Benzyl-2-{6,7-dimethoxy-1-[(2-methyl-1,3-benzoxazol-5-yl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (38)
To aminophenol 37 (56 mg, 0.12 mmol) in chloroform (10 mL) was added ethyl acetimidate hydrochloride (17 mg, 0.13 mmol). The reaction was heated to reflux for 16 hr. The solvent was removed under reduced pressure and the crude purified by chromatography on silica (0-10% MeOH in DCM) to give the desired benzoxazole (29 mg, 50%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.49 (s, 1H), 7.19 - 7.29 (m, 4H), 7.11 (d, J = 8.29 Hz, 1H), 6.82 - 6.90 (m, 2H), 6.71 (br. s., 1H), 6.60 (s, 1H), 6.49 (s, 1H), 4.34 (dd, J = 7.91, 15.26 Hz, 1H), 3.87 (s, 3H), 3.82 (s, 3H), 3.62 - 3.72 (m, 1H), 3.41 - 3.57 (m, 2H), 3.09 - 3.34 (m, 2H), 2.83 - 3.08 (m, 4H), 2.57 (s, 3H), 2.44 - 2.60 (m, 1H). m/z 486 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H32N3O4 [M+H]+ 486.2387, m/z found 486.2435.
N-Benzyl-2-(6,7-dimethoxy-1-{[3-(propan-2-yloxy)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (41)
This was made by the general procedure starting from 3-hydroxyphenylacetic acid to the phenol precursor in 4 steps in 6% overall yield. For the final step, the phenol (30 mg, 0.067 mmol), potassium carbonate (23 mg, 0.168 mmol) and tetrabutylammonium iodide (5 mg, 0.013 mmol) were combined in DMF (0.5 mL) and 2-bromopropane (12 mg, 9 μL, 0.101 mmol) was added and the reaction heated at 50 °C overnight. An additional 20 μL of 2-bromopropane was added and the reaction was heated at 50 °C for a further 24 hr. It was cooled, diluted with EtOAc, washed with NaHCO3 solution, water and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-70% EtOAc in hexane) to give the desired isoproxy derivative as an off-white solid (20 mg, 61%): mp 123-125 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.35 (m, 3H), 7.04 - 7.17 (m, 3H), 6.92 - 7.04 (m, 1H), 6.76 (d, J = 2.17 Hz, 2H), 6.60 - 6.73 (m, 1H), 6.58 (s, 1H), 6.43 (s, 1H), 4.38 - 4.54 (m, 2H), 3.83 - 3.89 (m, 3H), 3.76 - 3.82 (m, 3H), 3.60 - 3.71 (m, 2H), 3.34 - 3.52 (m, 1H), 3.07 - 3.32 (m, 2H), 2.79 - 3.03 (m, 4H), 2.42 - 2.55 (m, 1H), 1.26 - 1.32 (m, 6H). m/z 489 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H37N2O4 [M+H]+ 489.2748, m/z found 489.2802.
N-Benzyl-2-{6,7-dimethoxy-1-[(3-nitrophenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (42)
This was made by the general procedure starting from 3-nitrophenylacetic acid in 4 steps in 4% overall yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.03 (s, 1H), 7.86 (td, J = 0.99, 8.19 Hz, 1H), 7.43 (d, J = 7.35 Hz, 1H), 7.17 - 7.36 (m, 4H), 6.98 - 7.08 (m, 2H), 6.70 (t, J = 5.65 Hz, 1H), 6.60 (s, 1H), 6.43 (s, 1H), 4.33 (dd, J = 6.97, 14.88 Hz, 1H), 3.87 (s, 3H), 3.92 (d, J = 5.27 Hz, 0H), 3.81 (s, 3H), 3.71 (dd, J = 5.56, 9.32 Hz, 1H), 2.84 - 3.48 (m, 8H), 2.44 - 2.57 (m, 1H). m/z 476 (M+H). HRMS (ESI, CH3OH) m/z calcd for C27H30N3O5 [M+H]+ 476.2180, m/z found 476.2229.
2-{1-[(3-Aminophenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-benzylacetamide (43)
To the nitro derivative 42 (100 mg, 0.21 mmol) in ethanol (12 mL) was added hydrazine monohydrate (100 mg, 0.1 mL, 21 mmol) and the reaction warmed to 50 °C. Raney nickel (2800 type as a slurry in water, 50 mg) was added and the reaction stirred at 50 °C for 1 hr. The reaction was cooled, filtered through Celite and washed with ethanol. The solvent was removed under reduced pressure to give the amine (80 mg, 90%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.21 - 7.36 (m, 3H), 7.15 (d, J = 7.54 Hz, 2H), 6.94 - 7.10 (m, 2H), 6.56 - 6.65 (m, 2H), 6.43 - 6.54 (m, 3H), 4.47 (dd, J = 7.86, 14.93 Hz, 1H), 3.85 (d, J = 13.00 Hz, 6H), 3.76 (dd, J = 4.95, 14.93 Hz, 1H), 3.64 (dd, J = 5.89, 8.90 Hz, 1H), 3.36 - 3.54 (m, 3H), 3.23 - 3.35 (m, 1H), 3.06 - 3.19 (m, 1H), 2.76 - 3.00 (m, 4H), 2.43 - 2.55 (m, 1H). m/z 446 (M+H). HRMS (ESI, CH3OH) m/z calcd for C27H32N3O3 [M+H]+ 446.2438, m/z found 446.2487.
N-Benzyl-2-(1-{[3-(dimethylamino)phenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (44)
To a solution of amine 43 (80 mg, 0.18 mmol) in methanol (1 mL) was added formaldehyde (37% solution in water, 1.5 mL) and glacial acetic acid (39 mg, 37 μL, 0.65 mmol). To this was then added sodium cyanoborohydride (56 mg, 0.9 mmol) and the reaction stirred at RT for 2 hr. 1N HCl (0.1 mL) was added then the reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-50% EtOAc in hexane) to give the desired dimethylamine as an off-white solid (30 mg, 35%): mp 101-103 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.33 (m, 4H), 7.06 - 7.14 (m, 3H), 6.98 - 7.06 (m, 1H), 6.45 - 6.62 (m, 4H), 4.46 (dd, J = 8.19, 14.98 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.55 - 3.71 (m, 2H), 3.39 - 3.53 (m, 1H), 3.08 - 3.33 (m, 2H), 2.86 - 2.88 (m, 6H), 2.78 - 3.00 (m, 4H), 2.42 - 2.55 (m, 1H). m/z 496 (M+Na), 474 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H36N3O3 [M+H]+ 474.2751, m/z found 474.2803.
N-Benzyl-2-{6,7-dimethoxy-1-[(4-propoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (45)
This was prepared as per 41 except using 1-bromopropane to give the desired product as a white solid. Yield of final step 39%: mp 126-127 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.34 (m, 3H), 7.06 - 7.15 (m, 4H), 6.84 - 6.94 (m, 1H), 6.76 (d, J = 8.57 Hz, 2H), 6.58 (s, 1H), 6.45 (s, 1H), 4.47 (dd, J = 8.10, 14.98 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.77 (dt, J = 2.35, 6.59 Hz, 2H), 3.54 - 3.66 (m, 2H), 3.37 - 3.52 (m, 1H), 3.08 - 3.32 (m, 2H), 2.80 - 2.99 (m, 4H), 2.42 - 2.55 (m, 1H), 1.72 - 1.86 (m, 2H), 1.03 (t, J = 7.44 Hz, 3H). m/z 489 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H37N2O4 [M+H]+ 489.2748, m/z found 489.2804.
N-Benzyl-2-(6,7-dimethoxy-1-{[4-(propan-2-yloxy)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (46)
This was prepared as 41 starting from 4-hydroxyphenylacetic acid in 5 steps in 3% overall yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.34 (m, 3H), 7.14 (d, J = 7.63 Hz, 2H), 7.07 (d, J = 8.19 Hz, 2H), 6.94 - 7.03 (m, 1H), 6.76 (d, J = 8.10 Hz, 2H), 6.58 (s, 1H), 6.41 (s, 1H), 4.35 - 4.52 (m, 2H), 3.86 (s, 3H), 3.80 (s, 3H), 3.54 - 3.72 (m, 2H), 3.33 - 3.52 (m, 1H), 3.07 - 3.32 (m, 2H), 2.78 - 2.99 (m, 4H), 2.42 - 2.54 (m, 1H), 1.31 (d, J = 6.03 Hz, 6H). m/z 489 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H37N2O4 [M+H]+ 489.2748, m/z found 489.2807.
2-{1-[(4-Aminophenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-benzylacetamide (47)
This was made by the general procedure starting from 4-nitrophenylacetic acid in 4 steps in 86% overall yield to give the 4-nitrophenyl derivative. The nitro compound (0.89 g, 1.87 mmol) was dissolved in ethanol (30 mL) and to it was added hydrazine monohydrate (1 mL), the solution warmed to 50 °C and Raney nickel (2800 type as a slurry in water, 0.25 g) was added. The reaction was stirred at 50 °C until gas evolution ceased (~ 1 hr) then it was filtered through Celite and the solvent was removed under reduced pressure to give the desired amine as an off-white solid (0.79 g, 95%): mp 145-147 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.21 - 7.37 (m, 3H), 7.17 (d, J = 6.78 Hz, 2H), 6.97 (d, J = 8.10 Hz, 3H), 6.58 (s, 1H), 6.50 (d, J = 8.01 Hz, 2H), 6.46 (s, 1H), 4.47 (dd, J = 7.96, 14.93 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.72 (dd, J = 4.99, 14.79 Hz, 1H), 3.36 - 3.61 (m, 4H), 3.05 - 3.33 (m, 2H), 2.74 - 3.01 (m, 4H), 2.40 - 2.55 (m, 1H). m/z 468 (M+Na), 446 (M+H). HRMS (ESI, CH3OH) m/z calcd for C27H32N3O3 [M+H]+ 446.2438, m/z found 446.2476.
N-Benzyl-2-(1-{[4-(dimethylamino)phenyl]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (48)
This was made by the general procedure starting from 4-dimethylaminophenylacetic acid in 4 steps in 15% overall yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 3H), 7.04 - 7.14 (m, 3H), 6.92 - 7.02 (m, 1H), 6.54 - 6.65 (m, 3H), 6.48 (s, 1H), 4.40 - 4.53 (m, 1H), 3.80 - 3.90 (m, 6H), 3.35 - 3.64 (m, 3H), 3.06 - 3.33 (m, 2H), 2.78 - 3.00 (m, 10H), 2.42 - 2.55 (m, 1H). m/z 474 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H36N3O3 [M+H]+ 474.2751, m/z found 474.2746.
N-Benzyl-2-{1-[(4-acetamidophenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (49)
To a solution of amine 47 (25 mg, 0.056 mmol) and diisopropylethlamine (18 mg, 24 μL, 0.140 mmol) in DCM (1 mL) under N2 cooled in an ice bath was added acetyl chloride (9 mg, 8 μL, 0.112 mmol). The reaction was stirred in ice for 10 min, then at RT for 3 hr. The reaction was diluted with NaHCO3 solution and extracted 3 times with EtOAc. The combined extracts were washed with brine, dried over MgSO4 and the solvents were removed under reduced pressure. The crude was purified by chromatography on silica (0-75% EtOAc in hexane) to give the desired amide as an off-white solid (13 mg, 48%): mp 99-100 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.21 - 7.38 (m, 5H), 7.03 - 7.16 (m, 4H), 6.76 - 6.90 (m, 2H), 6.59 (s, 1H), 6.46 (s, 1H), 4.42 (dd, J = 7.77, 15.31 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.74 - 3.80 (m, 1H), 3.56 - 3.66 (m, 1H), 3.36 - 3.51 (m, 1H), 3.07 - 3.35 (m, 2H), 2.81 - 3.00 (m, 4H), 2.43 - 2.55 (m, 1H), 2.12 (s, 3H). m/z 488 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H34N3O4 [M+H]+ 488.2544, m/z found 488.2597.
N-Benzyl-2-[1-({4-[(hexylcarbamoyl)amino]phenyl}methyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (50)
To the amine 47 (27 mg, 0.061 mmol) in toluene (1 mL) was added n-hexylisocyanate (8.5 mg, 10 μL, 0.067 mmol) and the reaction heated to 75 °C for 3 hr. The reaction was cooled and the solvents were removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the desired urea as a white solid (33 mg, 94%): mp 81-84 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.22 - 7.35 (m, 4H), 7.05 - 7.13 (m, 5H), 6.82 - 6.93 (m, 1H), 6.59 (s, 1H), 6.45 (s, 1H), 5.97 (s, 1H), 4.55 (t, J = 5.56 Hz, 1H), 4.42 (dd, J = 7.77, 15.12 Hz, 1H), 3.86 (s, 3H), 3.81 (s, 3H), 3.73 - 3.80 (m, 1H), 3.61 (dd, J = 5.79, 9.00 Hz, 1H), 3.35 - 3.50 (m, 1H), 3.08 - 3.34 (m, 4H), 2.80 - 3.01 (m, 4H), 2.42 - 2.56 (m, 1H), 1.44 - 1.55 (m, 2H), 1.22 - 1.39 (m, 6H), 0.85 - 0.93 (m, 3H). m/z 573 (M+H). HRMS (ESI, CH3OH) m/z calcd for C34H45N4O4 [M+H]+ 573.3435, m/z found 573.3495.
N-Benzyl-2-(6,7-dimethoxy-1-{[4-(propan-2-yl)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (51)
This was made by the general procedure starting from 4-isopropylphenylacetic acid in 4 steps to give the desired product as a white solid in 65% overall yield: mp 97-99 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.36 (m, 3H), 7.03 - 7.15 (m, 6H), 6.58 (s, 1H), 6.35 (s, 1H), 4.37 (dd, J = 7.54, 15.07 Hz, 1H), 3.86 (s, 3H), 3.76 (s, 3H), 3.59 - 3.83 (m, 2H), 3.39 - 3.54 (m, 1H), 3.09 - 3.35 (m, 2H), 2.76 - 3.04 (m, 5H), 2.43 - 2.57 (m, 1H), 1.18 (dd, J = 5.18, 6.69 Hz, 6H). m/z 473 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H37N2O3 [M+H]+ 473.2799, m/z found 473.2858.
N-Benzyl-2-{6,7-dimethoxy-1-[(3,4,5-trimethoxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (52)
This was made by the general procedure starting from 3,4,5-trimethoxyphenylacetic acid in 4 steps to give the desired product as an orange solid in 7% overall yield: mp 127-128 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.10 - 7.39 (m, 6H), 6.59 (s, 1H), 6.32 - 6.41 (m, 3H), 4.46 - 4.57 (m, 1H), 3.86 (s, 3H), 3.78 (s, 3H), 3.78 (s, 3H), 3.77 (s, 6H), 3.81 (d, J = 2.26 Hz, 1H), 3.70 (dd, J = 5.84, 8.48 Hz, 1H), 3.12 - 3.45 (m, 3H), 2.78 - 3.04 (m, 4H), 2.45 - 2.59 (m, 1H). m/z 521 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H37N2O6 [M+H]+ 521.2646, m/z found 521.2683.
N-Benzyl-2-{1-[(3,4-dimethylphenyl)methyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (53)
This was made by the general procedure starting from 3,4-dimethylphenylacetic acid in 4 steps to give the desired product as an off-white solid in 37% overall yield: mp 112-114 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 3H), 6.95 - 7.08 (m, 4H), 6.83 - 6.95 (m, 2H), 6.59 (s, 1H), 6.48 (s, 1H), 4.44 (dd, J = 8.15, 15.12 Hz, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.40 - 3.67 (m, 3H), 3.06 - 3.32 (m, 2H), 2.80 - 3.01 (m, 4H), 2.43 - 2.55 (m, 1H), 2.13 (s, 6H). m/z 459 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H35N2O3 [M+H]+ 459.2642, m/z found 459.2695.
N-Benzyl-2-[6,7-dimethoxy-1-(naphthalen-2-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (54)
This was made by the general procedure starting from 2-naphthaleneacetic acid in 4 steps in 50% overall yield as an off-white: mp 69-72 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.68 - 7.84 (m, 3H), 7.66 (s, 1H), 7.41 - 7.53 (m, 2H), 7.35 (dd, J = 1.55, 8.34 Hz, 1H), 7.15 (dd, J = 1.79, 4.90 Hz, 2H), 6.64 - 6.76 (m, 3H), 6.61 (s, 1H), 6.49 (s, 1H), 4.13 - 4.25 (m, 1H), 3.87 (s, 3H), 3.70 - 3.81 (m, 4H), 3.45 - 3.60 (m, 1H), 3.05 - 3.32 (m, 4H), 2.83 - 3.03 (m, 3H), 2.45 - 2.58 (m, 1H). m/z 481 (M+H). HRMS (ESI, CH3OH) m/z calcd for C31H33N2O3 [M+H]+ 481.2486, m/z found 481.2493.
N-Benzyl-2-[6,7-dimethoxy-1-(quinolin-6-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (55)
This was made by the general procedure starting from 2-(quinolin-6-yl)acetic acid in 4 steps in 48% overall yield as a yellow glassy solid. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.90 (dd, J = 1.60, 4.24 Hz, 1H), 8.01 (dd, J = 9.09, 12.76 Hz, 2H), 7.54 - 7.62 (m, 2H), 7.36 (dd, J = 4.24, 8.29 Hz, 1H), 7.13 - 7.20 (m, 2H), 6.69 - 6.81 (m, 3H), 6.61 (s, 1H), 6.43 (s, 1H), 4.23 (dd, J = 8.01, 14.79 Hz, 1H), 3.87 (s, 3H), 3.74 (s, 3H), 3.69 - 3.84 (m, 1H), 3.42 - 3.58 (m, 1H), 3.06 - 3.35 (m, 4H), 2.84 - 3.02 (m, 3H), 2.46 - 2.60 (m, 1H). m/z 482 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H32N3O3 [M+H]+ 482.2438, m/z found 482.2483.
N-Benzyl-2-[6,7-dimethoxy-1-(1-phenylethyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (56)
This was made by the general procedure starting from 2-phenylpropionic acid in 4 steps in 29% overall yield as a yellow glassy solid. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.04 - 7.35 (m, 10H), 6.63 - 6.74 (m, 1H), 6.54 (s, 1H), 4.38 (dd, J = 7.63, 14.98 Hz, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.74 - 3.89 (m, 1H), 3.45 - 3.52 (m, 1H), 3.34 - 3.45 (m, 1H), 3.13 - 3.22 (m, 1H), 2.79 - 3.08 (m, 3H), 2.48 - 2.71 (m, 2H), 1.25 (d, J = 7.25 Hz, 1H). m/z 445 (M+H). HRMS (ESI, CH3OH) m/z calcd for C28H33N2O3 [M+H]+ 445.2486, m/z found 445.2496.
Pictet-Spengler route to 1-alkyl-tetrahydroisoquinolines. General procedure
N-Benzyl-2-[1-(3,4-dimethoxyphenyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (59)
3,4-Dimethoxyphenethylamine (0.10 g, 93 μL, 0.55 mmol) and 3,4-dimethoxybenzaldehyde (0.11 g, 0.66 mmol) were combined in dry toluene (0.55 mL). Trifluoroacetic acid (0.50 g, 0.33 mL, 4.41 mmol) was added and the reaction heated in the microwave at 140 °C for 30 min. The reaction was cooled, the solvent was removed under reduced pressure and water was added. The pH was adjusted to 8-9 with 2N NaOH solution then extracted three times with CH2Cl2. The combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure to yield the tetrahydroisoquinoline which was used in the next step without further purification.
The crude tetrahydroisoquinoline was combined with N-benzyl bromoacetamide (0.19 g, 0.82 mmol) and tetrabutylammonium iodide (41 mg, 0.11 mmol) in DMF (6 mL), diisopropylethylamine (0.18 g, 0.24 mL, 1.37 mmol) was added then the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent removed under reduced pressure. The crude was purified by chromatography on silica (0-75% EtOAc in hexane) to give the desired 1-phenyl derivative as an off-white solid (0.11 g, 41%): mp 140-141 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.47 - 7.58 (m, 1H), 7.25 - 7.38 (m, 3H), 7.21 (d, J = 7.44 Hz, 2H), 6.77 (s, 2H), 6.60 (d, J = 4.05 Hz, 2H), 6.14 (s, 1H), 4.35 - 4.55 (m, 3H), 3.89 (s, 3H), 3.86 (s, 3H), 3.60 (s, 6H), 3.33 (d, J = 16.48 Hz, 1H), 3.12 (dd, J = 4.05, 11.11 Hz, 1H), 2.93 - 3.06 (m, 2H), 2.65 - 2.83 (m, 2H). m/z 477 (M+H). HRMS (ESI, CH3OH) m/z calcd for C28H33N2O5 [M+H]+ 477.2384, m/z found 477.2438.
N-Benzyl-2-{1-[2-(3,4-dimethoxyphenyl)ethyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (60)
This was prepared via Bischler-Napieralski cyclization using the general procedures outlined for 12. The compound was obtained in 45% yield over 4 steps as a yellow glassy solid. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.70 (br. t, J = 5.70 Hz, 1H), 7.22 - 7.40 (m, 5H), 6.74 (d, J = 8.01 Hz, 1H), 6.52 - 6.61 (m, 3H), 6.45 (s, 1H), 4.48 - 4.55 (m, 2H), 3.85 (s, 3H), 3.84 (s, 3H), 3.82 (s, 6H), 3.54 (dd, J = 4.90, 7.82 Hz, 1H), 3.15 - 3.40 (m, 3H), 2.74 - 2.95 (m, 2H), 2.41 - 2.72 (m, 3H), 1.84 - 2.12 (m, 2H). m/z 505 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H37N2O5 [M+H]+ 505.2697, m/z found 505.2685.
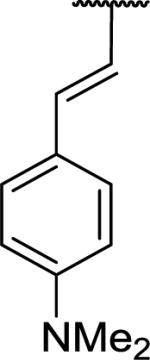
N-Benzyl-2-{1-[(E)-2-[4-(dimethylamino)phenyl]ethenyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (61)
Synthesized via Pictet-Spengler general method from 4-dimethylaminocinnamaldehyde. Yield 9%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.36 (m, 7H), 6.72 - 6.79 (m, 1H), 6.63 - 6.71 (m, 4H), 6.41 (d, J = 15.73 Hz, 1H), 5.66 (dd, J = 8.57, 15.73 Hz, 1H), 4.83 (d, J = 14.88 Hz, 1H), 4.31 (d, J = 8.67 Hz, 1H), 4.07 (d, J = 14.79 Hz, 1H), 3.84 (s, 3H), 3.79 (s, 3H), 3.73 (d, J = 15.07 Hz, 1H), 3.19 (dd, J = 1.46, 14.27 Hz, 1H), 3.00 (s, 6H), 2.49 - 2.75 (m, 4H). m/z 486 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H36N3O3 [M+H]+ 486.2751, m/z found 486.2817.
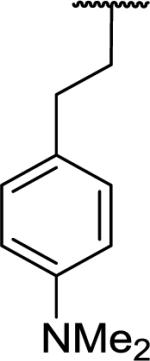
N-Benzyl-2-(1-{2-[4-(dimethylamino)phenyl]ethyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (62)
The olefin 61 (20 mg, 0.041 mmol) and palladium on carbon (10%, 20 mg) in ethanol (5 mL) were stirred under an atmosphere of hydrogen (35 psi) on a Parr shaker for 1.5 hr. The reaction was filtered through Celite, rinsed with ethanol and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-75% EtOAc in hexane) to give the saturated analog as a yellow oil (7 mg, 35%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.38 (m, 5H), 6.87 (d, J = 8.48 Hz, 2H), 6.76 - 6.82 (m, 1H), 6.64 - 6.75 (m, 5H), 5.00 (d, J = 14.98 Hz, 1H), 4.01 (br. s., 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.77 - 3.95 (m, 2H), 3.21 (d, J = 15.26 Hz, 1H), 2.92 (s, 6H), 2.81 - 2.88 (m, 1H), 2.59 - 2.72 (m, 2H), 2.24 - 2.56 (m, 2H), 1.79 (dt, J = 3.53, 8.08 Hz, 2H). m/z 488 (M+H). HRMS (ESI, CH3OH) m/z calcd for C30H38N3O3 [M+H]+ 488.2908, m/z found 488.2956.
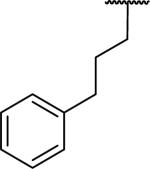
N-Benzyl-2-[6,7-dimethoxy-1-(3-phenylpropyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (63)
Synthesized via Pictet-Spengler general method from 4-phenylbutyraldehyde. Yield 69%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.70 (br. s., 1H), 7.15 - 7.38 (m, 8H), 7.05 (d, J = 7.72 Hz, 2H), 6.53 (s, 1H), 6.41 (s, 1H), 4.48 (d, J = 5.93 Hz, 2H), 3.82 (s, 3H), 3.79 (s, 3H), 3.47 (d, J = 4.14 Hz, 1H), 3.17 - 3.34 (m, 2H), 3.07 - 3.17 (m, 1H), 2.69 - 2.89 (m, 2H), 2.43 - 2.64 (m, 3H), 1.53 - 1.80 (m, 4H). m/z 459 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H35N2O3 [M+H]+ 459.2642, m/z found 459.269.
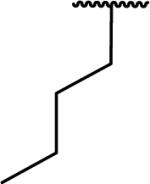
N-Benzyl-2-(1-butyl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (64)
Synthesized via Pictet-Spengler general method from valeraldehyde. Yield 68%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.76 (br. s., 1H), 7.28 - 7.41 (m, 5H), 6.55 (s, 1H), 6.48 (s, 1H), 4.51 (t, J = 5.70 Hz, 2H), 3.84 (s, 6H), 3.44 (dd, J = 4.85, 8.05 Hz, 1H), 3.12 - 3.36 (m, 3H), 2.71 - 2.92 (m, 2H), 2.44 - 2.57 (m, 1H), 1.51 - 1.75 (m, 2H), 1.14 - 1.34 (m, 4H), 0.80 (t, J = 6.78 Hz, 3H). m/z 397 (M+H). HRMS (ESI, CH3OH) m/z calcd for C24H33N2O3 [M+H]+ 397.2486, m/z found 397.253.
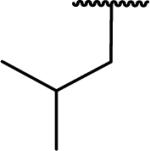
N-Benzyl-2-[6,7-dimethoxy-1-(2-methylpropyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (65)
Synthesized via Pictet-Spengler general method from isovaleraldehyde. Yield 100% as an off-white solid: mp 92-94 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.75 (br. s., 1H), 7.24 - 7.41 (m, 5H), 6.55 (s, 1H), 6.43 (s, 1H), 4.40 - 4.60 (m, 2H), 3.84 (s, 6H), 3.49 (dd, J = 4.57, 9.00 Hz, 1H), 3.09 - 3.41 (m, 3H), 2.73 - 2.99 (m, 2H), 2.45 (dd, J = 4.85, 16.44 Hz, 1H), 1.64 - 1.79 (m, 2H), 1.21 - 1.41 (m, 1H), 0.88 (dd, J = 2.07, 6.31 Hz, 6H). m/z 397 (M+H). HRMS (ESI, CH3OH) m/z calcd for C24H33N2O3 [M+H]+ 397.2486, m/z found 397.2544.
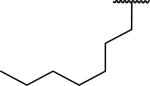
N-Benzyl-2-(1-heptyl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (66)
Synthesized via Pictet-Spengler general method from octyl aldehyde. Yield 89%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.76 (br. s., 1H), 7.21 - 7.42 (m, 5H), 6.55 (s, 1H), 6.47 (s, 1H), 4.51 (d, J = 5.84 Hz, 2H), 3.85 (s, 3H), 3.84 (s, 3H), 3.44 (dd, J = 4.99, 8.10 Hz, 1H), 3.10 - 3.36 (m, 3H), 2.70 - 2.90 (m, 2H), 2.44 - 2.58 (m, 1H), 1.52 - 1.76 (m, 2H), 1.10 - 1.39 (m, 10H), 0.88 (t, J = 6.88 Hz, 3H). m/z 439 (M+H). HRMS (ESI, CH3OH) m/z calcd for C27H39N2O3 [M+H]+ 439.2955, m/z found 439.3016.
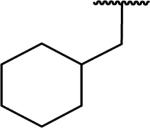
N-Benzyl-2-[1-(cyclohexylmethyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (67)
Synthesized via Pictet-Spengler general method from 2-cyclohexylacetaldehyde. Yield 69%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.77 (br. s., 1H), 7.27 - 7.42 (m, 5H), 6.55 (s, 1H), 6.43 (s, 1H), 4.54 - 4.65 (m, 1H), 4.37 - 4.49 (m, 1H), 3.84 (s, 6H), 3.50 - 3.60 (m, 1H), 3.09 - 3.42 (m, 3H), 2.72 - 2.99 (m, 2H), 2.46 (dd, J = 4.99, 16.48 Hz, 1H), 1.55 - 1.85 (m, 5H), 1.32 - 1.52 (m, 2H), 0.82 - 1.20 (m, 6H). m/z 437 (M+H). HRMS (ESI, CH3OH) m/z calcd for C27H37N2O3 [M+H]+ 437.2799, m/z found 437.2855.
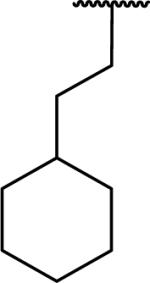
N-Benzyl-2-[1-(2-cyclohexylethyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (68)
Synthesized via Pictet-Spengler general method from 3-cyclohexylpropionaldehyde. Yield 65%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.75 (t, J = 4.99 Hz, 1H), 7.28 - 7.41 (m, 5H), 6.54 (s, 1H), 6.47 (s, 1H), 4.41 - 4.58 (m, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.41 (dd, J = 4.99, 7.91 Hz, 1H), 3.21 - 3.28 (m, 2H), 3.09 - 3.20 (m, 1H), 2.68 - 2.90 (m, 2H), 2.44 - 2.58 (m, 1H), 1.48 - 1.75 (m, 7H), 1.03 - 1.23 (m, 6H), 0.64 - 0.82 (m, 2H). m/z 451 (M+H). HRMS (ESI, CH3OH) m/z calcd for C28H39 N2O3 [M+H]+ 451.2955, m/z found 451.301.
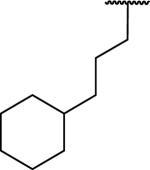
N-Benzyl-2-[1-(3-cyclohexylpropyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (69)
Synthesized via Pictet-Spengler general method from 4-cyclohexylbutyraldehyde. Yield 43%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.76 (t, J = 5.60 Hz, 1H), 7.23 - 7.41 (m, 5H), 6.54 (s, 1H), 6.47 (s, 1H), 4.50 (d, J = 5.93 Hz, 2H), 3.84 (s, 3H), 3.84 (s, 3H), 3.44 (dd, J = 4.85, 8.24 Hz, 1H), 3.11 - 3.35 (m, 3H), 2.70 - 2.91 (m, 2H), 2.43 - 2.57 (m, 1H), 1.48 - 1.75 (m, 9H), 1.02 - 1.20 (m, 6H), 0.68 - 0.86 (m, 2H). m/z 465 (M+H). HRMS (ESI, CH3OH) m/z calcd for C29H41 N2O3 [M+H] 465.3112, m/z found 465.3171.
2-(3-Methoxy-4-propoxyphenyl)-N-[2-(3-methoxy-4-propoxyphenyl)ethyl]acetamide (71)
To a solution of 4-hydroxy-3-methoxyphenethylamine hydrochloride 7 (2.24 g, 10.98 mmol), 4-hydroxy-3-methoxyphenylacetic acid (2.0 g, 10.98 mmol) and HBTU (4.58 g, 12.08 mmol) in dry DMF (60 mL) was added diisopropylethylamine (5.68 g, 7.7. mL, 43.92 mmol) and the reaction stirred under N2 at RT overnight. The reaction was diluted with EtOAc, washed with 1N
HCl, NaHCO3 solution and saturated brine, then dried over MgSO4 and the solvent removed under reduced pressure to give the amide.
To a solution of the amide in DMF (60 mL) was added potassium carbonate (9.08 g, 65.73 mmol) and 1-iodopropane (7.45 g, 4.3 mL, 43.82 mmol) and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure to give the dipropoxy amide as a yellow oil which solidified on standing (3.69 g, 81%). 1H NMR (300 MHz, CHLOROFORM-d) δ 6.80 (d, J = 8.67 Hz, 1H), 6.72 (d, J = 8.10 Hz, 1H), 6.61 - 6.69 (m, 3H), 6.49 (dd, J = 2.07, 8.10 Hz, 1H), 5.43 (br. t, J = 5.10 Hz, 1H), 3.96 (q, J = 6.78 Hz, 4H), 3.81 (s, 3H), 3.81 (s, 3H), 3.39 - 3.50 (m, 4H), 2.66 (t, J = 6.88 Hz, 2H), 1.79 - 1.92 (m, 4H), 1.01 - 1.08 (m, 6H).
6-Methoxy-1-[(3-methoxy-4-propoxyphenyl)methyl]-7-propoxy-1,2,3,4-tetrahydroisoquinoline (72)
This was prepared by the method used for 10, from amide 71. Yield 28%. 1H NMR (300 MHz, METHANOL-d4) δ 6.83 - 6.89 (m, 1H), 6.71 - 6.82 (m, 2H), 6.66 (s, 1H), 6.56 (s, 1H), 4.09 (t, J = 6.78 Hz, 1H), 3.91 (t, J = 6.59 Hz, 2H), 3.71 - 3.81 (m, 8H), 3.04 - 3.22 (m, 2H), 2.80 - 2.92 (m, 2H), 2.71 (t, J = 5.75 Hz, 2H), 1.66 - 1.85 (m, 4H), 1.03 (t, J = 7.44 Hz, 3H), 0.99 (t, J = 7.54 Hz, 3H). m/z 400 (M+H).
N-Benzyl-2-{6-methoxy-1-[(3-methoxy-4-propoxyphenyl)methyl]-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (73)
Amine 72 (0.85 g, 2.13 mmol), N-benzyl-2-bromoacetamide (0.58 g, 2.55 mmol) and potassium carbonate (0.59 g, 4.20 mmol) were combined in DMF (50 mL) and heated to 65 °C overnight. The reaction was cooled, diluted with water then extracted 3 times with EtOAc. The combined extracts were washed with brine, dried over MgSO4 and the solvent removed under reduced pressure. The crude material was purified by chromatography on silica (0-80% EtOAc in hexane) to give the desired product as a pale brown solid (0.67 g, 58%): mp 98-101 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.34 (m, 3H), 7.11 (d, J = 6.78 Hz, 2H), 6.96 - 7.06 (m, 1H), 6.62 - 6.73 (m, 3H), 6.58 (s, 1H), 6.47 (s, 1H), 4.49 (dd, J = 8.05, 14.93 Hz, 1H), 3.89 (t, J = 6.83 Hz, 2H), 3.84 (s, 3H), 3.76 - 3.83 (m, 5H), 3.57 - 3.70 (m, 2H), 3.34 - 3.48 (m, 1H), 3.11 - 3.33 (m, 2H), 2.79 - 2.98 (m, 4H), 2.42 - 2.54 (m, 1H), 1.76 - 1.92 (m, 4H), 1.04 (t, J = 7.39 Hz, 3H), 1.02 (t, J = 7.39 Hz, 3H). m/z 547 (M+H). HRMS (ESI, CH3OH) m/z calcd for C33H43N2O5 [M+H]+ 547.3167, m/z found 547.3231.
Calcium Mobilization Ke Assay for OX1 and OX2
Two individual stable cell lines were created by over-expressing human OX1 and OX2 receptors in CHO-RD-HGA16 (Molecular Devices) cells. The day before the assay, cells were plated into 96-well black-walled assay plates at 25,000 cells/well in Ham's F12 supplemented with 10% fetal bovine serum, 100 units of penicillin and streptomycin, and 100 μg/mL normocin™. The cells were incubated overnight at 37°C, 5% CO2. Prior to the assay, Calcium 5 dye (Molecular Devices) was reconstituted according to the manufacturer instructions. The reconstituted dye was diluted 1:40 in pre-warmed (37°C) assay buffer (1X HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37°C). Growth medium was removed and the cells were gently washed with 100 μL of pre-warmed (37°C) assay buffer. The cells were incubated for 45 minutes at 37°C, 5% CO2 in 200 μL of the diluted Calcium 5 dye. A single concentration of each test compound was prepared at 10x the desired final concentration in 2.5% BSA/8% DMSO/assay buffer. Serial dilutions of orexin A were prepared at 10x the desired final concentration in 0.25% BSA/1% DMSO/assay buffer, aliquoted into 96-well polypropylene plates, and warmed to 37°C. After the dye-loading incubation period, the cells were pre-treated with 25 μL of the test compounds and incubated for 15 min at 37°C. After the pre-treatment incubation period, the plate was read with a FlexStation II (Molecular Devices). Calcium-mediated changes in fluorescence were monitored every 1.52 seconds over a 60 second time period, with the FlexStation II adding 25 μL of the orexin A serial dilutions at the 19 second time point (excitation at 485 nm, detection at 525 nm). Peak kinetic reduction (SoftMax, Molecular Devices) relative fluorescent units (RFU) were plotted against the log of compound concentration. Data were fit to a three-parameter logistic curve to generate EC50 values (Prism, version 6.0, GraphPad Software, Inc., San Diego, CA). Ke values were calculated using the equation Ke = [L]/((EC50+/EC50−) – 1) where [L] is the concentration of test compound, EC50+ is the EC50 of orexin A with test compound, and EC50− is the EC50 of orexin A alone.
Behavioral studies
Animals
Sixteen adult male Sprague-Dawley rats (Harlan, Indianapolis, IN) (n = 8 per group) were housed individually on a 12/12-h light/dark cycle (behavioral experiments were conducted during the light period) with free access to water and food except during testing. Animals were maintained and experiments were approved by the Institutional Animal Care and Use Committee, University at Buffalo, the State University of New York, and with the 2011 Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources on Life Sciences, National Research Council, National Academy of Sciences, Washington DC).
Drugs
Drugs used in this study included cocaine hydrochloride (Research Technology Branch, National Institute of Drug Abuse, Rockville, MD, USA) and compound 73. Cocaine hydrochloride was dissolved in 0.9% physiological saline. Compound 73 was dissolved in a mixture of 1 part absolute ethanol, 1 part Emulphor-620 (Rhodia Inc.), and 18 parts physiologic saline. Doses were expressed as the weight of the forms listed above in milligrams per kilogram of body weight and drugs were administered intraperitoneally.
Experimental protocols
Locomotor activity was monitored by an infrared motor-sensor system (AccuScan Instruments, Columbus, OH) fitted outside clear acrylic chambers (40 × 40 × 30 cm) that were cleaned between test sessions. Locomotor activity (distance travelled) was analyzed with the Versa Max animal activity monitoring software (AccuScan Instruments, Columbus, OH).41 The dose-effect curve of cocaine was determined by using a cumulative dosing procedure as previously described.41, 42 For this experiment, vehicle or 10 mg/kg compound 73 was administered immediately prior to the start of the test session and different doses of cocaine (cumulative doses of 3.2, 10, 32 mg/kg) were given at times 20 min, 40 min and 60 min. The locomotor effects of each dose of cocaine were recorded for 20 min but for each dose the data from the first 5 min immediately after the drug injection were discarded due to the brief hyperactivity associated with handling and injection. For behavioral sensitization study, a similar protocol was used as described in our previous reports.41, 42 Briefly, an acute cocaine dose-effect curve with vehicle or 10 mg/kg compound 73 pretreatment was determined on day 1, which was followed by 7 days of daily 15 mg/kg cocaine in combination with vehicle or 10 mg/kg compound 73 and stayed in the test chambers for 1 hour. Thereafter, six days of drug-free period was implemented, which was followed by another cocaine dose-effect curve determination on day 15 during which no compound 73 was given.
Statistical analyses
The locomotion data were analyzed by two-way ANOVA (cocaine dose × compound 73 treatment) followed by post hoc Bonferroni's test. P < 0.05 was considered statistically significant.
Supplementary Material
Figure 1.

Orexin Antagonists
ACKNOWLEDGMENT
We are grateful to National Institute on Drug Abuse, National Institutes of Health, U.S. (Grants DA032837 and DA026582 to Y.Z. and Grant DA034806 to JXL) for the financial support of this research. We thank Tiffany Langston, Keith Warner and Dr. Angela Giddings for their valuable technical assistance.
ABBREVIATIONS
- BOP
(Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- HBTU
O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate
- HPLC
performance liquid chromatography
- OX1
orexin 1 receptor
- OX2
orexin 2 receptor
- SAR
structure-activity relationship
- TLC
thin layer chromatography
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. HPLC analysis of target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 2.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. Journal of Neuroscience. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. Distribution of orexin neurons in the adult rat brain. Brain Research. 1999;827:243–260. doi: 10.1016/s0006-8993(99)01336-0. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Hu Z, de Lecea L. The hypocretins/orexins: integrators of multiple physiological functions. Br. J. Pharmacol. 2014;171:332–350. doi: 10.1111/bph.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kukkonen JP, Leonard CS. Orexin/hypocretin receptor signalling cascades. Br. J. Pharmacol. 2014;171:314–331. doi: 10.1111/bph.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonard CS, Kukkonen JP. Orexin/hypocretin receptor signalling: a functional perspective. Br. J. Pharmacol. 2014;171:294–313. doi: 10.1111/bph.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. Journal of Comparative Neurology. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 9.Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Letters. 1998;438:71–75. doi: 10.1016/s0014-5793(98)01266-6. [DOI] [PubMed] [Google Scholar]
- 10.Kukkonen JP, Holmqvist T, Ammoun S, Akerman KE. Functions of the orexinergic/hypocretinergic system, American journal of physiology. Cell physiology. 2002;283:C1567–1591. doi: 10.1152/ajpcell.00055.2002. [DOI] [PubMed] [Google Scholar]
- 11.Mahler SV, Moorman DE, Smith RJ, James MH, Aston-Jones G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nat. Neurosci. 2014;17:1298–1303. doi: 10.1038/nn.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kukkonen JP. Physiology of the orexinergic/hypocretinergic system: a revisit in 2012, American journal of physiology. Cell physiology. 2013;304:C2–32. doi: 10.1152/ajpcell.00227.2012. [DOI] [PubMed] [Google Scholar]
- 13.Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol. Rev. 2009;61:162–176. doi: 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- 14.Mahler SV, Smith RJ, Moorman DE, Sartor GC, Aston-Jones G. Multiple roles for orexin/hypocretin in addiction. Prog. Brain Res. 2012;198:79–121. doi: 10.1016/B978-0-444-59489-1.00007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 16.Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, de Lecea L. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19168–19173. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence AJ, Cowen MS, Yang HJ, Chen F, Oldfield B. The orexin system regulates alcohol-seeking in rats. British journal of pharmacology. 2006;148:752–759. doi: 10.1038/sj.bjp.0706789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards JK, Simms JA, Steensland P, Taha SA, Borgland SL, Bonci A, Bartlett SE. Inhibition of orexin-1/hypocretin-1 receptors inhibits yohimbine-induced reinstatement of ethanol and sucrose seeking in Long-Evans rats. Psychopharmacology. 2008;199:109–117. doi: 10.1007/s00213-008-1136-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roecker AJ, Coleman PJ. Orexin receptor antagonists: medicinal chemistry and therapeutic potential. Curr Top Med Chem. 2008;8:977–987. doi: 10.2174/156802608784936746. [DOI] [PubMed] [Google Scholar]
- 20.Boss C, Brisbare-Roch C, Jenck F. Biomedical application of orexin/hypocretin receptor ligands in neuroscience. Journal of Medicinal Chemistry. 2009;52:891–903. doi: 10.1021/jm801296d. [DOI] [PubMed] [Google Scholar]
- 21.Gatfield J, Brisbare-Roch C, Jenck F, Boss C. Orexin receptor antagonists: a new concept in CNS disorders? ChemMedChem. 2010;5:1197–1214. doi: 10.1002/cmdc.201000132. [DOI] [PubMed] [Google Scholar]
- 22.Kodadek T, Cai D. Chemistry and biology of orexin signaling. Molecular bioSystems. 2010;6:1366–1375. doi: 10.1039/c003468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annual Review of Pharmacology and Toxicology. 2011;51:243–266. doi: 10.1146/annurev-pharmtox-010510-100528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mieda M, Sakurai T. Orexin (hypocretin) receptor agonists and antagonists for treatment of sleep disorders. Rationale for development and current status. CNS Drugs. 2013;27:83–90. doi: 10.1007/s40263-012-0036-8. [DOI] [PubMed] [Google Scholar]
- 25.Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br. J. Pharmacol. 2014;171:283–293. doi: 10.1111/bph.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang LP. Suvorexant: First Global Approval, Drugs. 2014 doi: 10.1007/s40265-014-0294-5. [DOI] [PubMed] [Google Scholar]
- 27.Lebold TP, Bonaventure P, Shireman BT. Selective orexin receptor antagonists. Bioorg. Med. Chem. Lett. 2013;23:4761–4769. doi: 10.1016/j.bmcl.2013.06.057. [DOI] [PubMed] [Google Scholar]
- 28.Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA, Jerman JC. SB-334867-A: the first selective orexin-1 receptor antagonist. Br. J. Pharmacol. 2001;132:1179–1182. doi: 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stasi PL, Rovati L. Spiro amino compounds suitable for the treatment of inter alia sleep disorders and drug addiction. 2011 WO2011006960, Rottapharm S.P.A.
- 30.Stasi LP, Artusi R, Bovino C, Buzzi B, Canciani L, Caselli G, Colace F, Garofalo P, Giambuzzi S, Larger P, Letari O, Mandelli S, Perugini L, Pucci S, Salvi M, Toro P. Discovery, synthesis, selectivity modulation and DMPK characterization of 5-azaspiro[2.4]heptanes as potent orexin receptor antagonists. Bioorg. Med. Chem. Lett. 2013;23:2653–2658. doi: 10.1016/j.bmcl.2013.02.093. [DOI] [PubMed] [Google Scholar]
- 31.Gozzi A, Turrini G, Piccoli L, Massagrande M, Amantini D, Antolini M, Martinelli P, Cesari N, Montanari D, Tessari M, Corsi M, Bifone A. Functional magnetic resonance imaging reveals different neural substrates for the effects of orexin-1 and orexin-2 receptor antagonists. PloS one. 2011;6:e16406. doi: 10.1371/journal.pone.0016406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steiner MA, Gatfield J, Brisbare-Roch C, Dietrich H, Treiber A, Jenck F, Boss C. Discovery and Characterization of ACT-335827, an Orally Available, Brain Penetrant Orexin Receptor Type 1 Selective Antagonist. ChemMedChem. 2013;8:898–903. doi: 10.1002/cmdc.201300003. [DOI] [PubMed] [Google Scholar]
- 33.Jiang R, Song X, Bali P, Smith A, Bayona CR, Lin L, Cameron MD, McDonald PH, Kenny PJ, Kamenecka TM. Disubstituted piperidines as potent orexin (hypocretin) receptor antagonists. Bioorganic & medicinal chemistry letters. 2012;22:3890–3894. doi: 10.1016/j.bmcl.2012.04.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perrey DA, German NA, Gilmour BP, Li JX, Harris DL, Thomas BF, Zhang Y. Substituted tetrahydroisoquinolines as selective antagonists for the orexin 1 receptor. Journal of Medicinal Chemistry. 2013;56:6901–6916. doi: 10.1021/jm400720h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perrey DA, Gilmour BP, Runyon SP, Thomas BF, Zhang Y. Diaryl urea analogues of SB-334867 as orexin-1 receptor antagonists. Bioorganic & medicinal chemistry letters. 2011;21:2980–2985. doi: 10.1016/j.bmcl.2011.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perrey DA, Gilmour BP, Thomas BF, Zhang Y. Toward the Development of Bivalent Ligand Probes of Cannabinoid CB1 and Orexin OX1 Receptor Heterodimers. ACS medicinal chemistry letters. 2014;5:634–638. doi: 10.1021/ml4004759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirose M, Egashira S, Goto Y, Hashihayata T, Ohtake N, Iwaasa H, Hata M, Fukami T, Kanatani A, Yamada K. N-acyl 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline: the first orexin-2 receptor selective non-peptidic antagonist. Bioorganic & medicinal chemistry letters. 2003;13:4497–4499. doi: 10.1016/j.bmcl.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 38.Koberstein R, Aissaoui H, Bur D, Clozel M, Fischli W, Jenck F, Mueller C, Nayler O, Sifferlen T, Treiber A, Weller T. Tetrahydroisoquinolines as Orexin Receptor Antagonists: Strategies for Lead Optimization by Solution-Phase Chemistry. Chimia. 2003;57:270–275. [Google Scholar]
- 39.Neubauer DN. Almorexant, a dual orexin receptor antagonist for the treatment of insomnia. Curr Opin Investig Drugs. 2010;11:101–110. [PubMed] [Google Scholar]
- 40.Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Thorn DA, Zhang C, Zhang Y, Li JX. The trace amine associated receptor 1 agonist RO5263397 attenuates the induction of cocaine behavioral sensitization in rats. Neurosci. Lett. 2014;566:67–71. doi: 10.1016/j.neulet.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thorn DA, Jing L, Qiu Y, Gancarz-Kausch AM, Galuska CM, Dietz DM, Zhang Y, Li JX. Effects of the trace amine-associated receptor 1 agonist RO5263397 on abuse-related effects of cocaine in rats. Neuropsychopharmacology. 2014;39:2309–2316. doi: 10.1038/npp.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.