

Abstract
Advances in maintaining multiple human tissues on microfluidic platforms has led to a growing interest in developing microphysiological systems for drug development studies. Determining the proper design principles and scaling rules for body-on-a-chip systems is critical for their strategic incorporation into physiologically based pharmacokinetic (PBPK)/pharmacodynamic model (PD) -aided drug development. While the need for a functional design considering organ-organ interactions has been considered, robust design criteria and steps to build such systems have not yet been defined mathematically. In this paper, we first discuss strategies for incorporating body-on-a-chip technology into current PBPK modeling-based drug discovery to provide a conceptual model. We propose two types of platforms that can be involved in different stages of PBPK modeling and drug development; these are a μOrgans-on-a-chip and a μHuman-on-a-chip. Then we establish design principles for both types of systems and develop parametric design equations that can be used to determine dimensions and operating conditions. In addition, we discuss the availability of the critical parameters required to satisfy the design criteria, consider possible limitations on estimating such parameter values and propose strategies to address such limitations. This paper is intended to be a useful guide to the researchers focused on designing microphysiological platforms for PBPK/PD based drug discovery.
Introduction
In the last decade, the advances in developing bio-mimetic in vitro culture models using microtechnology has led to a growing interest in building co-culture and multiple organ models often called microphysiological sytems or body-on-a-chip devices.1 Some groups have combined different organs, such as GI-tract and liver, on the same platform and have shown increased functionality of the tissues.1–3 The integration of an increasing number of functional organ-on-a-chip modules on one platform holds great promise for drug development purposes. Such a goal is not an easy task due to many challenges such as maintaining variety of cell types on one device, use of a re-circulating common medium, replicating inter-organ interactions, and transport of nutrients and soluble factors at a physiologically relevant level.
Physiologically based pharmacokinetic (PBPK) models are widely used to predict the pharmocokinetics of a drug. PBPK models can also be used in conjunction with pharmacodynamic (PD) models to predict drug efficacy and toxicity as well as dose adjustment. Shuler et al have proposed and demonstrated the potential use of multiple-organ-on-a-chip systems in PBPK/PD models for drug development.4 Determining the proper design principles and scaling rules for multiple-organs-on-a-chip systems is critical for their incorporation into PBPK/PD model-based drug development. There have been different broad approaches for scaling of multiple-organ-chips platforms.5 Some groups have considered the allometric ratios as the scaling factor of organ sizes.6 Allometric ratio-based scaling has been the most commonly used method due to availability of data and its simplicity in terms of implementation. Shuler’s group has pioneered the scaling of the organ models based on the residence time of the blood in each organ in the body.4 This approach takes into account the extent of reaction in tissues to replicate organ-organ interactions. More recently, Takayama’s group has drawn attention to the role of number of cells and metabolic activity in allometric scaling.5 Besides these current scaling approaches, there is a need for a more functional and robust scaling approach where other potentially critical parameters such as cell density, metabolic clearance or production rates and drug partitioning can be taken into account. The design principles of these systems need to be determined according to their strategic involvement in various stages of drug development. However, it is unclear how these microphysiological in vitro systems will best be incorporated in PBPK model -based drug development.
In this conceptual paper, we first discuss strategies for incorporating body-on-a-chip technology into current PBPK modeling-based drug discovery. We propose two types of platforms that can be involved in different stages of PBPK modeling and drug development. Then we suggest design principles for both types of systems and accordingly develop parametric design equations that can be used to determine dimensions and operating conditions. We also discuss the availability of the critical parameters required to satisfy the design criteria and discuss possible limitations and strategies to address the issue of parameter availability. This paper describes these design criteria and their potential application to the design of multiple-organs-on-a-chip platforms for PBPK/PD based drug discovery.
Incorporation of in vitro Microphysiological Systems in Drug Discovery and Development
Strategic use of Microphysiological Systems
Physiologically based pharmacokinetic (PBPK) models are used to quantitatively describe the absorption, distribution, metabolism and excretion (ADME) of a drug using a set of differential equations defined for different compartments (e.g. organs) in the body. PBPK models, as opposed to simplified single-compartment pharmacokinetic models, can be used to predict the spatial and temporal fate of administered compounds in the body. Several input parameters need to be measured or calculated to achieve a fully predictive PBPK model. These input parameters are typically the drug solubility (Sd) and permeability (Pd) through the barrier tissue (e.g. GI-tract), drug partitioning into each organ (Kp), blood to plasma partitioning (B:P), unbound fraction of the drug (fu), and intrinsic clearance rate (CLint) in the liver and kidneys. Recently, the number of studies using PBPK models for drug development has drastically increased due to the advances in mechanistic approaches to determine PBPK input parameters.7, 8 These mechanistic approaches as first proposed by Poulin et al8 and later modified by Rogers et al7 can predict selected critical input parameters, such as Kp, based on the previously known molecular composition of the tissues in the body. To estimate input parameters, several initial assumptions are usually made. In the tutorial paper of Jones et al, the assumptions and fundamental steps of PBPK model-based drug discovery are described in detail.9 Briefly, after the estimation of input parameters, a PBPK model is verified using the data collected from preclinical trials on animals. If the PBPK model fits the in vivo data, the same assumptions can be made more safely for clinical PBPK models using human specific in vitro data. Then the confirmed PBPK model is used to predict clinical PK by substituting the previously used input parameters with those estimated for humans.
Successful integration of in vitro microphysiological platforms in PBPK model-based drug discovery could accelerate the drug discovery process and reduce rates of drug attrition. Here we describe two types of platforms that can strategically be used at various stages of the drug discovery methodology. One of them is, as we called “μOrgans-on-a-chip” in this paper, a relatively basic platform that can be used in preclinical stages, and “μHuman-on-a-chip” which is a more comprehensive platform that can be used in early human trials and late stages of drug development. Both of these systems and their functions are described in more detail as follows:
μOrgans-on-a-chip (μOOC)
This chip is designed to be used in preclinical trials as a replacement or complement to animal models. The main purpose of animal experiments in PBPK modeling is to test if the assumptions made in the in vitro estimations, such as drug solubility and permeability through gut, are valid for a physiological system (e.g. animal). Typically, the pharmacokinetics of the drugs at this stage does not necessarily follow that of human’s. Thus, the μOOC does not need to perfectly mimic the drug concentration profiles or pharmacokinetics in humans, as previously demonstrated by Tatosian et al10, as long as appropriate parameters of a human PBPK model can be estimated. The primary design goal of this chip is to promote the functions of individual organs by emulating the interaction/communication between all or a relevant subset of organs similar to that in human body. The design criteria should be that the steady-state concentration values of critical intrinsic metabolites (glucose, O2, essential amino acids, cytokines etc.) in each organ should closely mimic the values in human body. Since the μOOC recapitulates the physiological crosstalk between organs, it can potentially be used to predict or validate the PBPK parameters more reliably, compared to individual organ-on-a-chip systems. This chip can be used in the transition stage from animal to early human trials or can directly be used as a preclinical PBPK simulation model. Since μOrgans-on-a-chip is not designed to work for all types of compounds and not to mimic fully human pharmacokinetics, it cannot be routinely used for drug dose adjustment or PD evaluations. In addition, μOOC, in terms of design constrains, serves as a starting point for the development of μHuman-on-chip.
μHuman-on-a-chip (μHOC)
This chip will be more comprehensive than μOOC and used in parallel to human trials for the purposes of dose adjustment, PD assessment, or patient specific screening (iPSCs). The μHOC will not only consider inter-organ interactions at steady-state but also have the capability to directly mimic human PBPK. The design criterion for the μHOC is that the time-dependent concentration profiles of the drug in the blood and tissues should very closely simulate those in human body. With this capability, μHOC will serve as an in vitro model of human PBPK, and unlike the μOOC, will also allow for direct interpretation of human pharmacodynamic data obtained using the chip. In the design, all parameters such as drug partitioning, drug unbound fraction, residence time, drug metabolism rate, drug permeability rate, cardiac output ratios have to be considered. After developing the first generation of the chip following the design steps proposed in this paper, the chip needs to be validated for a specific drug using the PK data obtained from early human trials.
This μHuman-on-a-chip proposed here is a drug specific PBPK/PD tool and should be particularly confirmed for the drug of interest. The challenges in developing a generic μHuman-on-a-chip that could work for all type of drugs are discussed under ‘Estimation and Availability of Design and Drug-specific Input Parameters’ section.
Description of Strategic Steps of Drug Development using in vitro Microphysiological Systems
In this section we describe the incorporation of μOOC and μHOC platforms into each strategic step of drug development process (Figure 1):
Figure 1.
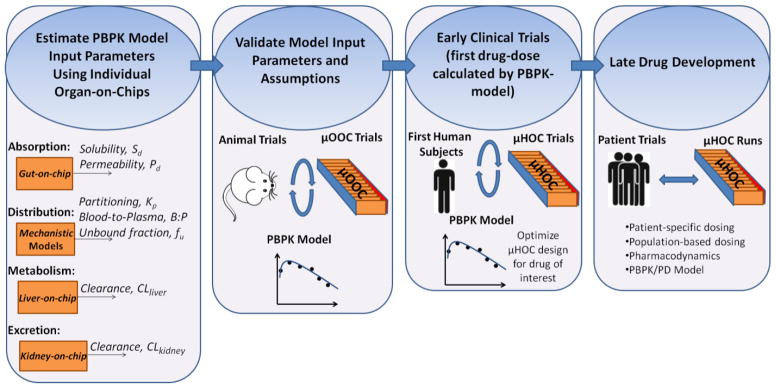
Strategic steps of drug development using microphysiological systems.
-
Estimate the ADME parameters for a specific drug candidate using single organ-on-a-chip systems.
Absorption: Absorption of a drug to the body can occur through physiologic barriers such as gastrointestinal-tract, lungs, and skin. Values of the solubility (Sd) and permeability (Pd) of a drug through these barriers must be estimated. Individual human-organ-on-a-chip platforms including lungs-on-a-chip,11 gut-on-a-chip,12 and skin-on-a-chip13, 14 can potentially be used to estimate Sd and Pd more reliably than conventional cultures of human/animal cells or animal cadavers.
Distribution: Following the absorption, the drug is distributed to each organ. The important parameters affecting the distribution process are unbound fraction of the drug, blood-to-plasma ratio and drug partitioning into the organs. The first two can be experimentally measured using the blood surrogate or a common culture medium of organs-on-a-chip system. The partitioning coefficient depends on the chemical composition of each organ, mostly on the concentration of lipoproteins. This parameter can be estimated after determining the molecular composition of each single organ-on-a-chip.
Metabolism: The drugs are primarily metabolized in the liver although other organs may be involved. The intrinsic clearance rate per microsome or hepatocyte can be estimated for any compound in vitro. These data can then be extrapolated to liver-on-a-chip system15, 16 using in vitro to in vivo extrapolation (IVIVE) method 17 and relevant scaling factors based on the measured levels of CYP enzymes or total number of hepatocytes in the liver-on-a-chip system. The intrinsic metabolites or nutrients can also be consumed by other organs or excreted from the body. This consumption is also called “clearance” throughout this paper and a critical parameter to maintain organ-organ interactions.
Excretion: Excretion of a compound from the body mainly occurs through the renal/biliary excretion. The total renal clearance (CLrenal) is a function of filtration rate, secreted clearance and reabsorption. These values can be estimated using the kidney-on-a-chip or kidney proximal tubule-on-a-chip systems.18 Subsequently, IVIVE method can be used to extrapolate these values to in vivo total kidney clearance of a compound.19
Design the “μOrgans-on-a-chip” using the estimated ADME parameters for one or more selected model molecules as an input into the proposed model equations described further in this paper (Table 1). The model molecule can be a nutrient (e.g. glucose, O2) that is consumed in all organs or a protein/amino acid that is critical for interactions between organs. For example, albumin may be a good candidate since it is involved in transport of molecules in circulation. However, designing the chip based on one single model molecule will not provide accurate levels of other critical molecules in each organ due to disproportional deviations of in vitro clearance rates of these molecules from clinical clearance rates. Alternatively, the design of the μOrgans-on-a-chip can also be determined based on an average of the values obtained by using ADME parameters of a selected pool of critical parameters. For example, ADME parameters can first be estimated for glucose, O2 and albumin. Then the design parameters determined using the ADME parameters of each molecule can be weight-averaged based on the degree of significance of each model molecule for targeted inter-organ interactions. Since this design method considers the contribution of more than one critical molecule, it may potentially allow for approximating the synergistic relation between organs more closely.
Validate the PBPK input parameters and assumptions using the μOrgans-on-a-chip in parallel with in vivo experiments. Various assumptions are made in step 1 while predicting ADME parameters. For example, absorption can be assumed to occur through passive or active diffusion through barrier tissue (e.g. gut). Similarly, the scaling factors used for extrapolation of CLint in the liver and kidney, the metabolite pathways, and distribution kinetics (e.g. perfusion- limited) are based on assumptions. If PBPK model does not provide a good correlation with the measured values, then the assumptions should be reevaluated to determine if a PBPK model consistent with μOrgans-on-a-chip and in vivo data can be constructed.
Use the experimentally confirmed PBPK model for determining suggested first clinical trial doses.
Run first clinical trials typically with healthy volunteers using the pre-determined doses, and obtain clinical pharmacokinetic data to correct the PBPK parameters.
Design the “μHuman-on-a-chip” using the validated ADME parameters for the drug of interest as an input to the proposed model equations further in this paper (Table 1). The μHuman-on-a-chip should be confirmed to have comparable PK to clinical data.
Utilize the “μHuman-on-a-chip” for analyses of Pharmacodynamics, development of PBPK/PD models, dose adjustments, and patient-specific or population-based dose optimizations. Since the μHuman-on-chips will have the capability to mimic clinical PK, they can also be used to estimate Pharmocodynamics. This allows for developing PBPK/PD mathematical models which could complement the μHuman-on-a-chip experiments. Moreover, μHuman-on-chips can potentially be used for patient-specific or population-based drug testing using iPSC-derived cells for each organ although current issues of genetic and epigenetic variations in iPSCs have to be addressed prior to moving forward with this goal.20
Table 1.
Simplified parametric design criteria
| Parametric Criteria | Parameters to determine | Known parameters | ||
|---|---|---|---|---|
| μHuman-organs-on-a-chip Criteria: | ||||
| 1. | ∅Chip = ∅Hum | ∅Chip for each organ | ∅Hum [24, 25] | |
| 2. | where Rint=CLint or Rint=RLint and |
, estimate in vitro ,fixed for the chip ,for each organ |
[24] [26] [26, 27] |
|
| Additional criterion for μHuman-on-a-chip: | ||||
| 3. | and n | τChip, for each organ and blood | τHum [calculated from 24, 25] | |
| Compound specific Criteria: | ||||
| 4. |
|
, for each organ B:PChip, fixed for the chip |
[7, 8] B:PHum[28] |
|
| 5. |
|
,for each organ | [7] | |
Design principles for multiple-organs/human-on-a-chip
There is a growing number of multiple-organs-on-a-chip platforms developed for drug testing purposes. These platforms are typically scaled based on two different approaches: Allometric scaling and residence-time based scaling. The allometric scaling has been the most commonly used method due to availability of data on allometric ratios and its simplicity in terms of implementation. Wikswo et al. have compiled the scaling factors in their review paper for human-on-a-chip systems and shown sample calculations for micro - and milli scale platforms.6 On the other hand, they also point out in their analyses a need for a functional approach where organ-organ interactions can be better mimicked and the difficulty of sustaining the allometric ratios upon scale-down. For example, some drugs, such as 5-flourouracil, are first metabolized by the liver to their active compounds. Therefore, if the liver-on-a-chip does not provide a physiological conversion of the drug, the other organs could not be exposed to realistic levels. Similarly, the cytokines produced by each organ should be present in the blood at physiological concentrations so that paracrine signaling between organs can be replicated. Shuler pioneered scaling organ-on-chips considering the amount of time that each organ is exposed to a molecule (a.k.a residence time).4 Residence time-based scaling is based on the degree of chemical conversion and has a great advantage over allometric scaling in terms of considering the extent of reaction in the tissue, and may better replicate organ-organ interactions.
Although residence time is a critical parameter for organ-organ interactions, the effect of several other parameters also need to be considered to achieve the physiological conversion/production rates of compounds (e.g. cytokines, nutrients, drugs etc.). For example, conventional adherent and three dimensional (3D) cell culture models typically do not replicate in vivo relevant volumetric cell densities mainly due to pseudo tissue architecture and the resulting limitations of nutrient transport.21, 22 This variation in cell densities can result in unrealistic conversion rates and non-physiologic levels of compounds on human-on-chips although the residence time can be modified to compensate for cultures with reduced cell density. Similarly, intrinsic metabolic rates of cells change when the cells are isolated from the body and no longer reside in their physiological microenvironment, which is an issue for any in vitro tissue mimic.23 This factor can also cause deviations in multiple-organ-on-a-chip systems in terms of achieving realistic levels of compounds in the blood. Takayama and colleagues have recently proposed a metabolically supported functional scaling approach which fundamentally follows allometric scaling rules, but at the same time, aiming to achieve in vivo relevant metabolic rates of cells by recapitulating the concentrations of nutrients, particularly oxygen.5 In addition to metabolic rates, other parameters such as drug partitioning in organs, cardiac output ratios, and diffusion distances are also equally important to achieve a physiologically relevant communication between organs. Even though the need for a functional design considering organ-organ interactions has been previously considered by others, robust design criteria and steps to build such systems have not yet been defined mathematically. In this section, we first set design criteria for two different systems (μOrgans-on-a-chip and μHuman-on-a-chip systems) described in the previous section. Then we derive general parametric equations that satisfy these criteria. Subsequently, we propose a simplified solution to the general equations to give insights into the design process and critical parameters.
(i) Design Criteria for μOrgans-on-a-Chip
μOrgans-on-a-Chip mimics a physiologically relevant interaction between organs and tissues. Unlike μHuman-on-chip, it does not necessarily need to mimic time-dependent drug concentration profiles in the blood or organs. We set the design criteria for μOrgans-on-a-Chip as: “At the organ level (not necessarily in the blood), the steady-state concentrations of unbound critical nutrients and/or cytokines ( ) that are responsible for organ-organ interactions should be equal to those in the body”
(Criterion 1)
| (Eqn. 1) |
(ii) Design Criteria for μHuman-on-a-Chip
μHuman-on-a-chip as described previously needs to closely mimic clinical pharmacokinetics to be used as complementary to or in replacement of human trials in late drug development stage. Therefore, we set the following two design criteria which should be satisfied to build μHuman-on-a-chip systems:
-
“The time-dependent drug concentration profiles in blood surrogate/medium should be equal to that in the body”
(Criterion 2)(Eqn. 2) -
“The time-dependent unbound drug concentration (Ct,u) profiles in each organ should be equal to that in the body”
(Criterion 3)(Eqn. 3)
(iii) Derivation of General Parametric Design Criteria
We used macroscopic mass balance equations to rewrite the criteria 1 to 3 in terms of the parameters determining the steady-state and time-dependent concentrations of compounds in tissue (Ct) and blood (Cb). The macroscopic balance over each organ can be written as:
| (Eqn. 4) |
where Vt is the volume of the organ, Qt is the blood flow rate in the organ, #cell is the total number of cells in the organ, and are the concentrations of the drug in the blood entering and leaving the organ, respectively. Rint is defined as the intrinsic reaction rate per cell and per drug concentration in the tissue. When the compound of interest is a nutrient, for example, the reaction term can be substituted with the intrinsic clearance rate (CLint). If the compound is a cytokine produced by a specific organ, the reaction term should be replaced with the intrinsic release rate (RLint) for that organ. Here, we consider the organs as a well-mixed reactor rather than a plugged flow reactor and also assume that the transport of molecules between the tissues and blood would follow perfusion-limited kinetics, which restricts the diffusion distances used in the organs-on-a-chip (see Section 4). This assumption is typically used in PBPK models and allows one to relate the steady state concentrations in the organ to blood. Writing the in terms of Ct yields:
| (Eqn. 5) |
where Kp is the tissue partitioning coefficient and defined as KP = Ct/Cblood; B:P is blood-to-plasma ratio of the compound. Since the criterion 1 is given for steady-state concentration, the time dependent term is equal to zero. Similarly, macroscopic mass balance can be made in the blood compartment to define Cblood as follows:
| (Eqn. 6) |
where Vblood is the total blood volume, QB is the total volumetric flow rate of blood or blood surrogate, and is the cardiac output ratio for the organ, i. The Cblood at steady-state can then be defined as:
| (Eqn. 7) |
Substituting with Cblood in Eqn.5 and solving it for Ct yields the equation below:
| (Eqn. 8) |
Using Eqn.8 and the fraction of unbound compound in tissue (ft,u) the Criterion 1 (equal steady-state concentrations) can be represented in a parametric form that can be used to design μOrgans-on-a-chip systems:
(General Parametric Criterion 1 for each organ)
| (Eqn. 9) |
Most of the human body parameters (right hand side of the equation) are known or can be determined for specific organ and specific compounds. Each chip parameter (left hand side of the equation) needs to be determined carefully to satisfy the equation for each organ. Due to the large number of parameters, there is more than one solution to this problem. Here, we propose one simple solution as summarized in Table 1 to give insights on the design process and critical parameters. The solution steps and derivation of the general parametric criteria for μHuman-on-a-chip systems are described in Supplementary Information.
Estimation and Availability of Design and Drug-Specific Input Parameters
The majority of the parameters, such as cardiac output ratio to each organ, total volumetric flow rate of the blood, the number of cells in each organ, residence times in each organ are readily available in the literature.24–27 The availability of reaction rate data depends on the compound of interest.26 The data can easily be found for common nutrients such as oxygen and glucose. However for specific cytokines or newly developed drugs it may be challenging to measure the production or clearance rate values in the body. In this case, IVIVE method can be used for initial estimations of in vivo clearance or release data. In Table 2, we demonstrate sample calculations for the brain and O2 as a model organ and molecule, respectively.
Table 2.
Sample calculations of chip design parameters using the available values for the human brain and O2 as the model molecule.
| Known Parameters | Equations Used for Estimation | Estimated Design Parameter | ||||||
|---|---|---|---|---|---|---|---|---|
| ∅Hum =0.12 a | ∅Chip = ∅Hum | ∅Chip =0.12 | ||||||
|
|
(using criterion 2 in Table 1) |
|
||||||
| τHum =1.9 min d [23] |
τChip = τHum
|
τChip=1.9 min VChip=33 μL |
Values are taken from the references cited
Blood volumetric flow is scaled down from based on a chosen scaling factor (αSF) of 1:50000
Value is taken from the reference cited although it highly depends on culture conditions. Therefore, this parameter needs to be measured using individual organ-on-a-chip platforms at relevant conditions.
Calculated from the reference cited
It should be noted that, some of the parameters such as were directly taken from previous studies although they depend on culture conditions. Therefore, these parameters need to be measured separately using individual organ-on-a-chip platforms at relevant conditions. In these calculations the O2 levels in the blood surrogate were assumed to be maintained at physiological O2 levels (5–12% O2). Other parameters such as Kp and ft,u can be estimated using mechanistic methods as discussed previously.
The total volume of organ chambers depends on the number of cells and residence times calculated for the chip using the design equations. In Table 2, the volume of the brain compartment on the chip was calculated as 33 μl, yielding a neuron density( ) of 3.3×108 cells/ml. It is important to note that the design equations were derived assuming high transport rate of molecules within tissues. Therefore, the organon-a-chip systems should be designed considering the mass transfer resistances, which can result from unstirred culture medium or a layer of extracellular matrix surrounding cells. The effect of these resistances on the concentration of dissolved molecules at cellular level was shown in adherent and 3D cultures of various cell types.29 In particular, dissolved oxygen concentrations at cellular level were shown to deviate dramatically from the ambient O2 levels due to the mass transfer resistances.22 This effect is expected to be more profound for proteins and therapeutic compounds, given their relatively high molecular weight, compared to O2. Also given that the typical microchannel settings used in the chips provide a significantly smaller surface-to-volume ratio compared to that in the body, it is challenging to achieve physiological mass transfer rates. This can potentially be addressed by the availability of three dimensional vascularization techniques which may allow for achieving physiological capillary surface-to-volume ratios.30
Another possible solution to this limitation could be minimizing the diffusional distances between the microchannels and tissues in the chip. In humans, the maximum distance of a highly metabolically active tissue from the nearest blood vessel is 100–200 μm,31 which can be used as a threshold value to set the diffusion distance from the supply channels to any portion of the tissue on the chip. This means the depth and the width of the organ chambers are limited by this threshold value. This constraint can be used along with the calculated total volume to optimize each dimension of the tissue compartment on the chip. In several systems, there is a porous membrane layered between the organ chamber and the connecting transport channels.32 In this type of design, the additional resistance coming from the membrane should also be taken into account when determining the dimensions of the system. On the other hand, based on the values and scaling factor of 1:50000 given in Table 2, a medium volume of 100 μl would be needed to meet the design criteria, whereas the total volume of the medium circulated in μOOC is typically larger due to technical challenges in terms of fluid handling and dead volumes. Although such unrealistically large medium volumes would delay the time to reach the steady-state concentrations in each tissue on the chip, it would still satisfy the Criterion 1 for μOOC, after the steady-state is reached. However, for Criteria 2 and 3 set for μHOC, this delayed steady-state would lead to unrealistic drug profiles in the blood. The general parametric equations we demonstrated in this paper would be helpful to address this issue by adjusting the other parameters in the equation accordingly. One possible approach, for example, would be increasing the volumetric flow rate of the medium to maintain and at the same time increasing the cell density in each tissue to generate higher production/consumption rate of compounds to be transferred to/from the circulating media.
Compound specific properties shown in Table 1 are functions of several other parameters and very critical to achieve a physiologically relevant model. For example, Kp and ft,u depend on volume fraction of intracellular and extracellular water and cells and the concentration of lipids and phospholipids.7 Therefore, the compositions of the microphysiological tissues on the chip should be carefully designed to satisfy the compound specific criteria. However, it may be challenging to achieve physiological extracellular matrix compositions and phospholipid content. One possible approach to this challenge would be revisiting the general parametric criteria to compensate for the variations in compound specific properties by adjusting other parameters such as cell density and residence time. Although this approach can work for drug-specific μHuman-on-a-chip systems, it is likely to fail when developing generic μHuman-on-a-chip systems which can be used for all types of drugs. This problem can eventually be overcome by advancements towards engineering bio-inspired synthetic scaffolds or using decellularized natural scaffolds to give the desired Kp and fu,t for each organ. Using such scaffolds mimicking physiological extracellular matrix would also allow for achieving more realistic cellular responses, such as metabolic rates. Another potential solution would be calculating the deviation that is caused by the variations in compound specific parameters and compensate for the effect of this deviation in the PBPK model. On the other hand, the other compound specific property, B:P, depends on the protein content of the blood or blood surrogate. This parameter should be estimated empirically for blood and blood surrogate/culture medium separately since the blood surrogate/culture medium typically do not involve red blood cells or physiological levels of proteins in human plasma.
Majority of the current organ-on-a-chip platforms in the literature are made of polydimethylsiloxane (PDMS), which has been explicitly shown to cause adsorption of biomolecules.33 Growing effort to address this problem has led to several promising solutions, such as modifying surface chemistry of PDMS to prevent adsorption34 or using alternative materials (e.g. polystyrene) to PDMS35. In our parametric design criteria, we did not take into account the adsorption of molecules on the walls of the chip. However, if the uncontrolled adsorption of molecules remains as an unresolved issue for human-on-a-chip systems, it needs to be considered in the parametric criteria as a separate factor affecting the concentrations of compounds in tissues and blood surrogate.
The in vitro clearance and production rate of compounds show variations from the in vivo values as we mentioned previously. This variation can also be disproportional for different drugs. For example, in vitro metabolism rate for drug A may be 10% higher than the in vivo, whereas the metabolism for drug B may be 20% higher than the in vivo value. This is another significant challenge to develop a generic μHuman-on-a-chip system. Recapitulation of an increasing number of biochemical and biomechanical factors in the cellular microenvironment may eventually lead cells to exhibit physiologically identical metabolic profiles and may allow for coping with this challenge.
Concluding Remarks
In this critical review, we proposed strategic ways of incorporating body-on-a-chip technology into PBPK-model aided drug discovery. We highlight the need for a functional design of human-on-a-chip platforms and proposed parametric design equations for systematic development of these platforms. The design principles of body-on-a-chip platforms completely depend on strategic involvement of the platforms into drug discovery process. The current multiple-organ-on-a-chip systems and previously described scaling methodologies can be considered as the steps toward building μOrgans-on-a-chip platforms rather than μHuman-on-a-chip, according to our classifications in this paper. μOrgans-on-a-chip platforms will be very useful at the preclinical drug development stages. However, for later stages there is a need for more comprehensive systems like μHuman-on-a-chip. It should be noted that, in this paper μHuman-on-a-chip was described and envisioned to directly provide physiological information mimicking human PK. In principle, platforms can also be designed to provide non-physiological data which then can be converted to physiologically relevant information using extrapolation principles although such techniques have not yet been formalized. It is currently unclear how closely such microphysiological platforms should mimic human PK to allow evaluation of PBPK parameters and to acquire human relevant PD data.
It is evident that the development of better organ-mimetic platforms recapitulating larger number of physiological factors in tissue microenvironment will advance the human-on-a-chip technology. Nevertheless, more thorough theoretical studies, critical discussions and conceptual design will be prerequisite for the effective adaptation of these platforms into drug discovery studies.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This project is funded by the National Center for Advancing Translational Sciences at the National Institutes of Health (UH2TR000156-01). We would like to thank John Graf at General Electric Healthcare for critical comments on the manuscript.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
References
- 1.Esch MB, Smith AS, Prot JM, Oleaga C, Hickman JJ, Shuler ML. Adv Drug Deliv Rev. 2014;69–70:158–169. doi: 10.1016/j.addr.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Midwoud PM, Merema MT, Verpoorte E, Groothuis GM. Lab Chip. 2010;10:2778–2786. doi: 10.1039/c0lc00043d. [DOI] [PubMed] [Google Scholar]
- 3.Zhang C, Zhao Z, Abdul Rahim NA, van Noort D, Yu H. Lab Chip. 2009;9:3185–3192. doi: 10.1039/b915147h. [DOI] [PubMed] [Google Scholar]
- 4.Sung H, Kam C, Shuler ML. Lab Chip. 2010;10:446–455. doi: 10.1039/b917763a. [DOI] [PubMed] [Google Scholar]
- 5.Moraes C, Labuz JM, Leung BM, Inoue M, Chun TH, Takayama S. Integr Biol (Camb) 2013;5:1149–1161. doi: 10.1039/c3ib40040a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wikswo JP, Curtis EL, Eagleton ZE, Evans BC, Kole A, Hofmeister LH, Matloff WJ. Lab Chip. 2013;13:3496–3511. doi: 10.1039/c3lc50243k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodgers T, Leahy D, Rowland M. J Pharm Sci. 2005;94:1259–1276. doi: 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- 8.Poulin P, Theil FP. J Pharm Sci. 2000;89:16–35. doi: 10.1002/(SICI)1520-6017(200001)89:1<16::AID-JPS3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 9.Jones H, Rowland-Yeo K. CPT Pharmacometrics Syst Pharmacol. 2013;2:e63. doi: 10.1038/psp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatosian DA, Shuler ML. Biotechnol Bioeng. 2009;103:187–198. doi: 10.1002/bit.22219. [DOI] [PubMed] [Google Scholar]
- 11.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahler GJ, Esch MB, Glahn RP, Shuler ML. Biotechnol Bioeng. 2009;104:193–205. doi: 10.1002/bit.22366. [DOI] [PubMed] [Google Scholar]
- 13.Atac B, Wagner I, Horland R, Lauster R, Marx U, Tonevitsky AG, Azar RP, Lindner G. Lab Chip. 2013;13:3555–3561. doi: 10.1039/c3lc50227a. [DOI] [PubMed] [Google Scholar]
- 14.Abaci HE, Gledhill K, Guo Z, Christiano AM, Shuler ML. Lab on a chip. 2015 doi: 10.1039/C4LC00999A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Lab Chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novik E, Maguire TJ, Chao P, Cheng KC, Yarmush ML. Biochem Pharmacol. 2010;79:1036–1044. doi: 10.1016/j.bcp.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rostami-Hodjegan A. Clin Pharmacol Ther. 2012;92:50–61. doi: 10.1038/clpt.2012.65. [DOI] [PubMed] [Google Scholar]
- 18.Jang KJ, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh KY, Ingber DE. Integr Biol (Camb) 2013;5:1119–1129. doi: 10.1039/c3ib40049b. [DOI] [PubMed] [Google Scholar]
- 19.Kunze A, Huwyler J, Poller B, Gutmann H, Camenisch G. J Pharm Sci. 2014;103:994–1001. doi: 10.1002/jps.23851. [DOI] [PubMed] [Google Scholar]
- 20.Liang G, Zhang Y. Cell Stem Cell. 2013;13:149–159. doi: 10.1016/j.stem.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lovett M, Lee K, Edwards A, Kaplan DL. Tissue Eng Part B Rev. 2009;15:353–370. doi: 10.1089/ten.teb.2009.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abaci HE, Truitt R, Tan S, Gerecht S. Am J Physiol Cell Physiol. 2011;301:C431–440. doi: 10.1152/ajpcell.00074.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hewitt NJ, Buhring KU, Dasenbrock J, Haunschild J, Ladstetter B, Utesch D. Drug Metab Dispos. 2001;29:1042–1050. [PubMed] [Google Scholar]
- 24.Price PS, Conolly RB, Chaisson CF, Gross EA, Young JS, Mathis ET, Tedder DR. Crit Rev Toxicol. 2003;33:469–503. [PubMed] [Google Scholar]
- 25.Davies B, Morris T. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 26.Herculano-Houzel S. PLoS One. 2011;6:e17514. doi: 10.1371/journal.pone.0017514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herculano-Houzel S. Front Hum Neurosci. 2009;3:31. doi: 10.3389/neuro.09.031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu S, Li S, Yang H, Lee F, Wu JT, Qian MG. Rapid Commun Mass Spectrom. 2005;19:250–254. doi: 10.1002/rcm.1777. [DOI] [PubMed] [Google Scholar]
- 29.Abaci HE, Truitt R, Luong E, Drazer G, Gerecht S. Am J Physiol Cell Physiol. 2010;298:C1527–1537. doi: 10.1152/ajpcell.00484.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moya ML, Hsu YH, Lee AP, Hughes CC, George SC. Tissue engineering Part C, Methods. 2013;19:730–737. doi: 10.1089/ten.tec.2012.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baish JW, Stylianopoulos T, Lanning RM, Kamoun WS, Fukumura D, Munn LL, Jain RK. Proc Natl Acad Sci U S A. 2011;108:1799–1803. doi: 10.1073/pnas.1018154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abaci HE, Devendra R, Smith Q, Gerecht S, Drazer G. Biomedical microdevices. 2012;14:145–152. doi: 10.1007/s10544-011-9592-9. [DOI] [PubMed] [Google Scholar]
- 33.Li N, Schwartz M, Ionescu-Zanetti C. Journal of biomolecular screening. 2009;14:194–202. doi: 10.1177/1087057108327326. [DOI] [PubMed] [Google Scholar]
- 34.Liu VA, Jastromb WE, Bhatia SN. Journal of biomedical materials research. 2002;60:126–134. doi: 10.1002/jbm.10005. [DOI] [PubMed] [Google Scholar]
- 35.Young EW, Berthier E, Guckenberger DJ, Sackmann E, Lamers C, Meyvantsson I, Huttenlocher A, Beebe DJ. Analytical chemistry. 2011;83:1408–1417. doi: 10.1021/ac102897h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.