

INTRODUCTION
Modern cell biology increasingly relies on molecular tools to facilitate the study of cellular processes. With the advent of recombinant DNA technology, reverse transcription–polymerase chain reaction (RT-PCR) to amplify almost any gene, and fluorescent proteins that can be fused to any desired target protein, the number and scope of functional studies designed to determine the roles of proteins within cells have exploded. Creating the appropriate fusion protein plasmid is a key step, but the DNA must also be delivered to the cell for expression and function studies. There are several common approaches for delivering DNA, including liposome- and polymer-mediated transfection, electroporation, and direct DNA delivery by microinjection. This article provides an overview of several of these nonviral gene delivery methods, with an emphasis on direct injection of plasmids into cells.
RELATED INFORMATION
Specific protocols are available for Liposome-Mediated Transfection (Dean and Gasiorowski 2011a) and Dendrimer-Mediated Transfection (Dean and Gasiorowski 2011b). In addition to techniques for Plating Cells for Microinjection (Dean and Gasiorowski 2011c), two methods are described to prepare injection pipettes (Preparing Injection Pipettes on a Flaming/Brown Pipette Puller [Dean and Gasiorowski 2011d] and Preparing Injection Pipettes on a PUL-1 Micropipette Puller [Dean and Gasiorowski 2011e]). Once the DNA samples are loaded (see DNA Sample Preparation and Loading Sample into Pipettes for Microinjection of Cells [Dean and Gasiorowski 2011f]), they can be delivered either by Microinjecting Cells Using a Pulsed-Flow Microinjection System (Dean and Gasiorowski 2011g) or by Microinjecting Cells Using a Constant-Flow Microinjection System (Dean and Gasiorowski 2011h).
NONVIRAL TRANSFECTION
A number of chemical and physical methods for introducing DNA and RNA into cells have been developed over the years. The common thread to all of the chemical techniques is that they rely on cationic carriers to complex with negatively charged nucleic acids for their uptake by cells. Because plasmids and RNAs are intrinsically negatively charged and the plasma membrane carries a net positive charge, cells are largely impermeable to uncomplexed nucleic acids. The steps of essentially all chemical methods for transfection include dilution/preparation of the carrier, formation of DNA/carrier complexes, addition to cells, and subsequent removal of the non-endocytosed excess complexes (Elouahabi and Ruysschaert 2005). The first-generation transfection reagents included calcium phosphate and diethylamino ethanol (DEAE)–dextran, which are inexpensive, extremely simple to use, and continue to show good efficacy in certain cell types.
With the introduction of liposomal reagents in the 1980s (Felgner et al. 1987), the use of calcium phosphate and DEAE-dextran has decreased. Cationic liposomes are the most widely used class of carrier for transfection. Among the available products, most of which differ by performance among cell types, there are Lipofectin (a 1:1 mixture of DOTMA [N-[1(2,3-dioleoyloxy)propyl]-N,N,N-trimethyl-ammonium chloride and DOPE [dioleoyl phosphatidylethanolamine]; Invitrogen), Transfectam (Promega), and DOTAP [1,2-dioleoyloxy-3-trimethylammonium propane]:DOPE. All these reagents are rather costly, so it can be cheaper to purchase purified lipids (DOTAP, DOPE, and DOTMA) and formulate transfection reagents in house. Avanti Polar Lipids, Inc. produces all of these lipids in very high purity, and the methods for formulation are simple: Add equimolar amounts of each lipid suspended in chloroform, mix, dry under inert gas, store, and reconstitute in an appropriate buffer.
Polymer-based systems can also be used to transfect cells, some of which rely on one type of molecule, whereas others contain a mixture of polymers and lipids. Reagents such as SuperFect or PolyFect (QIAGEN) use activated dendrimers, which are positively charged and resemble snowflakes in structure. Multicomponent reagents, including Lipofectamine 2000 (Invitrogen), FuGENE 6 (Roche), and TransIT (Mirus), use mixtures of polymers and lipids that work together to condense the DNA and form micelles for a more uniform size distribution of particles.
Protocols are available for Liposome-Mediated Transfection (Dean and Gasiorowski 2011a) and Dendrimer-Mediated Transfection (Dean and Gasiorowski 2011b). Often, the choice of transfection reagent comes down to personal preference or empirical data on its utility with a desired cell type. Determining which reagent is best for a particular set of experiments and cells often requires trial and error to optimize the conditions. It is also important to note that for many experiments, especially those relying on imaging and expression of fluorescently tagged proteins, it is not necessary to achieve 100% gene transfer efficiency because the transfected cells can be selected before imaging. In some cases, transfection efficiencies as low as 10% can be sufficient to make detailed studies possible.
Choosing Where in the Cell Cycle to Transfect
The success of these methods requires that cells be actively dividing to obtain high levels of transfection (Brunner et al. 2000). Plasmid-carrier complexes must be endocytosed, escape the endosome, traffic through the cytoplasm, and cross the nuclear envelope, all before any transcription can occur. The nuclear envelope represents a major barrier to DNA delivery, and the primary way that plasmids cross this barrier is during mitosis and the concomitant breakdown of the nuclear envelope. Thus, for most protocols, it is advisable to transfect cells when they are between 50% and 70% confluent and are very likely to undergo one round of division before the experimental analysis of gene expression.
Preparation of Plasmids
Although in the early days of transfections it was considered necessary to use highly purified plasmids obtained from cesium chloride density ultracentrifugation, the introduction of resin-based purification kits and columns over the past 15 years has greatly simplified DNA preparation for transfections. Any number of commercially available plasmid purification kits will generate sufficiently pure DNA to use in transfections, and the amount of endotoxin present in any of these kits (even those that are not considered endotoxin-free kits) is so low as to not cause problems with the cells. The only exception might be plasmids from minipreparation spin columns or other minipreparation kits. Although the DNA isolated from minipreparation-scale kits can be adequate for some transfections, results are inconsistent and often result in poor gene transfers or cell death.
Multicomponent-System-Mediated Transfection
As with dendrimers, a major advantage of multicomponent systems is that they can transfect cells in the presence of serum. As with the other gene delivery approaches, transfection conditions will need to be optimized and will vary slightly for each reagent and manufacturer. Common reagent preparation steps include diluting the DNA in serum-free medium, diluting the transfection reagent in medium (with or without serum), combining diluted DNA and reagent, incubating them for 10–30 min to allow complex formation, and finally adding growth medium for plating on washed cells. Because of the proprietary nature of these reagents, follow the specific protocol steps provided by the manufacturers.
ELECTROPORATION
Electroporation is the most widely used physical method to transfect populations of cells. When cells are exposed to brief electrical fields, transient membrane destabilization results, allowing uncomplexed plasmids or other nucleic acids to cross the plasma membrane (Escoffre et al. 2009). Once the electric field is removed, the membrane “seals,” trapping the nucleic acids inside the cell. If the applied electric field is too low, no membrane destabilization occurs and hence no DNA enters the cells. Conversely, if the applied field is too high, pores fail to reseal and cell death occurs. Thus, there is a fine balance between DNA delivery and cell death that must be optimized for each cell type that is being used. Electroporation is simple, rapid (<5 min from start to finish), and can yield very high transfection efficiencies (up to 95% of cells in some cases). However, each cell type requires its own set of pulse parameters, and optimization for new cell types can require much trial and error unless suboptimal levels of transfection are sufficient.
A number of commercially available buffers are available for electroporation, many of which boast 100% transfection efficiency, but again, no buffer is best for all cell types; optimization and testing must be performed on each cell type and electroporation system. Phosphate-buffered saline (PBS) and cell medium (modified Eagle’s medium, Dulbecco’s modified Eagle’s medium, etc.) with or without serum are also often good choices for an electroporation buffer, although the presence of serum can result in bubbling or popping at high field strengths.
Electroporators
Two types of electroporators are available: square wave and exponential decay. Both types are suitable for electroporation of mammalian cells in culture, and again, as with other gene delivery methods, optimization is necessary to determine which works best for the desired cell type. Square wave electroporators deliver pulses of a set voltage to cells for a defined amount of time. These pulses typically last 0.1–20 msec, and fields are usually 250–1000 V/cm (i.e., 100–400 V in a 0.4-cm cuvette). Exponential decay electroporators deliver a peak of energy that dissipates exponentially, giving a time constant (τ) that is a function of the resistance of the sample and the capacitance set on the instrument. This corresponds to the time necessary for the charge to decrease to ~37% of the initial voltage. Neither type of waveform is better than the other, so access to individual instruments or published protocols usually drives the choice of system.
There are two approaches for delivering nucleic acids into cells using electroporation. The first, and more commonly used, method involves mixing purified plasmid with cells in suspension (usually trypsinized cells), placing the suspension in a 0.4-cm gap electrode, and delivering the pulse(s). Immediately following electroporation, the cells are removed from the cuvette, complete growth medium is added, and everything is dispensed into the appropriate plates. This method provides relatively uniform delivery of DNA, and transfected cells are randomly and uniformly distributed in the plates for later analysis. The drawback to this approach is that genes are delivered to trypsinized cells that must then settle and attach to tissue culture dish substrates and reestablish appropriate cytoskeletal and cellular structures.
The second electroporation method delivers genes directly to adherent cells using a novel type of electrode called a Petri Pulser (BTX Instrument Division, Harvard Apparatus). This electrode is a series of parallel plates, each separated by several millimeters, that fits into a single well of a six-well plate or an individual 35-mm dish. Cells are washed with PBS, plasmid is added to the well in 1 mL of buffer/medium, and the electrode is placed into the well so that the plates rest ~0.5 mm above the monolayer of cells; the correct distance of the electrode above the cells that maximizes expression might have to be determined empirically. One square wave pulse of 10-msec duration at 100–160 V is delivered. The electrode is removed, and complete medium is added to the cells. If the electrodes touch the cells, they will be killed (evident by equally spaced rows of missing cells across the dish).
DNA MICROINJECTION
Nuclear versus Cytoplasmic Microinjection
Direct microinjection of genetic material into cells began in the 1970s with the demonstration that mRNA isolated from one cell type could be translated in other cells after it was microinjected into the recipients (Graessmann and Graessmann 1971; Gurdon et al. 1971; Lane et al. 1971). In 1980, it was shown that when a plasmid expressing thymidine kinase was microinjected into the nuclei of thymidine kinase-deficient mouse fibroblasts, 50%–100% of the cells showed enzyme activity 24 h post-injection (Capecchi 1980). In contrast, no gene expression was detected in more than 1000 cytoplasmically injected cells during the same time frame. Similar results have been obtained in numerous systems (Graessmann et al. 1989; Mirzayans et al. 1992; Thornburn and Alberts 1993; Zabner et al. 1995; Dean et al. 1999). Thus, the site of injection can be critical for expression.
Implicit in these experiments is the fact that the microinjected cells did not divide during the course of the experiments. During mitosis, the nuclear envelope breaks down, eliminating a major barrier to gene transfer. If plasmids are present in the cytoplasm, they have full access to the nuclear compartment during this stage of the cell cycle. Although these experiments would suggest that plasmids are incapable of entering the nuclei of nondividing cells, this is not the case. Data from a number of laboratories suggest that certain DNA sequences can promote nuclear entry and gene expression even in nondividing cells in a sequence-specific manner (Graessmann et al. 1989; Dean 1997; Dean et al. 1999; Vacik et al. 1999; Mesika et al. 2001). However, to obtain maximal expression of desired transgenes in cells that will not undergo mitosis during the course of the experiment, microinjection of the DNA into the nucleus is strongly recommended.
DNA Concentration
Gene expression is dependent on the copy number of the gene. As few as one to three cytomegalovirus (CMV) promoter-driven green fluorescent protein (GFP)-expressing plasmids injected into the nucleus of a cell can result in detectable, albeit low, GFP expression in a reasonable percentage of injected cells (Dean et al. 1999). Increasing the number of plasmids delivered to the cell increases both the percentage of cells expressing the gene product and the amount of gene product (Fig. 1; Graessmann et al. 1989; Dean et al. 1999; Ludtke et al. 2002). In typical experiments, DNA should be injected at a starting concentration of ~300 ng/μL but can be used at concentrations between 50 and 500 ng/μL if sufficient expression is not obtained at first. A typical microinjection system is designed to deliver <10% of the total cell volume, which corresponds to an injection volume of ~10−14–10−12 L (0.01–1.0 pL). Assuming a delivery volume of 0.1 pL and a concentration of 300 ng/μL, roughly 5000 copies of a 6-kb plasmid would be delivered to the cell. Greater concentrations of DNA can be delivered to cells, but two problems arise. First, gene expression saturates above several hundred to a thousand copies of DNA per cell, so the benefit of delivering more plasmids will be lost. Second, at concentrations above 1 mg/mL, DNA becomes technically difficult to inject because of the viscosity and aggregation within injection needles. Injecting too much DNA can lead to other problems associated with overexpression, including potential toxicity of the gene product. Thus, lower concentrations are a better starting point.
FIGURE 1.
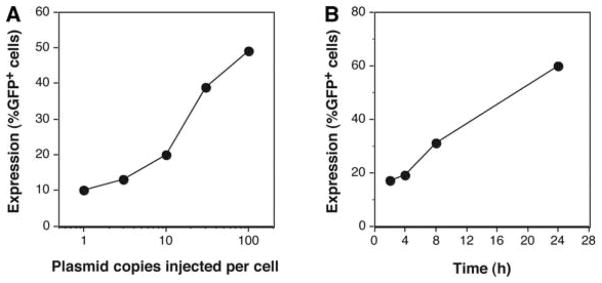
Dose dependency and time dependency of transgene expression in microinjected cells. (A) Human umbilical vein endothelial cells (HUVECs) were grown on etched coverslips and were microinjected with various copy numbers of pEGFP-N1 (Clontech) – expressing enhanced green fluorescent protein (eGFP) from the CMV immediate early promoter. Eight hours later, eGFP-expressing cells were counted and expressed as a percentage of cells injected. (B) HUVECs grown on etched coverslips were microinjected with 10 copies of pEGFP-N1, and GFP expression was assessed at the indicated times following injection. (Reprinted from Dean 2005 with permission.)
Timing of Gene Expression
Most studies assess gene expression at 4–24 h after nuclear injection. Unless very low copy numbers of plasmids are injected into the cell, significant expression can be detected by the earlier time point. One parameter that can affect the timing of gene expression is promoter strength: Weak promoters will usually take longer to produce sufficient protein to visualize. However, when using strong promoters, expression at early times after injection is readily detectable. Indeed, using a DNA concentration of 300 ng/μL, GFP expression from the CMV promoter (e.g., pEGFP-N1 from Clontech) can be visually detected in cells easily within 40 min of microinjection (Figs. 1, 2). Similar results have been obtained using plasmids expressing products from the simian virus 40 (SV40) early promoter and the CMV immediate early promoter/enhancer.
FIGURE 2.
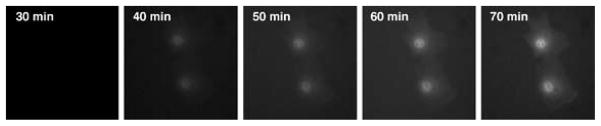
Early time course of gene expression in microinjected cells. TC7 cells (African green monkey kidney epithelial cells) were microinjected with pEGFP-N1 at 300 ng/μL and assessed for eGFP expression at the indicated times. All photographs were taken with the same exposure time. As can be seen, eGFP is first detected in these cells at 40 min post-injection, and the expression increases with time. (Reprinted from Dean 2005 with permission.)
Microinjection Needles
Microinjection needles can either be pulled from glass capillaries on a pipette puller in the laboratory or be purchased premade and sterile from a number of companies. The advantage to pulling needles in the laboratory is that a variety of different needle types can be pulled, depending on the samples and the cells being injected. Protocols for using two pipette pullers are available, one using a high-end Flaming/Brown pipette puller from Sutter Instruments (see Preparing Injection Pipettes on a Flaming/Brown Pipette Puller [Dean and Gasiorowski 2011d]) and one using a less expensive alternative that produces fine needles but requires a little more user input (see Preparing Injection Pipettes on a PUL-1 Micropipette Puller [Dean and Gasiorowski 2011e]). An added advantage of pulling needles in the laboratory is cost; once a pipette puller has been purchased, boxes of glass capillaries are inexpensive (a box of 500 usually costs <$50). The advantages to buying preformed and sterilized needles (~$5 per needle) include increased uniformity of needles from one to another, ease of use (open the packet, fill the needle, and inject), high quality, and not having to invest in a pipette puller.
When pulling needles, there are several variables that need to be addressed. These include filament design, heat, pull strength (tension), and delay time between heating and pulling. There are several types of filaments that will heat the capillaries, including box (surrounds the capillary in a box) and trough (a U shape) filaments. The trough is sufficient for most needle types on the less expensive pullers, but for high-end instruments, refer to the manufacturer for recommendations according to the desired pipettes. The heat setting will affect the length and the tip size of the needle; high heat will typically produce longer needles and finer tips. The pull strength will also affect length and tip size, with greater pull strength producing longer tapered needles with finer tips. Finally, shorter delay times between heating and pulling can result in longer tapers and finer needles. However, if the delay is too short, the glass will form fibers resembling glass wool instead of needles.
Micromanipulation/Microinjection Systems
There are two common types of microinjection systems, one that provides a constant flow of sample and the other that provides a pulsed flow. The former is very simple and can be assembled on a relatively low budget. In this system, a constant flow of sample is delivered from the tip of the pipette, and the amount of sample injected into the cell is determined by how long the pipette remains in the cell (Graessmann and Graessmann 1986). Although this means that each cell will receive a slightly different amount of sample, with practice, microinjections can become highly reproducible. A typical system is composed of a pressure regulator that can be adjusted for two pressures: back pressure and injection pressure (e.g., World Precision Instruments [WPI] pneumatic PicoPump PV830), a capillary holder, and a coarse and fine micromanipulator (e.g., Narishige Group, WPI or Stoelting Co.) (see Microinjecting Cells Using a Constant-Flow Microinjection System [Dean and Gasiorowski 2011h]). In this system, using a manual micromanipulator, the needle is positioned above the cell to be injected and lowered into the cell (Fig. 3). As the needle is lowered, the cell is slightly deformed because the tip is entering at an angle. Because sample is constantly flowing out of the needle, this might not be suitable for precious samples, although experience shows that even 5 μL of sample is more than enough to inject 1000 cells using this method (DA Dean, unpubl.).
FIGURE 3.

Microinjection systems. (Left) A traditional constant-pressure system using a manual micromanipulator. The needle is positioned over the site of injection (the nucleus) and lowered directly down into the cell. When the needle touches the cell at an angle, the membranes are distorted slightly until the needle enters the cell to deliver its contents. The needle is lifted out of the cell to complete the process. The volume delivered depends on the needle’s time inside the cell. (Right) The Eppendorf InjectMan system uses a motorized micromanipulator. After the needle has been positioned, the controller pulls the needle back in the x– y direction and lowers the needle on a diagonal so that the tip directly pierces the cell. After the contents have been delivered (based on the time set on the FemtoJet injector), the needle exits the cell on the same diagonal and returns to its original position. (Reprinted from Dean 2005 with permission.)
The second type of microinjection system uses a pulsed flow. The most commonly used pulsed flow system is the Eppendorf FemtoJet injector coupled with the Eppendorf InjectMan (see Microinject-ing Cells Using a Pulsed-Flow Microinjection System [Dean and Gasiorowski 2011g]). A pulsed-flow system provides much more control over the injection parameters, and hence, variability in injections is reduced. Another nice feature of this system is the dynamics of the injection itself. The needle is positioned over the site to be injected, and when the injection button is pressed, the needle is pulled back in the x–y direction to allow a diagonal insertion of the needle into the cell, causing a direct piercing of the cell (Fig. 3). This method is fast and likely does less damage to the cell than the constant-flow method.
References
- Brunner S, Sauer T, Carotta S, Cotten M, Saltik M, Wagner E. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Ther. 2000;7:401–407. doi: 10.1038/sj.gt.3301102. [DOI] [PubMed] [Google Scholar]
- Capecchi MR. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. 1980;22:479–488. doi: 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- Dean DA. Import of plasmid DNA into the nucleus is sequence specific. Exp Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]
- Dean DA. Gene delivery by direct injection and facilitation of expression by mechanical stretch. In: Goldman RD, Spector DL, editors. Live cell imaging: A laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2005. pp. 51–66. [Google Scholar]
- Dean DA, Gasiorowski JZ. Liposome-mediated transfection. Cold Spring Harb Protoc. 2011a doi: 10.1101/pdb.prot5583. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. Dendrimer-mediated transfection. Cold Spring Harb Protoc. 2011b doi: 10.1101/pdb.prot5584. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. Plating cells for microinjection. Cold Spring Harb Protoc. 2011c doi: 10.1101/pdb.prot5585. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. Preparing injection pipettes on a Flaming/Brown pipette puller. Cold Spring Harb Protoc. 2011d doi: 10.1101/pdb.prot5586. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. Preparing injection pipettes on a PUL-1 micropipette puller. Cold Spring Harb Protoc. 2011e doi: 10.1101/pdb.prot5587. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. DNA sample preparation and loading sample into pipettes for microinjection of cells. Cold Spring Harb Protoc. 2011f doi: 10.1101/pdb.prot5588. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. Microinjecting cells using a pulsed-flow microinjection system. Cold Spring Harb Protoc. 2011g doi: 10.1101/pdb.prot5589. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. Microinjecting cells using a constant-flow microinjection system. Cold Spring Harb Protoc. 2011h doi: 10.1101/pdb.prot5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean DA, Dean BS, Muller S, Smith LC. Sequence requirements for plasmid nuclear entry. Exp Cell Res. 1999;253:713–722. doi: 10.1006/excr.1999.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elouahabi A, Ruysschaert J-M. Formation and intracellular trafficking of lipoplexes and polyplexes. Mol Ther. 2005;11:336–347. doi: 10.1016/j.ymthe.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Escoffre JM, Portet T, Wasungu L, Teissié J, Dean D, Rols M-P. What is (still not) known of the mechanism by which electroporation mediates gene transfer and expression in cells and tissues. Mol Biotechnol. 2009;41:286–295. doi: 10.1007/s12033-008-9121-0. [DOI] [PubMed] [Google Scholar]
- Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graessmann A, Graessmann M. The formation of melanin in muscle cells after the direct transfer of RNA from Harding-Passey melanoma cells. Hoppe Seylers Z Physiol Chem. 1971;352:527–532. [PubMed] [Google Scholar]
- Graessmann M, Graessmann A. Microinjection of tissue culture cells using glass microcapillaries: Methods. In: Celis JE, et al., editors. Microinjection and organelle transplantation techniques: Methods and applications. Academic Press; London: 1986. pp. 3–37. [Google Scholar]
- Graessmann M, Menne J, Liebler M, Graeber I, Graessmann A. Helper activity for gene expression, a novel function of the SV40 enhancer. Nucleic Acids Res. 1989;17:6603–6612. doi: 10.1093/nar/17.16.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon JB, Lane CD, Woodland HR, Marbaix G. Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature. 1971;233:177–182. doi: 10.1038/233177a0. [DOI] [PubMed] [Google Scholar]
- Lane CD, Marbaix G, Gurdon JB. Rabbit haemoglobin synthesis in frog cells: The translation of reticulocyte 9 s RNA in frog oocytes. J Mol Biol. 1971;61:73–91. doi: 10.1016/0022-2836(71)90207-5. [DOI] [PubMed] [Google Scholar]
- Ludtke JJ, Sebestyén MG, Wolff JA. The effect of cell division on the cellular dynamics of microinjected DNA and dextran. Mol Ther. 2002;5:579–588. doi: 10.1006/mthe.2002.0581. [DOI] [PubMed] [Google Scholar]
- Mesika A, Grigoreva I, Zohar M, Reich Z. A regulated, NFκB-assisted import of plasmid DNA into mammalian cell nuclei. Mol Ther. 2001;3:653–657. doi: 10.1006/mthe.2001.0312. [DOI] [PubMed] [Google Scholar]
- Mirzayans R, Aubin RA, Paterson MC. Differential expression and stability of foreign genes introduced into human fibroblasts by nuclear versus cytoplasmic microinjection. Mutat Res. 1992;281:115–122. doi: 10.1016/0165-7992(92)90045-j. [DOI] [PubMed] [Google Scholar]
- Thornburn AM, Alberts AS. Efficient expression of miniprep plasmid DNA after needle micro-injection into somatic cells. Biotechniques. 1993;14:356–358. [PubMed] [Google Scholar]
- Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Ther. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]