

Abstract
Foxp3+ regulatory T (Treg) cells are essential to maintain immune homeostasis, yet controversy exists about the stability of this cell population. Bcl6-deficient (Bcl6−/−) mice develop severe and spontaneous T helper type 2 (Th2) inflammation and Bcl6-deficient Treg cells are ineffective at controlling Th2 responses. We used a lineage tracing approach to analyse the fate of Treg cells in these mice. In the periphery of Bcl6−/− mice, increased numbers of Foxp3-negative ‘exTreg’ cells were found, particularly in the CD25+ population. ExTreg cells from Bcl6−/− mice expressed increased interleukin-17 (IL-17) and extremely elevated levels of Th2 cytokines compared with wild-type exTreg cells. Although Treg cells normally express only low levels of cytokines, Treg cells from Bcl6−/− mice secreted higher levels of IL-4, IL-5, IL-13 and IL-17 than wild-type conventional T cells. Next, Treg-specific conditional Bcl6-deficient (Bcl6Foxp3−/−) mice were analysed. Bcl6Foxp3−/− mice do not develop inflammatory disease, indicating a requirement for non-Treg cells for inflammation in Bcl6−/− mice, and have normal numbers of exTreg cells. We induced Th2-type allergic airway inflammation in Bcl6Foxp3−/− mice, and found that while exTreg cytokine expression was normal, Bcl6-deficient Treg cells expressed higher levels of the Th2-specific regulator Gata3 than Bcl6+ Treg cells. Bcl6Foxp3−/− mice had increased numbers of Th2 cells after induction of airway inflammation and increased T cells in the bronchoalveolar lavage fluid. These data show both Treg-intrinsic and Treg-extrinsic roles for Bcl6 in controlling Treg cell stability and Th2 inflammation, and support the idea that Bcl6 expression in Treg cells is critical for controlling Th2 responses.
Keywords: Bcl6, ex-regulatory T cells, FoxP3, regulatory T cells, T helper type 2 differentiation
Introduction
Foxp3+ regulatory T (Treg) cells are essential for immunological tolerance and are required for curtailing inflammatory responses in a number of autoimmune diseases.1 Despite the central role of Treg cells in maintaining self-tolerance, there are unresolved questions about the stability and plasticity of the Treg phenotype in vivo. Most Treg cells maintain high Foxp3 levels following adoptive transfer in a non-pathogenic setting.2,3 Foxp3+ Treg cells can differentiate into cytokine-secreting Foxp3− T cells when cultured with cytokines in vitro.4–6 Further, studies employing Foxp3 reporter mice reveal that Foxp3 expression is unstable in inflammatory settings and Treg cells that lose Foxp3 expression, termed ‘exTreg’ cells, can acquire the cytokine production capabilities of effector T cells.7 Recently, in a mouse model of arthritis, interleukin-6 (IL-6) was shown to promote the conversion of Treg cells into T helper type 17 (Th17) -like pathogenic exTreg cells.8
Another example of apparent Treg cell plasticity is the finding that CD4+ CD25− Foxp3+ Treg cells can convert to Bcl6-expressing follicular helper T cells in mouse Peyer's patches.9 Intriguingly, transforming growth factor-β and retinoic acid present in the gut can induce miR-10a, a microRNA that targets Bcl6, so maintaining Treg cell stability and preventing Treg cell conversion to follicular helper T cells.10 These studies showing Treg plasticity contrast with studies showing that Foxp3+ Treg cells are extremely stable in vivo, under a variety of conditions.11–13 A recent publication reported that exTreg cells are not derived from stable thymus-derived Treg cells, but rather from a conventional CD4 T-cell population that transiently up-regulates Foxp3 following activation in the periphery.14 The extent to which these transient and unstable Foxp3+ cells act as Treg cells in vivo remains unclear. Further, the relationship of these transient Foxp3-expressing T cells to Treg cells induced in the periphery (peripheral Treg cells) is not known.12 Generally it is accepted that peripheral Treg cells are more unstable than thymus-derived Treg cells.12 The available data show that 90–95% of thymus-derived Foxp3+ T cells are extremely stable, whereas Foxp3+ T cells formed in the peripheral lymphoid organs contain a high fraction of unstable Foxp3+ T cells.4,13,15 Further, unstable Treg cells are particularly enriched in the CD25low Treg population, while stable Treg cells are CD25high.8 CD25low Treg cells may represent recently emerged peripheral Treg cells that are not fully committed to the Treg lineage and are still plastic16.
Bcl6-deficient mice develop a spontaneous and severe Th2-type inflammatory disease including myocarditis and pulmonary vasculitis,17–20 and Bcl6-deficient Treg cells fail to control Th2 inflammation.21 Bcl6 is required to repress Gata3 activity in Treg cells, and Bcl6-deficient Treg cells display an intrinsic increase in Th2 gene and microRNA-21 (miR-21) expression.21,22 Bcl6-deficient Treg cells from mixed bone marrow chimeras displayed a weaker expression of Th2 genes than Treg cells from Bcl6-deficient mice, indicating that a combination of wild-type Treg cells and the lack of Th2 inflammation in these mice was suppressing the up-regulation of Th2 cytokines by Bcl6-deficient Treg cells.21 Although Bcl6-deficient Treg cells had a strong Th2 gene expression bias, these cells did not show any reduction or loss of the classical Treg gene signature. Further, Bcl6-deficient Treg cells exhibited normal suppressive activity in vitro and in an in vivo colitis model.21 Hence, Bcl6-deficient Treg cells are largely normal, but the presence of Th2 inflammation induces abnormally strong Th2 gene expression. One explanation for the failure of Bcl6-deficient Treg cells to control Th2 inflammation is that the strong inflammatory environment in Bcl6-deficient mice promotes Th2 cytokine expression by Treg cells, short-circuiting the suppression of Th2 responses. Another possibility is that Bcl6 is required to stabilize Treg cells in the presence of Th2 inflammation, and Bcl6-deficient Treg cells exposed to Th2 inflammation undergo loss of Foxp3 expression and reprogramming of the cells to a Th2 effector fate.
To test this hypothesis, we developed a system whereby we could track exTreg cells in Bcl6-deficient mice. We find that in a Th2-type inflammatory environment, Bcl6-deficient Treg cells lose Foxp3 expression at a higher rate than wild-type Treg cells; however, in a non-inflammatory environment, Bcl6-deficient Treg cells are as stable as wild-type Treg cells. We further analyse the intrinsic role for Bcl6 in Treg cells for controlling Treg stability by testing Treg-specific Bcl6-deficient mice in an induced model of Th2 inflammation. Our data show that Bcl6 maintains Treg stability by both Treg-extrinsic and Treg-intrinsic pathways, and further define the role for Bcl6 in Treg cells for controlling Th2 responses.
Materials and methods
Mice
Bcl6−/− and Bcl6+/− mice on a mixed C57BL/6-129Sv background have been previously described.17,18 Foxp3-GFP-Cre and Rosa-YFP mice were obtained from Jackson Laboratories (Bar Harbor, ME). Bcl6+/− mice were mated consecutively to these two strains to produce the FCRY (Foxp3-gfp-Cre × Rosa-Yfp) strain. Mice were genotyped for iCre and the Rosa-YFP alleles according to PCR protocols from Jackson Laboratories. Bcl6−/− mice were used between 3 and 6 weeks of age; wherever possible, Bcl6+/+ mice were littermate controls. Bcl6−/− mice were used at a young age because of their consistent early death on this strain background. Conditional Bcl6fl/fl mice were recently described.23 Rosa-RFP (td-Tomato) mice were obtained from Jackson Laboratories and mated to Bcl6fl/fl mice already mated to Foxp3-GFP-Cre mice. Genotyping for the floxed Bcl6 allele was performed as described,23 and genotyping for iCre and Rosa-RFP was performed according to PCR protocols from Jackson Laboratories. Mice were bred under specific pathogen-free conditions at the laboratory animal facility at the Indiana University School of Medicine and were handled according to protocols approved by the Indiana University School of Medicine Animal Use and Care Committee.
Antibodies, flow cytometry and cell sorting
Flow cytometry analysis of Treg surface markers was performed using anti-CD4 antibody (RM4-5; BD Biosciences, San Jose, CA) and anti-CD25 antibody (PC61.5; eBioscience, San Diego, CA). Both Treg and exTreg cells were primarily tracked by GFP and YFP/RFP expression. Flow analysis was carried out on an LSR machine (Becton Dickinson, Franklin Lakes, NJ) and data were analysed using FLOWJO software (Tree Star, Inc., Ashland, OR). For isolation of stable Treg and exTreg cells, CD4+ CD25+ T cells were purified from Bcl6+/+ and Bcl6−/− FCRY mice using magnetic beads (Miltenyi Biotech, San Diego, CA), followed by Fluorescence Activated Cell Sorting (FACS) for pure GFP+ YFP+ (or GFP+ RFP+) stable Treg cells and GFP− YFP+ (or GFP− RFP+) exTreg cells using a FACSAria cell sorter (Becton Dickinson). Conventional T (Tconv) cells were CD4+ CD25− T cells obtained during magnetic selection of CD4+ CD25+ T cells. T cells were stimulated for 16 hr with anti-CD3 and anti-CD28 antibodies (BD Biosciences), as described previously,21 before mRNA isolation. Intracellular staining of Foxp3 and Gata3 protein was performed on spleen cells stimulated with PMA plus ionomycin as described elsewhere.21.
Gene expression analysis
Total cellular RNA was prepared using the Trizol method (Life Technologies, Grand Island, NY), and cDNA was prepared with the Transcriptor First Strand cDNA synthesis kit (Roche, Indianapolis, IN). Quantitative PCR were run by assaying each cDNA sample in triplicates with Taqman assays (ABI) for each specific gene. Primer sequences for Taqman assays are proprietary. Levels of mRNA expression were normalized to β-tubulin (Tubb5) mRNA levels, and differences between samples were analysed using the ΔΔCt method. Gene expression levels of reference samples were averaged at the ΔCt level of analysis, and then individual levels of expression for each sample were calculated and these levels were averaged to show in the graphs. Hence, the average normalized reference point can vary from a perfect 1·0, depending on the variation within the set of reference control samples from the average ΔCt.
Cytokine analysis
T cells were stimulated for 16 hr with anti-CD3 and anti-CD28 antibodies (BD Biosciences), and then supernatants were collected, as described.21 Cytokine levels in the media supernatants were determined by multiplex ELISA using the Milliplex MAP Kit (Millipore, Billerica, MA) as described.24 Intracellular cytokine staining was performed as described,21,23 on total spleen cells stimulated with PMA plus ionomycin.
Airway inflammation model
Bcl6+ Foxp3-Cre+ ROSA-RFP (control) mice and BCL6fl/fl Foxp3-Cre+ ROSA-RFP (Bcl6Foxp3−/−) mice were sensitized intraperitoneally with ovalbumin (OVA) (Sigma, St Louis, MO) adsorbed to alum (Sigma) at a dose of 20 μg OVA/2 mg alum on days 0 and 7 of the protocol.24 Starting on day 14, mice were challenged intranasally with OVA for 5 consecutive days (100 μg/day). Mice were killed by intraperitoneal injection of pentobarbital (5 mg/mouse) 48 hr after the final intranasal challenge on day 18. The trachea was cannulated and lungs were lavaged three times with 1 ml PBS to collect the bronchoalveolar lavage (BAL). Cells recovered in BAL fluid were counted with a haemocytometer. Eosinophils, neutrophils, T cells, B cells and mononuclear cells in the BAL fluid were distinguished by cell size and by expression of CD3, B220, CCR3, CD11c and MHC class II, and percentages of each cell type were assessed by flow cytometry as described previously.24 Paraffin-embedded lung sections were stained with haematoxylin & eosin for evaluation of the infiltration of inflammatory cells by light microscopy. Inflammation was scored on a scale of 0–5, where 0 = no inflammation of lung airways, 1 = faint inflammatory infiltrate around airways, 2 = light inflammatory infiltrate around airways, 3 = medium inflammatory infiltrate around airways, 4 = heavy inflammatory infiltrate around airways with inflammatory cells spreading into some portions of lung parenchyma, 5 = heavy inflammatory infiltrate around airways with inflammation spreading into most of the lung parenchyma.
Statistical analysis
P-values were calculated using Student's t-test or analysis of variance. All calculations were made using GraphPad Prism software (La Jolla, CA). A P-value < 0·05 was considered to show a significant difference.
Results
Stability of Treg cells in the absence of Bcl6
In order to perform lineage tracing of Bcl6−/− Foxp3+ Treg cells in the context of Th2 inflammation, we crossed Bcl6−/− mice with Foxp3-GFP-Cre × Rosa26-YFP mice (Fig.1a). In these mice, the Foxp3-GFP-Cre+ Treg cells excise the loxP-flanked stop codon in front of the YFP gene, allowing for constitutive YFP expression from the Rosa26 promoter. The Foxp3-Cre activity permanently labels the Foxp3+ T cells and their progeny.7 This dual reporter mouse strain enables tracing of Foxp3 induction and down-regulation concurrently by two-colour flow analysis of GFP and YFP expression (Fig.1b, c). Because of a delay between Foxp3 transcription (GFP expression) and excision of the loxP-flanked stop cassette in the Rosa26-YFP transgene by Cre, the GFP+ YFPlo−neg population represents cells that recently up-regulated Foxp3 expression. Hence, GFP+ YFP+ cells represent ‘stable’ mature Treg cells and GFP– YFP+ cells represent ‘exTreg’ cells. Using this system, we then monitored the percentages of stable Treg cells and exTreg cells in the thymus and peripheral lymphoid organs in Foxp3-GFP-Cre Rosa26-YFP Bcl6−/− [FCRY knockout (KO)] mice relative to control Foxp3-GFP-Cre Rosa26-YFP Bcl6+/+ [FCRY wild-type (WT)] mice. Similarly to Bcl6−/− mice,17–20 FCRY KO mice developed eosinophilic myocarditis and lung vasculitis at a high rate and died at an early age (data not shown). Hence, these mice develop severe Th2 inflammation typical of the Bcl6−/− mouse phenotype. Analysis of the thymus in the FCRY WT and FCRY KO mice demonstrated comparable percentages of stable Treg and exTreg cells, thereby suggesting that the Bcl6−/− Treg cells are not intrinsically biased towards losing Foxp3 expression or do not lose Foxp3 shortly after their development (Fig.2). We next looked in the peripheral Treg compartment. Our previous data showed that the percentage of total Foxp3+ Treg cells was not significantly altered in Bcl6−/− mice;21 however, analysis of stable Treg and exTreg populations in the peripheral lymphoid organs (spleen and lymph nodes) demonstrated a significant increase in the percentage of exTreg cells in the FCRY KO mice relative to FCRY WT mice (Fig.3a, b). Although Treg cells are largely CD25+, previous work has shown that exTreg cells are found at a higher frequency in the CD4+ CD25− cell population than in the CD4+ CD25+ cell population.8 Indeed, for FCRY WT mice, the average percentage of exTreg cells was increased with the total CD4+ gate compared with the CD4+ CD25+ gate (Fig.3a, b). However, this pattern was not observed with FCRY KO mice, as comparable percentages of exTreg cells were seen using either the total CD4+ or the CD4+ CD25+ gate (Fig.3a, b). Strikingly, within the CD4+ CD25+ cell subset, the percentage of exTreg cells in FCRY KO spleen and lymph nodes was two to three times greater than in FCRY WT mice. This result indicates that the Th2 inflammatory environment in Bcl6−/− mice potently de-stabilizes Treg cells and markedly promotes the loss of Foxp3 expression at the CD25+ Treg stage.
Figure 1.
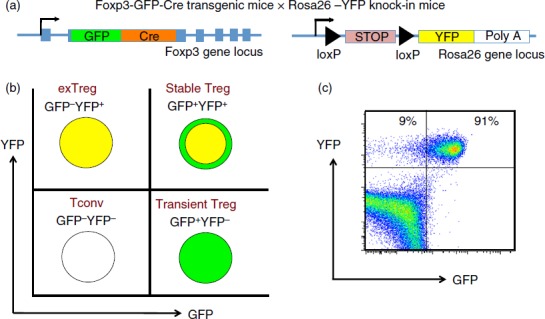
A genetic system for tracking regulatory T (Treg) cell stability. (a) Mouse gene constructs used in generation of Foxp3-GFP-Cre-Rosa-YFP (FCRY) mice. (b) expected fluorescence patterns for conventional T (Tconv) cells, transient Treg, stable Treg and exTreg cells. (c) Sample flow plot for GFP by YFP in spleen cells from a FCRY mouse.
Figure 2.
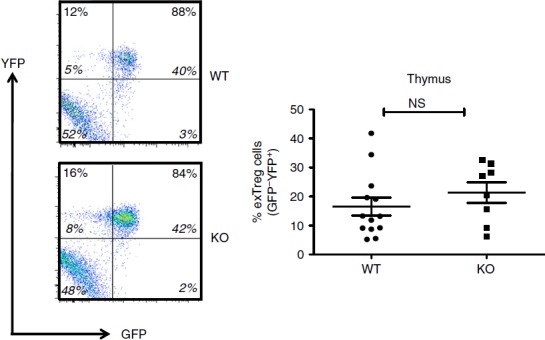
Regulatory T (Treg) cell stability in the thymus of Bcl6−/− mice. Data from the thymuses of Bcl6+/+ (wild-type; WT) and Bcl6−/− (knockout; KO) Foxp3-GFP-Cre-Rosa-YFP (FCRY) mice, with representative flow plots on left and graphed data on right. Thymocytes were gated for surface Treg markers (CD4+ CD25+) before GFP versus YFP analysis. The % of exTreg cells is calculated by the % GFP− YFP+ cells within the total YFP+ population. The numbers in large bold font in upper left and upper right quadrants are % of exTreg cells and % stable Treg cells within the YFP+ population, and these percentages add up to 100%. The smaller numbers in italic font are the % of cells for each of the four quadrants and all four of these numbers add up to 100%. Graphs show average exTreg cells ± SE, with n = 8 to n = 12 mice of each genotype. Data from four separate experiments were combined into each graph. Each symbol represents one mouse. Student's t-tests were used to calculate statistical significance and P-values. NS, not significant (P > 0·05).
Figure 3.
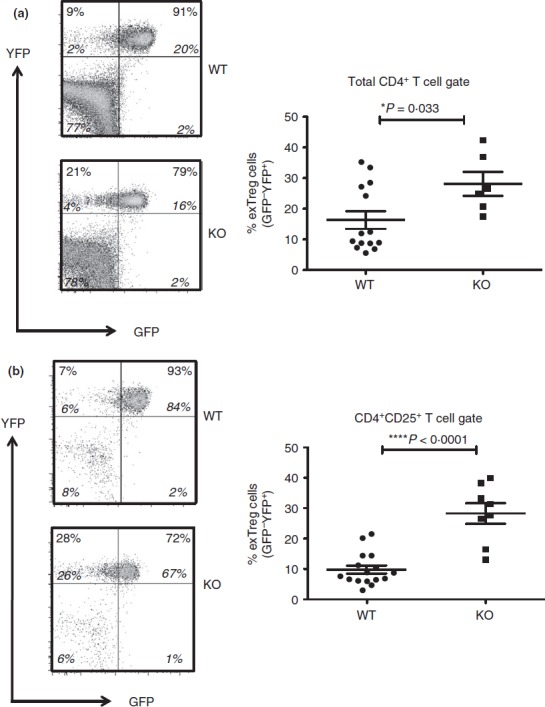
Regulatory T (Treg) cell stability in the periphery of Bcl6−/− mice. Data from the spleen of Bcl6+/+ (wild-type; WT) and Bcl6−/− (knockout; KO) Foxp3-GFP-Cre-Rosa-YFP (FCRY) mice, with representative flow plots on left and graphed data on right. Splenocytes were gated for either CD4+ (a) or CD4+ CD25+ (b) cells before GFP versus YFP analysis. The % of exTreg cells is calculated by the % GFP– YFP+ cells within the total YFP+ population. The numbers in large bold font in upper left and upper right quadrants are % exTreg cells and % stable Treg cells within the YFP+ population, and these percentages add up to 100%. The smaller numbers in italic font are the % of cells for each of the four quadrants and all four of these numbers add up to 100%. Graphs show average exTreg cells ± SE, with n = 8 to n = 12 mice of each genotype. Data from four separate experiments were combined into each graph. Each symbol represents one mouse. Student's t-tests were used to calculate statistical significance and P-values. NS, not significant (P > 0·05), *P < 0·05, ****P < 0·001.
Potent Th2 responses by conventional T cells, Treg cells and exTreg cells in the absence of Bcl6
ExTreg (GFP− YFP+) cells have been reported to exhibit an activated-memory phenotype, with reduced expression of Treg signature genes, high CD44 expression and acquisition of effector cytokine-producing ability [interferon-γ (IFN-γ) or IL-17A], depending on the microenvironment.7 To assess the effect of the Th2 inflammatory environment on the Treg and exTreg phenotype in Bcl6−/− mice, we sorted CD4+ CD25− Tconv cells, GFP+ YFP+ stable Treg cells and GFP− YFP+ exTreg cells from FCRY WT and FCRY KO mice, activated them in vitro and assessed their gene expression and cytokine secretion signature. Consistent with the well-demonstrated role for Bcl6 in inhibiting Th2 differentiation,21,25 we noted increased expression of Th2 genes (Gata3, Il4, Il5, Il13) in FCRY KO Tconv cells, relative to FCRY WT Tconv cells (Fig.4). This increased transcription of Th2 genes translated into increased secretion of Th2 cytokines by FCRY KO Tconv cells as shown in Fig.5. Although Il17a message and IL-17A expression were increased in FCRY KO Tconv cells, Ifng message and IFN-γ secretion were not significantly different. Secretion of IL-10 was augmented in the Bcl6−/− Tconv cells (Fig.5), consistent with previous work showing repression of IL-10 by Bcl6.23,25
Figure 4.
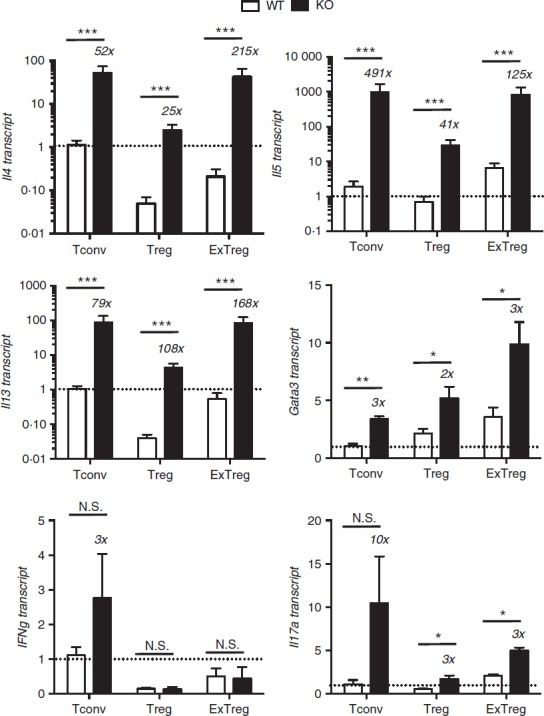
Cytokine gene expression in Bcl6−/− regulatory T (Treg) and exTreg cells. CD4+ CD25− conventional T (Tconv) cells and CD4+ CD25+ cells from Bcl6+/+ (wild-type; WT) and Bcl6−/− (knockout; KO) FCRY mice were obtained by magnetic bead selection. CD4+ CD25+ cells were further separated into Treg cells (GFP+ YFP+) and exTreg cells (GFP− YFP+) by cell sorting. Isolated cells were stimulated 16 hr with anti-CD3 and anti-CD28 antibodies before harvest for RNA isolation. Gene expression was assessed by quantitative PCR. Data shown are representative of two different experiments. Graphs show relative gene expression, and bars are average fold ± SE and wild-type Tconv cells set as 1. n = 3 to n = 5 of each genotype. Numbers on top of bars indicate fold-increase of KO to WT. Student's t-tests were used to calculate statistical significance and P-values. NS, not significant (P > 0·05), *P < 0·05, **P < 0·01, ***P < 0·001.
Figure 5.
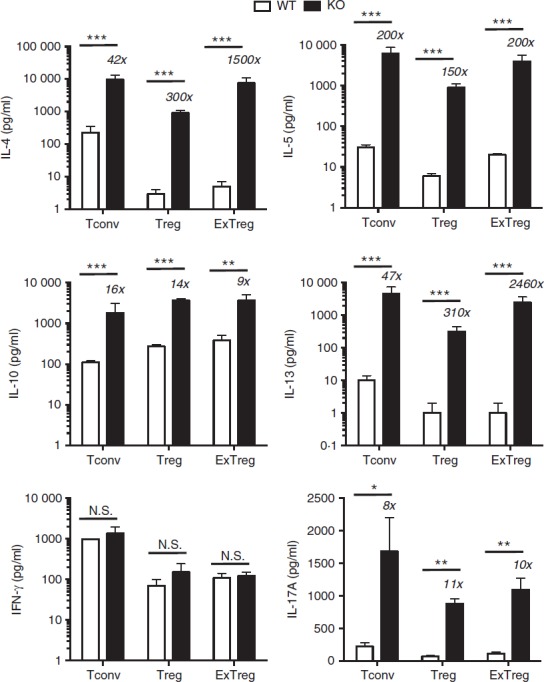
Cytokine secretion by Bcl6−/− regulatory T (Treg) cells and exTreg cells. CD4+ CD25− conventional T (Tconv) cells and CD4+ CD25+ cells from Bcl6+/+ (wild-type; WT) and Bcl6−/− (knockout; KO) FCRY mice were obtained by magnetic bead selection. CD4+ CD25+ cells were further separated into Treg cells (GFP+ YFP+) and exTreg cells (GFP– YFP+) by cell sorting. Isolated cells were stimulated for 16 hr with anti-CD3 and anti-CD28 antibodies, at which time cell supernatants were taken. Cytokine levels were determined by multiplex ELISA. Graphs show relative cytokine secretion, and bars are average fold ± SE and wild-type Tconv cells set as 1. n = 3 to n = 5 of each genotype. Numbers on top of bars indicate fold-increase of KO to WT. Student's t-tests were used to calculate statistical significance and P-values. NS, not significant (P > 0·05), *P < 0·05, **P < 0·01, ***P < 0·001.
In the stable Treg cell fraction, overall cytokine gene expression was reduced in both Bcl6+/+ and Bcl6−/− Treg cells compared with the respective Tconv cells, consistent with a role of Foxp3 in limiting effector cytokine expression in Treg cells. However, Treg cells from the FCRY KO mice exhibited a strong Th2 gene expression bias (Fig.4); specifically, Il4, Il5, Il13 and Gata3 mRNAs were strongly elevated in Bcl6−/− Treg cells, confirming that Bcl6 represses Th2 gene expression in Treg cells.21 The increased Th2 gene expression in Bcl6−/− Treg cells corresponded with a very strong increase in Th2 cytokine secretion, such that Bcl6−/− Treg cells produced 300-fold higher levels of IL-4 and IL-13 than Bcl6+/+ Treg cells (Fig.5). Even more strikingly, Bcl6−/− Treg cells secreted more IL-4 (4× higher), IL-5 (30× higher) and IL-13 (30× higher) than Bcl6+/+ Tconv cells (Fig.5). These data are consistent with the ability of Bcl6−/− Treg cells to promote rather than suppress Th2-type inflammation in an allergic airway disease model.21 Interleukin-10 secretion was increased by Bcl6−/− Treg cells (Figs5 and 6), consistent with earlier data that Bcl6 represses Il10 mRNA in Treg cells as well as in Tconv cells.21
Figure 6.
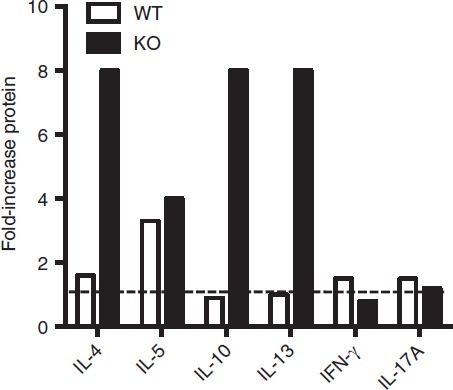
Up-regulation of cytokine genes by ex-regulatory T (exTreg) cells. Data from Fig.5 was further analysed to show increases in cytokine secretion going from Treg to exTreg cells for either Bcl6+/+ (wild-type; WT) or Bcl6−/− (knockout; KO) FCRY mice. Graph shows fold increase of cytokine expression for exTreg cells compared with Treg cells of each genotype.
In the exTreg cell fraction, there was a continued trend of much greater Th2 gene and cytokine expression from Bcl6−/− cells (Figs4 and 5). Secretion of IL-17 was also increased from Bcl6−/− exTreg cells but that of IFN-γ was not. Consistent with a loss of Foxp3 repression, Th2 cytokine expression by Bcl6−/− exTreg cells was much higher than Th2 cytokine expression from Bcl6−/− Treg cells and was in the range of Th2 cytokine expression by Bcl6−/− Tconv cells (Figs4, 5 and 6). Bcl6+/+ exTreg cells showed little to no increase in IFN-γ, IL-5 and IL-17 secretion compared with Bcl6+/+ Treg cells, but did show a notable increase in IL-4, IL-5 and IL-13 secretion (Fig.6). The increase in Th2 cytokines by Bcl6−/− exTreg cells compared with Bcl6−/− Treg cells, especially for IL-4, was overall far greater than the corresponding increase of cytokines by Bcl6+/+ exTreg cells compared with Bcl6+/+ Treg cells (Fig.6). Hence, these data show that Bcl6 is a strong and specific repressor of Th2 gene expression, not only in Tconv cells, but also in Treg and exTreg cells.
Treg-specific conditional knockout mice reveal Treg-intrinsic and Treg-extrinsic roles for Bcl6
We next wished to address whether the increased loss of Foxp3 expression and increased expression of cytokines in Bcl6−/− Treg and exTreg cells was due to inflammatory disease in the mice, or due to an intrinsic effect of loss of Bcl6 on Treg cell stability. We therefore used conditional knockout mice for Bcl623 and Foxp3-GFP-Cre to conditionally delete Bcl6 in the Treg lineage (Fig.7a). These Bcl6Foxp3−/− mice were then mated to mice with a similar conditional Rosa26 allele as shown in Fig.1, except expressing RFP instead of YFP. In these Foxp3-GFP-Cre Rosa26-RFP (FCRR) mice, stable Treg cells are GFP+ RFP+ and exTreg cells are GFP− RFP+. There was no difference in the overall numbers of Treg cells in Bcl6Foxp3−/− mice compared with control mice (Fig.7b and data not shown). In contrast to Bcl6−/− mice, there was also no difference in the generation of exTreg cells between Bcl6Foxp3−/− and control FCRR mice in the steady state (Fig.7b, c). Also, unlike Bcl6−/− mice, Bcl6Foxp3−/− mice do not develop inflammatory disease or significant Th2 cell skewing (data not shown). Hence, it is likely that Treg-extrinsic factors, such as the strong Th2 inflammatory environment in Bcl6−/− mice, promote the increase in exTreg cells in Bcl6−/− mice. To test whether Th2 inflammation could preferentially destabilize Bcl6-deficient Treg cells, we induced Th2-type allergic airway inflammation in control and Bcl6Foxp3−/− FCRR mice, where the antigen OVA in alum adjuvant is used for systemic priming and subsequent airway challenge.24 However, while induction of Th2 inflammation in both control and Bcl6Foxp3−/− FCRR mice led to a significant increase in exTreg cells compared with the steady state (Fig.7c), there was still no difference in exTreg generation between control and Bcl6-deficient Treg cells (Fig.7c). The data in Fig.7(b) show exTreg cells gated on total CD4+ T cells, and the percentage of exTreg cells was not increased when a CD4+ CD25+ gate was used (Fig.7d). These data indicate that the Th2-type inflammatory environment in Bcl6−/− mice destabilizes Treg cells by a mechanism unique from the Th2-type inflammatory environment created in the OVA-Alum allergic airway disease model.
Figure 7.
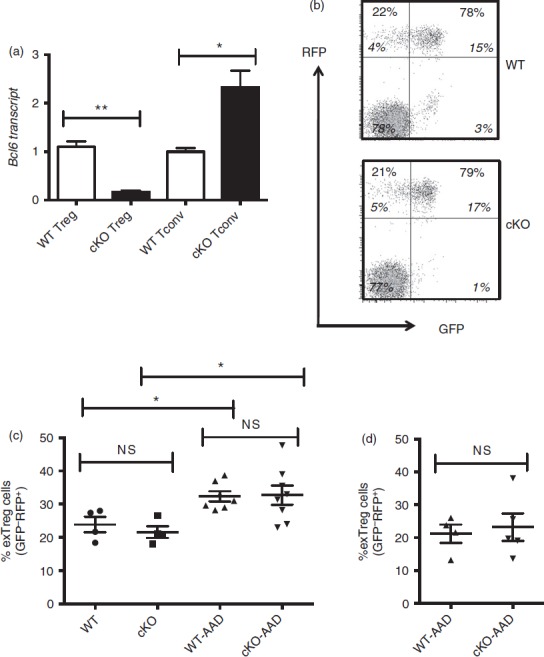
Regulatory T (Treg) stability in Treg-specific conditional Bcl6−/− mice. (a) Quantitative PCR analysis of Bcl6 transcript between Treg-specific conditional Bcl6−/− (Bcl6Foxp3−/−) (conditional knockout; cKO) mice and control Bcl6+/+ (Bcl6Foxp3+/+) (wild-type; WT) mice. CD4+ CD25+ FoxP3+ (Treg) and CD4+ CD25− (conventional T; Tconv) cells were prepared by MACS separation and FACS isolation, then activated for 16 hr with anti-CD3 and anti-CD28 before RNA and cDNA preparation. Relative gene expression is shown, where Bcl6 expression is normalized to tubb5. Data are average expression ± SE from three WT and six cKO mice. The increase in Bcl6 mRNA expression in cKO Tconv cells probably relates to an increase in Bcl6-expressing follicular helper T cells in these mice (unpublished data). (b) Data from the spleens of Treg-specific conditional Bcl6−/− (Bcl6Foxp3−/−) (cKO) mice and control Bcl6+/+ (Bcl6Foxp3+/+) (WT) mice, mated to a Foxp3-GFP-Cre-Rosa26-RFP (FCRR) background. Representative flow plots of splenocytes gated for CD4+ expression before GFP versus RFP analysis. RFP was used instead of YFP for these mice, but the gating pattern for Treg and exTreg cells is otherwise the same as for FCRY mice. The numbers in large bold font in upper left and upper right quadrants are % of exTreg cells and % of stable Treg cells within the RFP+ population, and these percentages add up to 100%. The smaller numbers in italic font are the % of cells for each of the four quadrants and all four of these numbers add up to 100%. The % of exTreg cells is calculated by the % GFP− RFP+ cells divided by the total RFP+ population. (c) ExTreg percentages in the steady state (WT and cKO) or after induction of allergic airway inflammation [WT-AAD and cKO-AAD; allergic airway disease (AAD)]. Cells shown are gated on total CD4+ T cells. (d) Percentages of exTreg cells after induction of allergic airway inflammation (WT-AAD and cKO-AAD). Cells shown are gated on CD4+ CD25+ T cells. Graphs show average ± SE, with n = 4 to n = 7 mice of each type. Data are shown is representative of two different experiments. Each symbol represents one mouse. Analysis of variance was used to calculate statistical significance and P-values. NS, not significant (P > 0·05), *P < 0·05, **P < 0·01
We had previously shown that Bcl6-deficient Treg cells from Bcl6−/− mice were unable to suppress Th2 inflammation and rather actually augmented Th2 inflammation.21 We therefore wondered if Bcl6-deficient Treg cells affected the Th2 response in Bcl6Foxp3−/− mice in the OVA-Alum allergic airway disease model. T cells from control and Bcl6Foxp3−/− mice induced for allergic airway disease were assessed for intracellular Gata3 and IL-4 expression as markers of Th2 responses. As shown in Fig.8(a), Tconv cells from Bcl6Foxp3−/− mice, had significantly increased numbers of Gata3+ and IL-4-expressing cells than control mice, consistent with the idea that Bcl6-deficient Treg cells are defective in controlling Th2 responses. The percentage of Tconv cells expressing IFN-γ was not different from control and Bcl6Foxp3−/− mice, indicating Th2 specificity to the defect in the Bcl6Foxp3−/− mice. We next analysed Treg cells in control and Bcl6Foxp3−/− mice in the OVA-Alum allergic airway disease model. The overall number of Treg cells in Bcl6Foxp3−/− mice was significantly higher than in control mice, indicating increased expansion of Bcl6-deficient Treg cells in response to Th2 inflammation (Fig.8b). Even more striking, the fraction of Gata3+ cells within the Bcl6-deficient Treg population was twofold higher than in control mice (Fig.8b). These data support the model that Bcl6 function in Treg cells is required for suppression of a Th2 phenotype within Treg cells.21 However, unlike in Bcl6−/− mice, Treg cells from Bcl6Foxp3−/− mice did not express detectable levels of cytokines (data not shown). To determine if exTreg cells in Bcl6Foxp3−/− mice had skewing towards the Th2 lineage, as we observed with Bcl6−/− mice, we sorted exTreg cells from control and Bcl6Foxp3−/− FCRR mice induced to develop allergic airway disease. As shown in Fig.8(c), exTreg cells from Bcl6Foxp3−/− FCRR mice showed no signs of abnormal Th2 skewing. Bcl6-deficient Treg cells did express significantly higher IL-10 mRNA, consistent with IL-10 being a direct target of Bcl6-mediated repression. Overall, these data show that Bcl6 expression in Treg cells is required to maintain normal Th2 responses, but that the extremely strong Th2 responses in Treg and exTreg cells in Bcl6−/− mice is due to the abnormal inflammatory background.
Figure 8.

T-cell responses in regulatory T (Treg) cell-specific conditional Bcl6−/− mice after induction of allergic airway disease. Data from the spleens of Treg-specific conditional Bcl6−/− (Bcl6Foxp3−/−) (conditional knockout; cKO) mice and control Bcl6+/+ (Bcl6Foxp3+/+) mice, mated to a Foxp3-GFP-Cre-Rosa26-RFP (FCRR) background and induced to develop allergic airway inflammation by priming with ovalbumin (OVA) -Alum and repeated nasal OVA challenge. (a, b) Spleen cells were stimulated with PMA plus ionomycin along with Golgi-plug for 6 hr, stained for CD4, Foxp3, Gata3, interleukin-4 (IL-4) and interferon-γ (IFN-γ), then analysed by flow cytometry. (c) Since cell fixation destroys the GFP signal, and precludes exTreg cell analysis by ICS, live GFP– RFP+ exTreg cells were obtained by sorting, then stimulated for 16 hr with anti-CD3 and anti-CD28 antibodies before harvest for RNA isolation. Gene expression was assessed by quantitative PCR. n = 4 to n = 7. NS, not significant (P > 0·05), *P < 0·05, **P < 0·01, ****P < 0·001.
Lastly, we analysed the level of airway inflammation in control and Bcl6Foxp3−/− mice induced in the allergic airway disease model. As shown in Fig.9(a), strong airway inflammation was induced in this model. However, there was no significant increase in the severity of lung inflammation in Bcl6Foxp3−/− mice (Fig.9b) and the overall cell infiltrate into bronchoalveolar spaces as tested by BAL, was not significantly different (data not shown). However, when the BAL cells were analysed more carefully for specific types of immune cells, although most cell types were at similar levels between control and Bcl6Foxp3−/− mice, we observed a significant increase in the numbers of T cells in BAL from Bcl6Foxp3−/− mice (Fig.9c). Hence, Bcl6-deficient Treg cells are defective in controlling numbers of T cells in the lung during Th2 inflammation. Overall, these data show that specific loss of Bcl6 in Treg cells has specific and significant influences on the inflammatory response in a Th2 model of airway inflammation.
Figure 9.
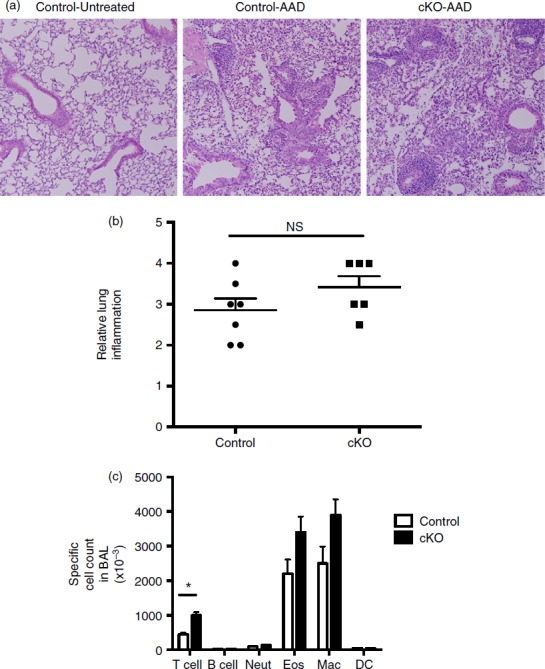
Inflammatory responses in regulatory T (Treg) cell-specific conditional Bcl6−/− mice after induction of allergic airway disease. Treg-specific conditional Bcl6−/− (Bcl6Foxp3−/−) (conditional knockout; cKO) mice and control Bcl6+/+ (Bcl6Foxp3+/+) mice were induced to develop allergic airway inflammation. Mice were primed with ovalbumin (OVA) antigen in Alum adjuvant intraperitoneally, and then challenged with OVA intranasally for 5 consecutive days, as described.21 (a) Representative lung histology on haematoxylin & eosin stained sections showing airway inflammation after disease induction. Control-Untreated is a control Bcl6+/+ mouse without any disease induction. Control-allergic airway disease (AAD) and cKO-AAD are control and cKO mice, respectively, induced to develop allergic airway disease. Strong infiltrates of inflammatory cells are observed around the airways of the AAD mice (b) Lung inflammation scores for Control-AAD and cKO-AAD mice, where the severity of the inflammatory infiltrate was analysed as described in Materials and methods. Each point shows one mouse. (c) Quantification of brochoalveolar lavage (BAL) cells obtained from Control-AAD and cKO-AAD mice. BAL cells were counted and assessed for specific cell types by flow cytometry. Graph shows average number of specific BAL cell types in Control-AAD and cKO-AAD mice. Neut, neutrophils; Eos, eosinophils; Mac, macrophage; DC, dendritic cells. n = 6 or n = 7. The experiment was performed twice with similar results obtained each time. NS, not significant (P > 0·05), *P < 0·05.
Discussion
We show that the severe Th2-type inflammation in Bcl6−/− mice not only promotes the formation of conventional Th2 responses, but also leads to the formation of increased numbers of Th2 cytokine-secreting exTreg cells. We also show for the first time that Bcl6−/− Treg cells can secrete Tconv-like levels of Th2 cytokines; these data help to explain the inability of Bcl6−/− Treg cells to control Th2-type inflammation.21 These data are novel in that they show that exTreg cells that formerly expressed Foxp3 can also become Th2-like, and not only develop Th1 and Th17 effector capacity, as has been previously shown.7 We further compare exTreg generation in wild-type and mutant mice, and observe that enhanced formation of exTreg cells and increased formation of Th2 cytokine-producing exTreg cells occurs only in the inflammatory Bcl6−/− background. Hence, while significant numbers of exTreg cells may be generated in the wild-type steady-state environment, exTreg cells only produce high levels of effector cytokines in the inflammatory environment.
A previous study showed that human Treg cells that lose Foxp3 after in vitro activation, preferentially develop into Th2 cytokine-producing cells.26 Another set of studies using a mouse Treg model showed that attenuated Foxp3 expression caused Treg cells to preferentially differentiate into Th2 cytokine-producing cells.27,28 Curiously, conversion of Treg cells into Th2 cytokine-producing cells was not seen if Foxp3 expression was completely lost.27 The ubiquitin ligase Itch has been shown to control Th2 responses, and mice with Itch deleted specifically in Treg cells develop Th2 cytokine-expressing Treg and exTreg cells similar to what we observe with Bcl6-deficient mice.29 However, in contrast to our findings, the Th2 pathway controlled by Itch is purely Treg-intrinsic, and further exTreg cells are not generated at increased proportions.29 Our current data indicate that the Th2-type inflammation observed in Bcl6−/− mice can strongly activate Th2 cytokine expression in both Treg and exTreg cells, by a Treg-extrinsic pathway. This Treg-extrinsic pathway may be relevant to human disease and can help explain why Treg cells cannot control the Th2 inflammation that occurs in allergic disease.
The stability of Treg cells has been a controversial topic in the past, but the recent consensus is that the large majority of Treg cells, particularly thymus-derived Treg cells, are very stable and maintain suppressor activity for their lifetime.13,15 If a small fraction of Treg cells loses Foxp3 expression, these cells can expand rapidly in vivo, due to loss of Foxp3-mediated inhibition of proliferation. Indeed, the overall percentage of stable Treg cells is not decreased in Bcl6−/− mice, indicating that exTreg cells may be formed from relatively few Treg cells. Furthermore, the bias towards self-reactivity of Treg cells can augment the responses of exTreg cells.16 The predominant question is whether exTreg cells, as observed here, and in other studies,7,14 represent the loss of Foxp3 by bona fide Treg cells or whether they represent Tconv cells that transiently induced endogenous Foxp3 and so induced Foxp3-Cre recombinase, leading to activation of the lineage tracer (e.g. YFP) expression.14 Once these cells lose the transient Foxp3 expression, they would be indistinguishable from true exTreg cells. The exTreg population we observe in Bcl6−/− mice may be a mixture of both true exTreg and Tconv cells that transiently induced Foxp3 and then lost expression. The strong Th2-type inflammation in Bcl6−/− mice, plus the loss of Bcl6 in Treg cells, may lead to Treg instability and increased production of true exTreg cells. It is also possible that in Bcl6−/−mice, a very high fraction of conventional T cells transiently induce Foxp3 (and hence turn on marker expression), and then lose Foxp3 expression over time, while also differentiating into Th2-secreting cells. However, an argument against this possibility is that pro-inflammatory cytokines appear to inhibit transient activation-induced Foxp3 expression,14 and so transient induction of Foxp3 in Tconv cells should be suppressed in Bcl6−/− mice with ongoing inflammatory disease. Consistent with this, we have observed lower Foxp3 expression in activated Tconv cells from Bcl6−/− mice.21 Another argument in favour of GFP− YFP+ T cells from Bcl6−/− mice being true exTreg cells is that they have a slightly different gene expression pattern from conventional CD4 T cells, in that they express markedly less IFN-γ and more Gata3 compared with total Tconv cells (Fig.4). However, this difference may be somewhat illusory if the exTreg cells are derived preferentially from effector Th2 cells generated in Bcl6−/− mice. Hence, exTreg cells may produce very similar levels of Th2 cytokines as Tconv cells that are predominantly Th2 differentiated.
Strikingly, the exTreg cells from both Bcl6+/+ and Bcl6−/− mice do not up-regulate IFN-γ compared with Treg levels, in contrast to the exTreg data from Zhou et al.7 This difference may be due to the NOD mouse background used in the Zhou et al. study, and the development of Th1-type autoimmune inflammation in the NOD mouse model. Also, unlike Zhou et al., we did not observe up-regulation of IL-17 in Bcl6+/+ Treg cells. Interleukin-17 was expressed at higher levels by Bcl6−/− exTreg cells than Bcl6+/+ exTreg cells, but IL-17 was not higher in Bcl6−/− exTreg cells compared with Bcl6−/− Treg cells. Hence, the main increase in cytokine expression from Treg to exTreg cells in the Bcl6−/− mice is restricted to Th2 cytokines.
Our data with Bcl6Foxp3−/− mice show that in Bcl6−/− mice, the in vivo environment leads to the greatly enhanced generation of Th2 cytokine-expressing exTreg cells, and that this in vivo environment in Bcl6−/− mice is unique and cannot be mimicked by induction of allergic Th2 airway inflammation. We are unable to determine definitively if there is increased instability of true Treg cells in Bcl6−/− mice, or whether there is a large fraction of Th2-type Tconv that transiently up-regulate Foxp3 to induce marker expression, and then lose Foxp3 at a high rate in the ongoing inflammatory response. Since ‘stable’ Treg cells in Bcl6−/− mice express high levels of Th2 cytokines, it is likely that Th2 cytokine-expressing exTreg cells in these mice are derived from the stable Treg cells and are therefore true exTreg cells. Also, the fact that exTreg generation was enhanced in control mice induced to develop allergic airway disease (Fig.7b) supports the idea that true wild-type Treg cells can be destabilized to develop into exTreg cells in an inflammatory response. As noted above, transient Foxp3 expression should be suppressed by inflammation in Bcl6−/− mice. The inflammatory disease in Bcl6−/− mice starts at an early age and is complex in aetiology. While the large generation of Th2 cytokine-expressing exTreg cells may be due to long-term exposure of Treg cells to Th2 inflammation, a more interesting possibility is that there are unique inflammatory factors circulating in Bcl6−/− mice, not normally found in Th2 inflammation, that lead to potent Treg cell instability. A further complicating factor is the intrinsic function of Bcl6 in Treg cells, and whether loss of Bcl6 in Treg cells, together with the severe Th2 inflammation in Bcl6−/− mice, leads to an additive or even synergistic effect on Treg cell instability.
Lastly, our data definitively show a Treg-intrinsic function for Bcl6 in controlling Treg responses. Although the Treg-intrinsic role of Bcl6 on Th2 responses is minor compared with the Treg-extrinsic effects of the inflammation in Bcl6−/− mice, as shown by functional studies,21 we still find a significant role for Bcl6 in Treg cells for controlling Gata3 expression, T-cell proliferation and Th2 responses. Our most unexpected finding is the increased Treg cell numbers found in spleens of Bcl6Foxp3−/− mice as well as the increased T-cell numbers in the BAL of the mice with induced allergic airway disease. These data indicate a specific inability of Bcl6-deficient Treg cells to control T-cell numbers in the face of Th2 inflammation.
Conclusion
These studies provide convincing evidence of a role of Bcl6 in controlling Th2 inflammatory responses by both Treg-intrinsic and Treg-extrinsic pathways.
Acknowledgments
We would like to thank Susan Rice and Kim Stoner of the IU Cancer Center Flow cytometry facility for help with cell sorting and flow cytometry gating. This work was supported by NIAID grants 1R21AI079349, 1R21AI090150 and 1R21AI092212 to ALD, and an American Heart Association pre-doctoral fellowship 10PRE4620001 to DVS.
Disclosures
The authors declare no conflict of interest with this work.
References
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–5. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. 2009;106:1903–8. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+ CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–9. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–7. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Okamoto K, Sawa S, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20:62–8. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science. 2009;323:1488–92. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kanno T, Nakayamada S, et al. TGF-beta and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol. 2012;13:587–95. doi: 10.1038/ni.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–71. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Stephan S, Bluestone JA. Peripherally induced tregs - role in immune homeostasis and autoimmunity. Front Immunol. 2013;4:232. doi: 10.3389/fimmu.2013.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. 2013;13:461–7. doi: 10.1038/nri3464. [DOI] [PubMed] [Google Scholar]
- Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–75. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol. 2013;4:152. doi: 10.3389/fimmu.2013.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joller N, Kuchroo VK. Good guys gone bad: exTreg cells promote autoimmune arthritis. Nat Med. 2014;20:15–17. doi: 10.1038/nm.3439. [DOI] [PubMed] [Google Scholar]
- Dent AL, Hu-Li J, Paul WE, Staudt LS. T helper type 2 inflammatory disease in the absence of IL-4 and STAT6. Proc Natl Acad Sci USA. 1998;95:13823–8. doi: 10.1073/pnas.95.23.13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–92. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- Ye BH, Cattoretti G, Shen Q, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2- type inflammation. Nat Genet. 1997;16:161–70. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Fukuda T, Hatano M, et al. The role of Bcl6 in mature cardiac myocytes. Cardiovasc Res. 1999;42:670–9. doi: 10.1016/s0008-6363(99)00007-3. [DOI] [PubMed] [Google Scholar]
- Sawant DV, Sehra S, Nguyen ET, et al. Bcl6 controls the th2 inflammatory activity of regulatory T cells by repressing gata3 function. J Immunol. 2012;189:4759–69. doi: 10.4049/jimmunol.1201794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant DV, Wu H, Kaplan MH, Dent AL. The Bcl6 target gene microRNA-21 promotes Th2 differentiation by a T cell intrinsic pathway. Mol Immunol. 2013;54:435–42. doi: 10.1016/j.molimm.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, Dent AL. Insights into the role of Bcl6 in follicular helper T cells using a new conditional mutant mouse model. J Immunol. 2013;191:3705–11. doi: 10.4049/jimmunol.1300378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Sehra S, Goswami R, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol. 2010;11:527–34. doi: 10.1038/ni.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusam S, Toney LM, Sato H, Dent AL. Inhibition of Th2 differentiation and GATA-3 expression by BCL-6. J Immunol. 2003;170:2435–41. doi: 10.4049/jimmunol.170.5.2435. [DOI] [PubMed] [Google Scholar]
- Hansmann L, Schmidl C, Kett J, Steger L, Andreesen R, Hoffmann P, Rehli M, Edinger M. Dominant Th2 differentiation of human regulatory T cells upon loss of FOXP3 expression. J Immunol. 2012;188:1275–82. doi: 10.4049/jimmunol.1102288. [DOI] [PubMed] [Google Scholar]
- Wang Y, Souabni A, Flavell RA, Wan YY. An intrinsic mechanism predisposes Foxp3-expressing regulatory T cells to Th2 conversion in vivo. J Immunol. 2010;185:5983–92. doi: 10.4049/jimmunol.1001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–70. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- Jin HS, Park Y, Elly C, Liu YC. Itch expression by Treg cells controls Th2 inflammatory responses. J Clin Invest. 2013;123:4923–34. doi: 10.1172/JCI69355. [DOI] [PMC free article] [PubMed] [Google Scholar]