

Abstract
The kinesin superfamily of microtubule associated motor proteins share a characteristic motor domain which both hydrolyses ATP and binds microtubules. Kinesins display differences across the superfamily both in ATP turnover and in microtubule interaction. These differences tailor specific kinesins to various functions such as cargo transport, microtubule sliding, microtubule depolymerization and microtubule stabilization. To understand the mechanism of action of a kinesin it is important to understand how the chemical cycle of ATP turnover is coupled to the mechanical cycle of microtubule interaction. To dissect the ATP turnover cycle, one approach is to utilize fluorescently labeled nucleotides to visualize individual steps in the cycle. Determining the kinetics of each nucleotide transition in the ATP turnover cycle allows the rate-limiting step or steps for the complete cycle to be identified. For a kinesin, it is important to know the rate-limiting step, in the absence of microtubules, as this step is generally accelerated several thousand fold when the kinesin interacts with microtubules. The cycle in the absence of microtubules is then compared to that in the presence of microtubules to fully understand a kinesin’s ATP turnover cycle. The kinetics of individual nucleotide transitions are generally too fast to observe by manually mixing reactants, particularly in the presence of microtubules. A rapid mixing device, such as a stopped-flow fluorimeter, which allows kinetics to be observed on timescales of as little as a few milliseconds, can be used to monitor such transitions. Here, we describe protocols in which rapid mixing of reagents by stopped-flow is used in conjunction with fluorescently labeled nucleotides to dissect the ATP turnover cycle of a kinesin.
Keywords: Chemistry, Issue 92, Kinesin, ATP turnover, mantATP, mantADP, stopped-flow fluorescence, microtubules, enzyme kinetics, nucleotide
Introduction
The kinesins are a superfamily of proteins characterized by a highly conserved motor domain1. The kinesin motor domain interacts with microtubules in a nucleotide-dependent manner. Members of the kinesin family display a range of functions from translocating kinesins, which carry cargo in a directed fashion along microtubules, to non-translocating microtubule end-binding kinesins, which regulate microtubule dynamics2,3.
Kinesins use the turnover of adensosine-5’-triphosphate (ATP) to regulate their interaction with the microtubule cytoskeleton4-6. The conformation of the kinesin motor domain and therefore its affinity for the microtubule depends on its nucleotide state: ATP, ADP, ADP.Pi or no nucleotide can be bound (Figure 1). To understand the molecular mechanism of a kinesin, an understanding of the relationship between ATP turnover and microtubule interaction is required. Therefore, a major goal in the kinesin field is to characterize the functional properties of different nucleotide states and the kinetics of the transitions between them both in the presence and absence of microtubules.
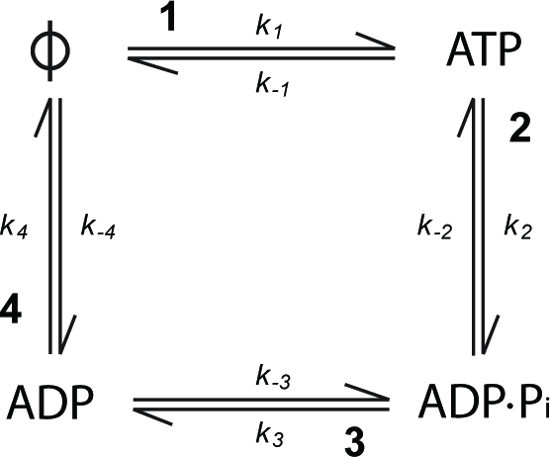 Figure 1. The simplest ATP turnover cycle for a kinesin consists of four possible states: nucleotide-free (Φ), ATP-bound, ADP.Pi-bound and ADP-bound. Each of the four transitions is characterized by a forward and backward rate constant (kx and k-x respectively).
Figure 1. The simplest ATP turnover cycle for a kinesin consists of four possible states: nucleotide-free (Φ), ATP-bound, ADP.Pi-bound and ADP-bound. Each of the four transitions is characterized by a forward and backward rate constant (kx and k-x respectively).
To understand how the chemical cycle of ATP turnover is coupled to the mechanical cycle of microtubule interaction, one must determine which step in the ATP turnover cycle is rate limiting in the absence of microtubules and then determine how the cycle is altered by the presence of microtubules. The simplest ATP turnover cycle consists of four individual chemicals steps: (1) ATP binding to the empty site (Φ denotes the empty site, i.e., nucleotide-free kinesin); (2) ATP cleavage; (3) phosphate dissociation and (4) ADP dissociation (Figure 1). The rate-limiting step sets the rate constant for the complete ATP turnover cycle. Therefore, to determine which step is rate limiting each of the individual transitions must be measured and compared to the rate constant for the complete cycle. Methods to measure the overall ATP turnover cycle rate constant are not described here but can be found in reference 7. Here, we describe methods by which rate constants for individual chemical steps can be determined. There are a number of methods available to study individual transitions in the turnover cycle of a nucleotide hydrolyzing protein. The methods presented here take advantage of the properties of nucleotides labeled with the fluorophore methylanthraniloyl, usually referred to as mant, which display a change in fluorescence intensity upon binding to protein8. The mant group is used because its small size means that, when coupled to the ribose (Figure 2A), it generally has little or no effect on the kinetics of nucleotide binding or hydrolysis9-12. Here, we present protocols describing the use of mant-labeled nucleotides, in conjunction with stopped-flow fluorescence, to allow observation of nucleotide binding and dissociation in the ATP turnover cycle of a kinesin.
Protocol
1. Determination of the Fluorescence Intensity Change upon Binding of Mant-labeled Nucleotide.
Add 1 μM mant-labeled nucleotide to 1 μM kinesin (final concentrations) in an appropriate reaction buffer. Typically a buffer that benefits microtubule growth and stability is used (see discussion). A commonly used buffer contains 80mM piperazine-bis-2-ethanesulfonic acid (PIPES) pH6.9, 1mM MgCl2 and 1mM ethylene glycol-bis-2 aminoether-tetraacetic acid (EGTA). Note: The concentration of kinesin stated in all protocols refers to the concentration of nucleotide binding sites (i.e., motor domains). Different kinesins exist as monomers, dimers or tetramers and so can contain more than one nucleotide-binding site per molecule.
After an incubation period (1-3 min) and using a standard fluorimeter, measure the emission spectrum for the mant-labeled nucleotide-kinesin complex. Use an excitation wavelength of 365nm and measure emitted light from 400-500 nm in 1nm increments.
Make a solution of 1mM mant-labeled nucleotide in the same reaction buffer used in Step 1.1.
Measure the emission spectrum for the mant nucleotide only sample, as previously described (see Step 1.2).
Normalize the spectra to the signal at the wavelength of peak fluorescence intensity of the spectrum of mant nucleotide in solution (i.e., without kinesin) by dividing through by this value.
Plot the normalized spectra (e.g., Figure 2B) to observe fluorescence intensity differences between the mantATP-bound and/or mantADP-bound kinesin and mant nucleotide in solution.
2. Measurement of the Association and Dissociation Rate Constants for mantATP
To a solution of up to 20 μM kinesin (see discussion) in any appropriate Mg2+-free buffer (e.g., 80mM PIPES pH6.9, 1mM EGTA, 0.1% Tween20), add 1 mM (final concentration) ethylenediaminetetraacetic acid (EDTA) and 1 mM (final concentration) dithiothreitol (DTT). Note: Addition of Tween20 to the buffer is recommended as it reduces/prevents loss of kinesin protein on the column used for buffer exchange (Steps 2.3 and 3.3)
Incubate the solution at 25 °C for 15 min.
- Separate free nucleotide and EDTA from the kinesin using a gravity flow G-25 sephadex gel filtration column according to manufacturer’s instructions (see list of materials).
- Equilibrate the column with at least 3 column volumes of an appropriate Mg2+-free buffer (see Step 2.1). Load the kinesin-containing solution onto the column. Prepare a collection tube by adding an appropriate volume of 100mM magnesium chloride solution to the empty tube, such that the final concentration of magnesium chloride is 1 mM after collection of the kinesin solution. Immediate addition of magnesium chloride helps stabilise the nucleotide-free kinesin.
- Elute the kinesin using an appropriate Mg2+-free buffer. Kinesins are unstable when free of nucleotide so it is important to work quickly. Store the nucleotide-free kinesin on ice and use within 1-2 hr.
During or prior to preparation of nucleotide-free kinesin, prepare a mantATP concentration series in the same reaction buffer as the final solution of nucleotide-free kinesin. Determine the range of concentrations of mantATP required according to the available concentration of kinesin with a typical series ranging from 0.1 to 50 μM (see step 2.5 and discussion).
Prepare a concentration series of nucleotide-free kinesin. For each concentration of mantATP (Step 2.4), a concentration of nucleotide-free kinesin should be prepared such that the mant-labeled nucleotide is in 5 to 10-fold molar excess over kinesin nucleotide binding sites.
Set the excitation wavelength on the stopped-flow fluorimeter to 365 nm and collect emitted light at >400 nm using a long-pass filter.
Starting with the lowest concentration of mantATP, mix the mantATP with an appropriate concentration of nucleotide-free kinesin in a 1:1 v/v ratio using a stopped-flow fluorimeter (Figure 3A and 3B Inset). Note: The reactants are diluted by 50% upon mixing. Keep a note of the nucleotide concentration both pre and post mixing (i.e., 3.0/1.5 μM) to avoid confusion.
Fit the change in fluorescence intensity measured at each concentration of mantATP to an exponential function plus a line of constant negative slope to account for photobleaching of the mant group (i.e., Fluorescence = A0.exp(kobs.t)+(m.t+c), where A0 is the amplitude, kobs is the rate constant and t is time, see Figure 3B and discussion). From these fits determine a rate constant (kobs) at each concentration of mantATP. Each kobs is a convolution of an association and dissociation rate constant (Figure 1, k1 and k-1).
Deconvolve the association and dissociation rate constants by plotting kobs against mantATP concentration (e.g., Figure 3C). Fit these data to a linear function (i.e., kobs = k1[mantATP] + k-1) to obtain the gradient and y-axis intercept. The gradient represents the rate constant for mantATP association (Figure 1, k1) with the units M-1s-1. The intercept represents the dissociation rate constant (Figure 1, k-1) with units of s-1 (see discussion). Note: This approach can also be applied to determine the ADP association and dissociation rate constants (Figure 1, k-4 and k4) by using mantADP as the nucleotide.
3. Measurement of the Dissociation Rate Constant for mantADP
To a solution of 2 μM kinesin in a suitable reaction buffer (e.g., 80 mM PIPES pH6.9, 1mM MgCl2, 1mM EGTA, 0.1% Tween20), add 50 μM mantADP (final concentration).
Incubate at 25 oC for 30 min.
Remove excess mantADP and other free nucleotide by buffer exchanging the kinesin into the chosen reaction buffer using a G-25 sephadex column (see list of materials) according to manufacturer’s instructions. The kinesin motor domain should now be loaded with mantADP.
Set the excitation wavelength on the stopped-flow fluorimeter to 365 nm and collect emitted light at >400 nm using a long-pass filter.
Mix the mantADP.kinesin complex (~1μM) with a 50-fold or greater molar excess of unlabeled ATP in a 1:1 v/v ratio using a stopped-flow fluorimeter (Figure 4A and 4B Inset).
Fit the decrease in fluorescence intensity observed over time to a single exponential function plus a line of constant negative slope to account for photobleaching of the mant group (i.e., Fluorescence = A0.exp(kobs.t)+(m.t+c), where A0 is the amplitude, kobs is the rate constant and t is time, see Figure 4B and discussion). The kobs determined from this fit is the rate constant for dissociation of ADP (Figure 1, k4) with units of s-1 (see discussion).
Representative Results
An increase in fluorescence intensity of the mant group is typically observed upon binding of mant-labeled nucleotide to a kinesin. However, the magnitude of such a signal change is dependent on the particular kinesin in question. Therefore, it is useful to perform equilibrium measurements to confirm both the presence and magnitude of a fluorescence intensity change before progressing to kinetic assays. Representative fluorescence spectra for mantATP and mantADP, both in solution and in complex with the kinesin-13, MCAK, are shown in Figure 2B. These data highlight the fluorescence intensity increase displayed by both mantATP and mantADP upon binding to MCAK. The change in fluorescence intensity upon binding of mant nucleotide to a kinesin is used to report on the association and dissociation of both ATP and ADP (Sections 2 and 3). In the case of MCAK, a fluorescence intensity difference is observed between mantATP.kinesin and mantADP.kinesin9.
Figure 3A depicts the reaction that occurs upon mixing mantATP with nucleotide-free kinesin. Representative data illustrating the increase in fluorescence intensity that occurs upon binding of mantATP to the kinesin-13, MCAK is shown in Figure 3B. Data of the type shown in Figure 3B is collected at a range of mantATP concentrations. A plot of the rate constants (kobs) determined versus the mantATP concentration is shown in Figure 3C. Fitting these data to a linear function (i.e., kobs = k1[mantATP] + k-1) allows deconvolution of the rate constants that contribute to kobs (Step 2.8). Therefore, enabling the determination of both the association and the dissociation rate constant for mantATP (Figure 1, k1 and k-1 respectively).
Figure 4A depicts the reaction which occurs upon mixing mantADP.kinesin with an excess of unlabeled ATP. The data in Figure 4B shows the fluorescence intensity decrease observed for mantADP upon dissociation from the kinesin-13, MCAK. By fitting this data to an exponential function (Step 3.6) the rate constant for dissociation of mantADP is determined directly.
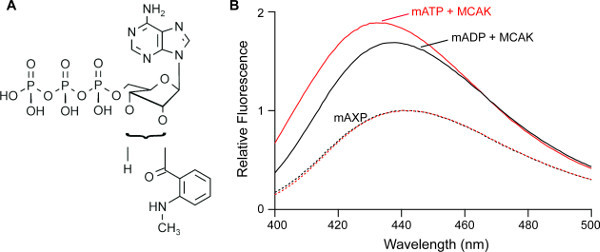 Figure 2. Nucleotides labeled with the fluorophore methylanthraniloyl (mant) are used to isolate individual steps in the ATP turnover cycle. (A) Structure of mantATP showing the position of the mant fluorophore conjugated to either the 2’ or 3’ position on the ribose. (B) Normalized fluorescence spectra for mantATP and mantADP in solution (mAXP) and mantATP and mantADP in complex with the kinesin MCAK (mATP+MCAK and mADP+MCAK). The mantATP spectra are normalized to the fluorescence at max for mantATP in solution and the mantADP spectra are normalized to fluorescence at max for mantADP in solution. The spectra of the mant nucleotides mixed with MCAK are collected at 1 min post mixing. The concentration of reagents and fluorimeter settings are as described in protocol steps 1.1 and 1.2.
Figure 2. Nucleotides labeled with the fluorophore methylanthraniloyl (mant) are used to isolate individual steps in the ATP turnover cycle. (A) Structure of mantATP showing the position of the mant fluorophore conjugated to either the 2’ or 3’ position on the ribose. (B) Normalized fluorescence spectra for mantATP and mantADP in solution (mAXP) and mantATP and mantADP in complex with the kinesin MCAK (mATP+MCAK and mADP+MCAK). The mantATP spectra are normalized to the fluorescence at max for mantATP in solution and the mantADP spectra are normalized to fluorescence at max for mantADP in solution. The spectra of the mant nucleotides mixed with MCAK are collected at 1 min post mixing. The concentration of reagents and fluorimeter settings are as described in protocol steps 1.1 and 1.2.
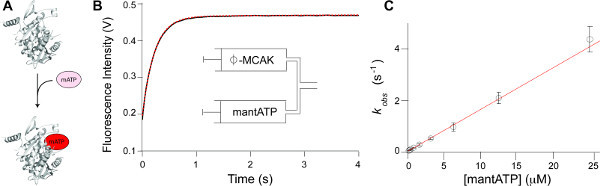 Figure 3. Measurement of the association rate constant of mantATP. (A) Schematic depicting the reaction which takes place post mixing by stopped-flow: mantATP (mATP) binds to nucleotide-free kinesin. The fluorescence intensity of the mant group increases upon binding to the protein. (B) Fluorescence signal change (black) observed upon mixing mantATP (25 M) with MCAK (5 M nucleotide-free, ). This data is fit (red dashed) to a single exponential function plus a line of constant negative slope, which accounts for photobleaching of the mant fluorophore (see discussion). Inset: Contents of the syringes prior to mixing on the stopped-flow fluorimeter. (C) Rate constants (kobs) determined for the reaction of mantATP with nucleotide-free MCAK plotted against concentration of mantATP. This plot will have a linear region with the gradient being the association rate constant (Figure 1, k1) with units of M-1 s-1 and the y-intercept being the dissociation rate constant (Figure 1, k-1) with units of s-1. Please click here to view a larger version of this figure.
Figure 3. Measurement of the association rate constant of mantATP. (A) Schematic depicting the reaction which takes place post mixing by stopped-flow: mantATP (mATP) binds to nucleotide-free kinesin. The fluorescence intensity of the mant group increases upon binding to the protein. (B) Fluorescence signal change (black) observed upon mixing mantATP (25 M) with MCAK (5 M nucleotide-free, ). This data is fit (red dashed) to a single exponential function plus a line of constant negative slope, which accounts for photobleaching of the mant fluorophore (see discussion). Inset: Contents of the syringes prior to mixing on the stopped-flow fluorimeter. (C) Rate constants (kobs) determined for the reaction of mantATP with nucleotide-free MCAK plotted against concentration of mantATP. This plot will have a linear region with the gradient being the association rate constant (Figure 1, k1) with units of M-1 s-1 and the y-intercept being the dissociation rate constant (Figure 1, k-1) with units of s-1. Please click here to view a larger version of this figure.
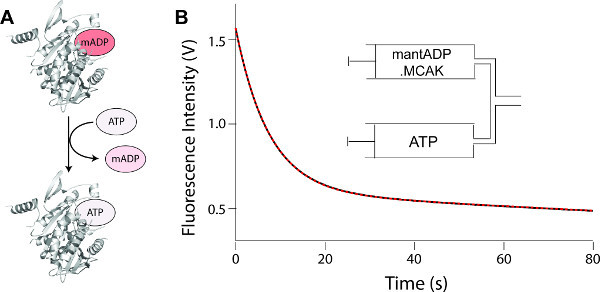 Figure 4. Measurement of the dissociation rate constant for mantADP.
(A) Schematic depicting the reaction which takes place post mixing by stopped-flow: kinesin preloaded with mantADP is diluted into an excess of unlabeled ATP. Dissociating mantADP is replaced by unlabeled ATP, thereby preventing the back reaction of mantADP association (see discussion). The fluorescence intensity of the mant group decreases upon dissociation from the protein. (B) Fluorescence decrease observed upon the dissociation of mantADP from the kinesin MCAK. The fluorescence signal (black) is fit (red dashed) to a single exponential plus a line of constant negative slope to account for photobleaching of the mant group (see discussion). The rate constant determined is the rate constant for dissociation of mantADP (Figure 1, k4). Inset: Contents of the syringes prior to mixing on a stopped-flow fluorimeter. Please click here to view a larger version of this figure.
Figure 4. Measurement of the dissociation rate constant for mantADP.
(A) Schematic depicting the reaction which takes place post mixing by stopped-flow: kinesin preloaded with mantADP is diluted into an excess of unlabeled ATP. Dissociating mantADP is replaced by unlabeled ATP, thereby preventing the back reaction of mantADP association (see discussion). The fluorescence intensity of the mant group decreases upon dissociation from the protein. (B) Fluorescence decrease observed upon the dissociation of mantADP from the kinesin MCAK. The fluorescence signal (black) is fit (red dashed) to a single exponential plus a line of constant negative slope to account for photobleaching of the mant group (see discussion). The rate constant determined is the rate constant for dissociation of mantADP (Figure 1, k4). Inset: Contents of the syringes prior to mixing on a stopped-flow fluorimeter. Please click here to view a larger version of this figure.
Discussion
Here, we present protocols for observation and analysis of the kinetics of transitions between nucleotide states for a kinesin. These protocols utilize the rapid mixing method stopped-flow in conjunction with fluorescently labeled nucleotides. Methods of this type have been used extensively to study nucleotide binding and dissociation for a variety of kinesins9-11,13-16. The methods described do not allow observation of phosphate release (Figure 1, Step 3). However, stopped-flow fluorescence can be used to observe this transition by using a phosphate sensing protein to couple the production of phosphate to a fluorescence intensity change17. The methods presented use nucleotides labeled with the small fluorophore methylanthraniloyl (mant), which generally exhibit an increase in fluorescence intensity when bound to protein. Furthermore, the mant fluorophore, when conjugated to either the 2’ or 3’ position on the ribose, often has little or no effect on the binding, dissociation or hydrolysis of the nucleotide9-12. Mant-labeled nucleotides are readily available from commercial sources (see list of materials) and are provided as mixtures of the 2’ and 3’ conjugate as they quickly re-equilibrate when purified to a single isomer8. Mant-labeled nucleotides make excellent reporter molecules by which the kinetics of nucleotide binding and dissociation can be observed.
The use of fluorescently labeled nucleotides means that the read out of the assays described is a real time change in fluorescence intensity. This makes these assays ideal for stopped-flow fluorescence, which allows rapid mixing of reagents and real time observation of changes in fluorescence associated with the subsequent reaction. The combination of stopped-flow as the mixing technique and fluorescence as the method of detection produces a system that is relatively sample efficient. For example, to acquire the data shown in Figure 3C requires 0.8 - 1.0 mg of purified kinesin-13, MCAK, and to acquire the data shown in Figure 4B requires ~0.3 mg of purified kinesin-13, MCAK. One drawback of the use of mant as a fluorophore is that it is prone to photobleaching. This can be observed in isolation from kinesin reaction kinetics by mixing a solution of mant-labeled nucleotide with reaction buffer, in the absence of kinesin, in the stopped-flow. A slow decrease in fluorescence intensity is observed as the mant group becomes bleached. Over the range of timescales used in the protocols described photobleaching of the mant group is well described by a linear function. Therefore, a simple way to correct data for photobleaching is to fit to an exponential function with a baseline described by a linear function (i.e., m.t + c) rather than the more common flat baseline, described by a single parameter.
mantATP binding
When measuring rate constants for association and dissociation of mantATP (Section 2) the ADP, which remains associated with the kinesin nucleotide-binding site in the absence of microtubules, must be removed. This allows mantATP association and dissociation (Figure 1, step 1) to be observed in isolation from ADP dissociation. To achieve this the kinesin is incubated with at least a 50-fold excess of EDTA over nucleotide binding sites (Step 2.1). The EDTA sequesters Mg2+ ions causing Mg2+ to dissociate from the nucleotide-binding pocket, which results in release of the nucleotide11. Therefore, when carrying out steps 2.1 - 2.3, it is vital to use a buffer free of Mg2+. The nucleotide-free kinesin protein is buffer exchanged to separate it both from free nucleotide and from EDTA. To accomplish this separation, a disposable gravity-flow gel filtration column is sufficient and is simple and rapid to use (see list of materials). The interaction of mantATP with nucleotide free kinesin is carried out at a range of mantATP concentrations (Step 2.4) to enable deconvolution of the association and dissociation rate constants (Step 2.8). The theory behind experiments of this type is well described in reference 18. In performing these experiments one begins with the lowest concentration of mantATP. This removes the necessity for wash steps between different concentrations. It will likely be necessary to adjust the sensitivity of the stopped-flow fluorimeter as the mantATP concentration is increased. The mantATP concentration must always be in at least 5-fold excess over the concentration of kinesin nucleotide binding sites to maintain pseudo first order conditions. However, as the concentration of mantATP is increased, the signal change can become swamped as the fraction of nucleotide that binds to kinesin decreases. Therefore, the concentration of the kinesin is also increased for each new concentration of mantATP such that the concentration of mantATP is never greater than 10-fold molar excess over nucleotide binding sites (Step 2.6).
mantADP Dissociation
Although association and dissociation rate constants for mantADP can be determined by the same method described for mantATP (Section 2). Dissociation of ADP from a kinesin can be directly observed by preloading the nucleotide-binding site with mantADP (Steps 3.1–3.3) The mantADP.kinesin complex is then mixed with an excess of unlabeled ATP (step 3.5). The excess unlabeled ATP prevents rebinding of mantADP to the kinesin and thereby allows the dissociation reaction (Figure 1, k4) to be observed in isolation from association of mantADP. In this assay the concentration of unlabeled ATP must be at least a 50-fold molar excess of nucleotide binding sites. The theory behind this assay is well described in reference 18. This direct method gives a more accurate value for the dissociation rate constant than the extrapolation to the y-axis described in Step 2.8.
Inclusion of Microtubules
The methods described can also be used to determine the kinetics of nucleotide binding and dissociation in the presence of microtubules. Microtubules are introduced to the nucleotide containing syringe prior to mixing in the stopped-flow:the lower syringe in Figure 3B and 4B(Insets). In this configuration, the kinesin will meet the nucleotide at the same time as it meets the microtubules. Stabilized microtubules are used, where the lifetime of the tubulin polymer is extended by the use of either a stabilizing chemical, such as taxol, or a nonhydrolysable GTP analogue, such as guanosine-5’-[(α,β)-methyleno]triphosphate (GMPCPP, see list of materials). It is possible to use two cycles of tubulin polymerization with GMPCPP to make microtubules that can be stored for many months in liquid nitrogen9,19. The use of pre-prepared microtubules significantly reduces the practical difficulties in performing these assays. To include microtubules in the assays, it is important to use a reaction buffer suitable for microtubule stability. The buffer of choice is PIPES based, with the most commonly used buffer containing 80 mM PIPES pH 6.9, 1 mM MgCl2 and 1 mM EGTA (known as BRB80)20,21.
The methods described are not restricted to use with kinesins, but can be adapted and applied to any nucleotide binding protein. For example, similar methods have been applied to the ATP turnover cycle of members of the myosin family of molecular motors12,22 and the use of mant labeled guanosine nucleotides further extends methods of this type to GTP hydrolyzing proteins23,24.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work is supported by the Biotechnology and Biological Sciences Research Council (BBSRC), the Royal Society and the University of Nottingham.
References
- Marx A, Hoenger A, Mandelkow E. Structures of kinesin motor proteins. Cell Motil Cytoskeleton. 2009;66:958–966. doi: 10.1002/cm.20392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Friel CT, Howard J. Coupling of kinesin ATP turnover to translocation and microtubule regulation: one engine, many machines. J. Muscle Res. Cell Motil. 2012;33:377–383. doi: 10.1007/s10974-012-9289-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M, Zhang X, Park CG, Park HW, Endow SA. A structural pathway for activation of the kinesin motor ATPase. EMBO J. 2001;20:2611–2618. doi: 10.1093/emboj/20.11.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Akimaru E, Akiba T, Endow SA, Amos LA. Large conformational changes in a kinesin motor catalyzed by interaction with microtubules. Mol. Cell. 2006;23:913–923. doi: 10.1016/j.molcel.2006.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa M, et al. Switch-based mechanism of kinesin motors. Nature. 2001;411:439–445. doi: 10.1038/35078000. [DOI] [PubMed] [Google Scholar]
- Friel CT, Bagshaw CR, Howard J. Analysing the ATP turnover cycle of microtubule motors. Methods Mol. Biol. 2011;777:177–192. doi: 10.1007/978-1-61779-252-6_13. [DOI] [PubMed] [Google Scholar]
- Bagshaw C. ATP analogues at a glance. J. Cell Sci. 2001;114:459–460. doi: 10.1242/jcs.114.3.459. [DOI] [PubMed] [Google Scholar]
- Friel CT, Howard J. The kinesin-13 MCAK has an unconventional ATPase cycle adapted for microtubule depolymerization. EMBO J. 2011;30:3928–3939. doi: 10.1038/emboj.2011.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SP, Webb MR, Brune M, Johnson KA. Pathway of processive ATP hydrolysis by kinesin. Nature. 1995;373:671–676. doi: 10.1038/373671a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu A, Taylor EW. A kinetic study of the kinesin ATPase. J. Biol. Chem. 1992;267:11352–11359. [PubMed] [Google Scholar]
- Woodward SK, Eccleston JF, Geeves MA. Kinetics of the interaction of 2'(3')-O-(N-methylanthraniloyl)-ATP with myosin subfragment 1 and actomyosin subfragment 1: characterization of two acto-S1-ADP complexes. Biochemistry. 1991;30:422–430. doi: 10.1021/bi00216a017. [DOI] [PubMed] [Google Scholar]
- Hackney DD. Pathway of ADP-stimulated ADP release and dissociation of tethered kinesin from microtubules. Implications for the extent of processivity. Biochemistry. 2002;41:4437–4446. doi: 10.1021/bi0159229. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Cross RA. Kinetics and motility of the Eg5 microtubule motor. Biochemistry. 1996;35:2365–2373. doi: 10.1021/bi952318n. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Porche K, Rayment I, Gilbert SP. The ATPase pathway that drives the kinesin-14 Kar3Vik1 powerstroke. J. Biol. Chem. 2012;287:36673–36682. doi: 10.1074/jbc.M112.395590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer ML, Gilbert SP, Johnson KA. Pathway of ATP hydrolysis by monomeric and dimeric kinesin. Biochemistry. 1998;37:800–813. doi: 10.1021/bi9711184. [DOI] [PubMed] [Google Scholar]
- Brune M, Hunter JL, Corrie JE, Direct Webb MR. real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 1994;33:8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- Goodrich JA, Kugel JF. Binding and Kinetics for Molecular Biologists. Vol. 4. Cold Spring, NY: Cold Spring Harbour Laboratory Press; 2006. Chapter 4; pp. 69–97. [Google Scholar]
- Gell C, et al. Microtubule dynamics reconstituted in vitro and imaged by single-molecule fluorescence microscopy. Methods Cell Biol. 2010;95:221–245. doi: 10.1016/S0091-679X(10)95013-9. [DOI] [PubMed] [Google Scholar]
- Olmsted JB, Borisy GG. Ionic and nucleotide requirements for microtubule polymerization in vitro. Biochemistry. 1975;14:2996–3005. doi: 10.1021/bi00684a032. [DOI] [PubMed] [Google Scholar]
- Brinkley W. Microtubules: a brief historical perspective. J. Struct Biol. 1997;118:84–86. doi: 10.1006/jsbi.1997.3854. [DOI] [PubMed] [Google Scholar]
- Ostap EM, Pollard TD. Biochemical kinetic characterization of the Acanthamoeba myosin-I ATPase. J. Cell Biol. 1996;132:1053–1060. doi: 10.1083/jcb.132.6.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccleston JF, Moore KJ, Brownbridge GG, Webb MR, Lowe PN. Fluorescence approaches to the study of the p21ras GTPase mechanism. Biochem. Soc. Trans. 1991;19:432–437. doi: 10.1042/bst0190432. [DOI] [PubMed] [Google Scholar]
- Ahmadian MR, Wittinghofer A, Herrmann C. Fluorescence methods in the study of small GTP-binding proteins. Methods Mol. Biol. 2002;189:45–63. doi: 10.1385/1-59259-281-3:045. [DOI] [PubMed] [Google Scholar]