

Abstract
Objective:
We assessed the mutation frequency in nicotinic acetylcholine receptor (nAChR) subunits CHRNA4, CHRNB2, and CHRNA2 in a cohort including autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) and sporadic nocturnal frontal lobe epilepsy (NFLE). Upon finding a novel mutation in CHRNA2 in a large family, we tested in vitro its functional effects.
Methods:
We sequenced all the coding exons and their flanking intronic regions in 150 probands (73 NFLE, 77 ADNFLE), in most of whom diagnosis had been validated by EEG recording of seizures. Upon finding a missense mutation in CHRNA2, we measured whole-cell currents in human embryonic kidney cells in both wild-type and mutant α2β4 and α2β2 nAChR subtypes stimulated with nicotine.
Results:
We found a c.889A>T (p.Ile297Phe) mutation in the proband (≈0.6% of the whole cohort) of a large ADNFLE family (1.2% of familial cases) and confirmed its segregation in all 6 living affected individuals. Video-EEG studies demonstrated sleep-related paroxysmal epileptic arousals in all mutation carriers. Oxcarbazepine treatment was effective in all. Whole-cell current density was reduced to about 40% in heterozygosity and to 0% in homozygosity, with minor effects on channel permeability and sensitivity to nicotine.
Conclusion:
ADNFLE had previously been associated with CHRNA2 dysfunction in one family, in which a gain of function mutation was demonstrated. We confirm the causative role of CHRNA2 mutations in ADNFLE and demonstrate that also loss of function of α2 nAChRs may have pathogenic effects. CHRNA2 mutations are a rare cause of ADNFLE but this gene should be included in mutation screening.
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is characterized by clusters of motor seizures arising during non-REM sleep, usually occurring in individuals of normal intellect.1 Ictal semiology includes a wide spectrum of motor manifestations, ranging from brief motor events to major episodes. Seizures usually appear within the first 2 decades of life and may disappear in adulthood.2 Carbamazepine, oxcarbazepine, and topiramate have proven effective in both ADNFLE and sporadic nocturnal frontal lobe epilepsy (NFLE) in uncontrolled trials.3–6
ADNFLE is the first epilepsy syndrome whose genetic bases have been identified, albeit reported mutations only explain a minority of cases.7 Overall, a limited number of mutations affecting neuronal nicotinic acetylcholine receptor (nAChR) subunits have been associated with the syndrome, including 6 in CHRNA4 (α4), 6 in CHRNB2 (β2), and 1 in CHRNA2 (α2). Functional studies indicate gain of function as the main pathogenic mechanism.8 Additional mutations leading to a similar phenotype have been identified in the KCNT1 gene, which encodes for a potassium channel subunit (4 mutations), and in the DEPDC5 gene, which is involved in G-protein signaling pathways (5 mutations).2,9,10 A causative role for the corticotrophin-releasing hormone (CRH) gene (1 mutation in the coding sequence, 4 noncoding) has also been suggested.11
We identified a missense mutation affecting the first amino acid of the second transmembrane domain of CHRNA2 in a large family in which affected individuals exhibited sleep-related seizures with paroxysmal arousals. This is the second point mutation of CHRNA2 to be associated with ADNFLE. Functional studies of the mutant receptor in human embryonic kidney (HEK) cells revealed reduced current density, demonstrating a loss-of-function effect.
METHODS
Subjects.
After identifying CHRNA2 as a causative gene for ADNFLE,12 we systematically tested this gene, along with the CHRNA4 and CHRNB2, in patients exhibiting a related phenotype. Overall we tested 150 consecutive probands (78 males, 72 females; 73 sporadic, 77 familial) referred for a clinical and genetic diagnosis between 2006 and 2013. In 74% of patients, diagnosis was validated by video-EEG recording of seizures.
We found the p.Ile297Phe mutation in CHRNA2 in the proband of a family in which 8 individuals over 3 generations were affected (figure 1A).
Figure 1. Genetic and molecular characterization of p.Ile297Phe mutation.

(A) Pedigree of the family showing that the mutation segregates in all affected members. Squares = males; circles = females; filled black circles = patients with ADNFLE; black arrow = proband; asterisks = individuals screened for the c.889A>T mutation. (B) Basic structure of nAChRs and position in each subunit of all reported mutations. Orange ellipse represents the ligand; circles represent amino acids in which mutations have been found in CHRNA4 (green), CHRNB2 (light blue), and CHRNA2 (pink). p.Ile297Phe (p.I297F) mutation is indicated in red, bold, and with a bigger font size. TM1, TM2, TM3, and TM4 indicate the 4 transmembrane domains of each nAChR subunit, and the second transmembrane domain, which is directly implicated in the gating structure, is colored in light gray. Upper right part of the panel represents the subunits assembly viewed from the top. Yellow circle represents a positively charged ion that moves through the channel. (C) In silico multiple sequence alignment, performed using Jalview software, reveals that the mutation affected an amino acid that is conserved among the species. According to the physicochemical properties of the wild-type and mutant amino acids, the mutation resulted in the substitution of an aliphatic, hydrophobic amino acid (isoleucine, I, colored in pink) with an aromatic one (phenylalanine, F, colored in orange). Black arrow indicates the position of the mutation. ADNFLE = autosomal dominant nocturnal frontal lobe epilepsy; nAChR = nicotinic acetylcholine receptor.
Standard protocol approvals, registrations, and patient consents.
All participants and their parents/legal guardians gave informed consent. The study was approved by the human research ethics committees of the Meyer Children's Hospital. A consent to disclose form was obtained for videos of any recognizable patient.
CHRNA2 mutation screening.
Genomic DNA was extracted from blood leucocytes using an automated isolation robot (QIASymphony; Qiagen, Hilden, Germany).
Based on known mutation rates,8,13 we screened in succession CHRNA4, CHRNB2, and CHRNA2.
Exons covering all the coding regions of CHRNA2 (Entrez gene ID: 1135; accession number: NM_000742.3) and their flanking intronic regions were amplified with primers designed using Primer3 Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/). Genomic DNA template (50 ng) was amplified using FastStart Taq DNA Polymerase (Roche, Mannheim, Germany). PCR products were checked by 1.5% agarose gel electrophoresis, purified using ExoSAP-IT (Affymetrix, Santa Clara, CA) and analyzed by direct sequencing, on both strands, using the BigDye Terminator V1.1 chemistry (Life Technologies, Grand Island, NY), on an ABI Prism 3130XL automated capillary sequencer (Life Technologies). Primers are available on request.
Video-EEG recordings.
All affected individuals underwent whole-night video-polysomnographic recordings (EB Neuro, Florence, Italy) with an extended EEG montage, electrooculography, ECG, chin and tibialis anterior muscles EMG, and chest and abdominal respiratory movements. During EEG recordings, none of the patients were receiving antiepileptic medication.
Cell culture and transfection procedure.
Either wild-type (WT) or mutant α2 constructs were synthesized in vitro (Biomatik, Cambridge, Canada) and transiently cotransfected with the β2 or β4 subunit in HEK cells (TsA subclone; American Type Culture Collection, Manassas, VA) using standard procedures. Cells were cultured in DMEM-F12 (Dulbecco's modified Eagle medium F12) (HyClone Laboratories, Logan, UT) supplemented with 10% fetal calf serum (HyClone) and 2 mM l-glutamine, at 37°C and 5% CO2.
Cells were seeded in 35-mm culture dishes and transfected with Lipofectamine 2000 (Life Technologies). cDNA ratios were α2:β4 of 1:1, for WT receptors; α2-Ile297Phe:β4 of 1:1 for homozygous mutant receptor; and α2:α2-Ile297Phe:β4 of 1:1:2, for the simulated heterozygous state. DNA concentration in the transfection mixture was 4 μg of nAChR subunit constructs plus 0.6 μg of expression vector for the enhanced green fluorescent protein E-GFPpcDNA3 (Clontech Laboratories, Mountain View, CA). E-GFP expression allowed easier detection of transfected cells. Cells were incubated with the transfection mixture for 4 to 6 hours. This procedure was slightly modified when testing the α2β2 form, which yields very low functional expression in mammalian cell lines (see the results section). We increased receptors' expression using the procedure of Cooper et al.14 In particular, 24 hours before recording, transfected cells were transferred to lower temperature (29°C–30°C, in 5% CO2).
Patch-clamp experiments.
We applied the whole-cell configuration of patch-clamp. Currents were registered 36 to 72 hours after transfection, with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA), at 22°C to 24°C. Micropipettes (2–3 MΩ) were pulled from borosilicate capillaries with a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument Co., Novato, CA). Cell capacitance and series resistance were compensated (up to approximately 75%). Cells were inspected with an Eclipse TE200 microscope (Nikon Corporation, Tokyo, Japan) equipped with a TE-FM epifluorescence attachment for detection of fluorescent cells. Currents were low-pass-filtered at 2 kHz and acquired online at 5 to 10 kHz with pClamp 9 hardware and software (Molecular Devices). Drugs were applied with an RSC-160 Rapid Solution Changer (Bio-Logic Science Instruments, Claix, France).
Solutions and drugs.
Unless otherwise specified, chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The extracellular solution contained the following (in mM): NaCl 130, KCl 5, CaCl2 2, MgCl2 2, HEPES 10, and d-glucose 5 (pH 7.3). Patch pipettes contained the following (in mM): K-aspartate 120, NaCl 10, MgCl2 2, CaCl2 1.3, EGTA-KOH 10, HEPES-KOH 10, and MgATP 1 (pH 7.3). Stock solutions of nicotine (10 mM) were prepared weekly in an extracellular solution and kept refrigerated. Extracellular solutions with the appropriate nicotine concentrations were prepared daily; pH was always rechecked after nicotine addition.
Analysis of patch-clamp data.
Data were analyzed with Clampfit 9.2 (Molecular Devices) and OriginPro 9 (OriginLab, Guangzhou, China). The concentration-response data were fitted to a single-term Hill-Langmuir equation:
 |
where Imax is the maximal current, IL is the current at a given concentration of agonist L, EC50 is the half-effective L concentration, and nH is the Hill coefficient (expressing the degree of apparent cooperativity). To estimate the reversal potential (Vrev) of WT and heterozygous currents, current–voltage (I/V) relations were obtained by applying voltage ramps from −60 to +10 mV, with 100 μM nicotine or without it. The background current was subtracted from the current measured in the presence of nicotine. To decrease the fluctuations in Vrev measurements, 3 voltage ramps were generally applied before and during nicotine application. Since our aim was to determine Vrev, we deemed as irrelevant the small differences (<10%) between current amplitudes obtained with the consecutive voltage ramps caused by slow desensitization of the α2β4 receptor. From each of the resulting I/V relations, we estimated Vrev visually after fitting the I/V curve with a polynomial function (e.g., Haghighi and Cooper,15 2000). No correction of liquid-junction potential was applied to any of the voltage values provided above.
Data are generally given as mean values ± SEM, with “n” representing the number of determinations in different cells. Statistical significance was determined with Student t test for unpaired samples, with the level of significance set at p < 0.05.
RESULTS
CHRNA2 mutation screening.
Direct sequencing of PCR-amplified products of CHRNA2 demonstrated the c.889A>T (p.Ile297Phe) mutation in 1/150 probands in our cohort (≈0.6% of the whole cohort: 95% confidence interval 0.017%–3.658%, 1.2% of familial cases). Segregation analysis demonstrated that all 7 affected individuals available for molecular screening were heterozygous for the mutation, whereas the proband's healthy grandmother was mutation-negative (figure 1A). The p.Ile297Phe mutation affects the first amino acid of the second transmembrane domain of the protein (figure 1B). Bioinformatic analysis using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and MutationTaster (http://www.mutationtaster.org/) programs predicted a pathogenic role of the Ile297Phe substitution.
To exclude that p.Ile297Phe mutation may be a rare, benign polymorphism, we interrogated both the EVS (http://evs.gs.washington.edu) and the ExAC (http://exac.broadinstitute.org) databases, comprising exome data of 6,503 controls and 61,486 individuals sequenced as part of various disease-specific and population genetic studies. Overall, these large datasets list 262 CHRNA2 missense variations, of which 106 are predicted as probably damaging but do not include the p.Ile297Phe mutation, supporting its pathogenic role.16
In silico analysis with the Jalview software17 revealed that the p.Ile297Phe mutation, which causes the substitution of an aliphatic, hydrophobic amino acid (isoleucine) with an aromatic one (phenylalanine), occurred in an evolutionarily conserved residue (figure 1C).
Clinical and neurophysiologic studies in affected individuals carrying the p.Ile297Phe CHRNA2 mutation.
Clinical information on affected individuals is summarized in the table. The family pedigree is reported in figure 1A. Clinical findings were homogeneous across affected individuals and characterized by frequent paroxysmal motor events during sleep. Mean age at onset was 6.2 years (median 4.5, range 3–16). In all patients, several seizures per night occurred with no correlation with any specific sleep phase. In undiagnosed, untreated patients (II-2, II-4, and III-2), seizures persisted in adulthood with consequent, frequent traumatic injuries.
Table.
Clinical information of affected family members
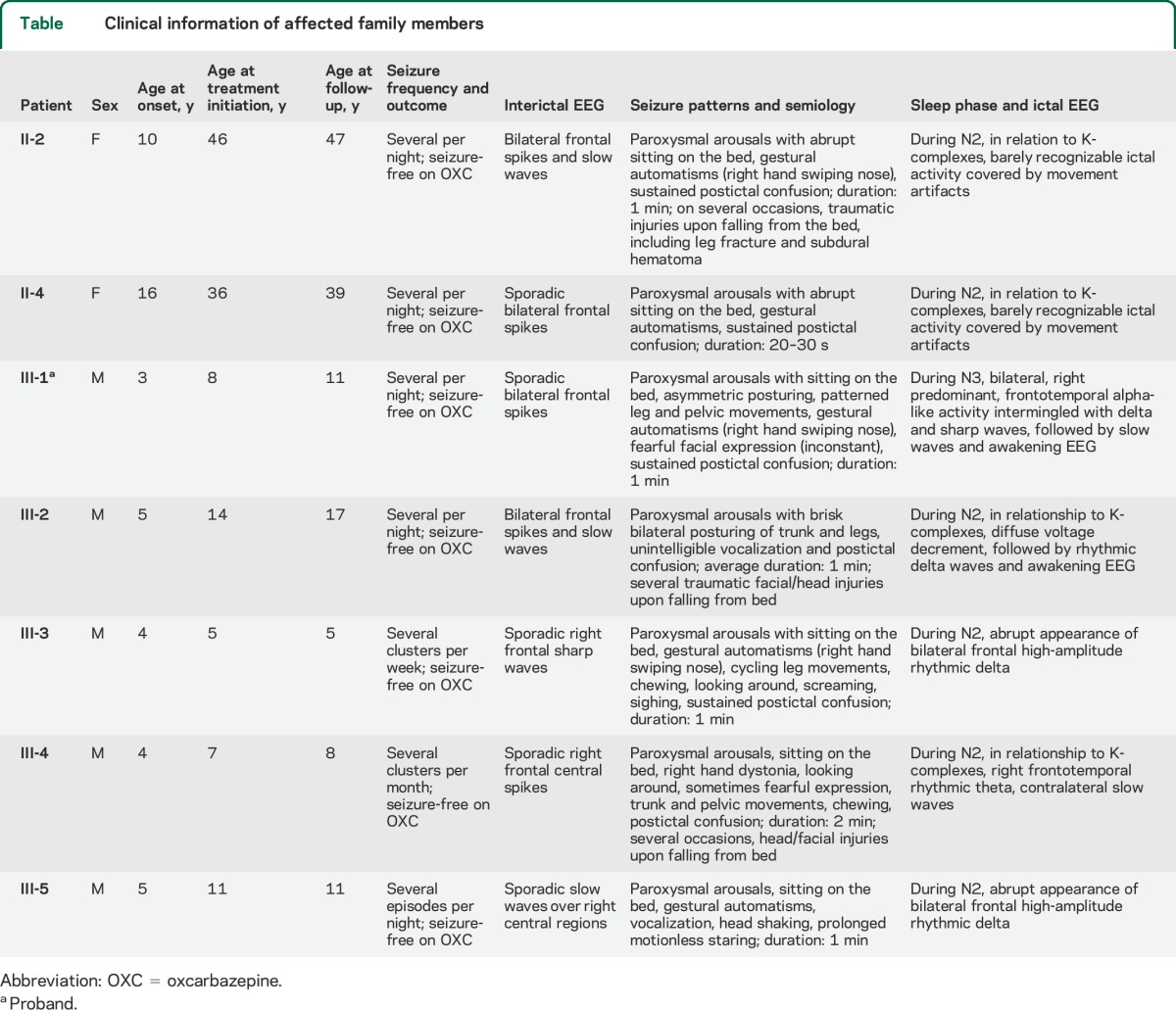
Personal history was unremarkable except for a diagnosis of attention deficit hyperactivity disorder (patient III-5) and other sleep disorder (somniloquium, patient III-2). Brain MRI was unremarkable in all.
Clinical seizures were consistent with epileptic paroxysmal arousals, with sudden motor manifestations, including bilateral tonic or dystonic posturing, with patterned automatic activities (table, and videos 1–4 on the Neurology® Web site at Neurology.org). After ictal motor manifestations and EEG activity subsided, the patients remained unresponsive, or uttered unintelligible words, for up to several minutes. No recall of subjective symptoms was possible. Ictal and interictal EEG data are summarized in the table and shown in figures 2 and e-1. Oxcarbazepine monotherapy was effective in all patients.
Figure 2. Polygraphic video-EEG recordings of nocturnal seizures in 4 patients.
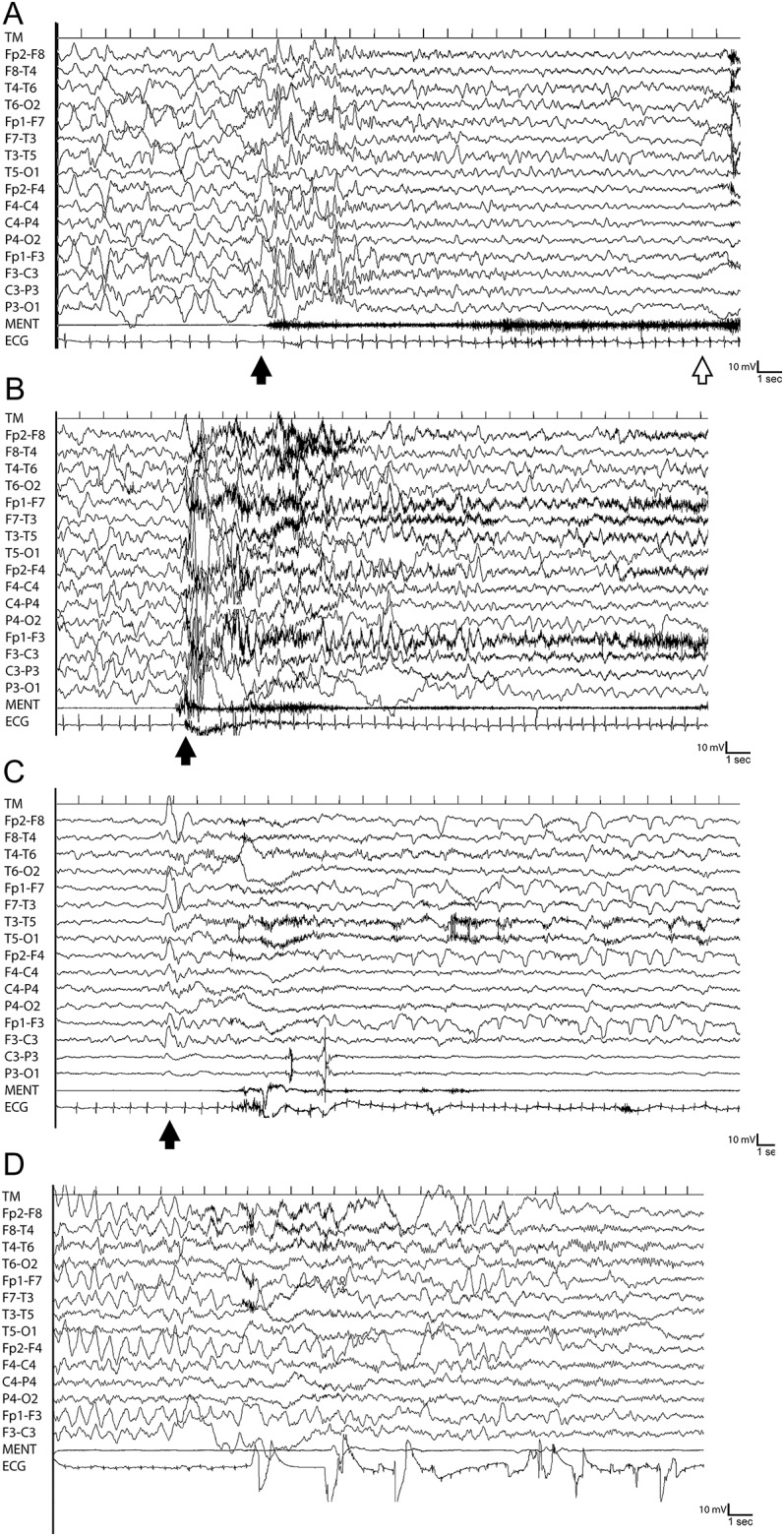
See the table for details of clinical semiology and ictal EEG. (A) Patient III-1. The black arrow indicates abrupt appearance of bilateral spikes prevalent over the anterior regions during slow-wave sleep, followed after a few seconds by diffuse, slow-wave activity. Clinically, the patient awakened and exhibited bilateral dystonic posturing. The white arrow indicates the end of the clinical seizure. (B) Patient III-3: paroxysmal arousal. The black arrow indicates the onset of bilateral, frontal predominant, high-amplitude rhythmic delta waves. Clinically, the patient manifested a paroxysmal arousal with sitting on the bed, gestural automatisms, cycling leg movements, chewing, screaming, sighing, and postictal confusion. (C) Patient III-4. The black arrow indicates the appearance of right frontotemporal rhythmic theta activity and contralateral slow waves, in relationship to a K-complex. Clinically, the patient exhibited a paroxysmal arousal with right hand dystonia, sitting on the bed, fearful expression, oral automatisms, trunk and pelvic movements, and postictal confusion. (D) Patient III-5: paroxysmal arousal. Artifact-free ictal EEG was only available after a few seconds from seizure onset and disclosed a high-amplitude, bilateral, frontal predominant, slow-wave activity. Clinically, the patient manifested a paroxysmal arousal, with gestural automatism, vocalization, head shaking, and motionless staring.
Functional studies.
In the primate brain, the α2 subunit exhibits a wide distribution that largely overlaps with that of the α4, β2, and β4 subunits.12,18,19 We first tested the effect of coexpressing α2 and α2-Ile297Phe with β4 in HEK cells. Figure 3A shows whole-cell current traces elicited by the indicated concentration of nicotine, applied for 2 to 3 seconds to cells maintained at −70 mV (i.e., close to the neuronal Vresting). The maximal current was tested at regular intervals to rule out nAChR rundown, which is sometimes observed in cell lines. Nicotine was preferred to the physiologic agonist acetylcholine (ACh) to avoid applying muscarinic receptor's blockers, whose effect on different nAChR subtypes is only partially defined.20 No current expression was observed in cells transfected with the homozygous α2-Ile297Pheβ4 form. Figure 3B shows the average nAChR current density (i.e., the maximal current divided by cell capacitance) in experiments performed with the indicated subunit combinations. The current density of the heterozygous form was approximately 40% of that observed in WT channels, whereas the homozygous form produced no measurable currents. No significant difference between WT and heterozygous receptors was observed in the kinetics of channel desensitization and deactivation (data not shown). Statistics are given in the figure legend.
Figure 3. Functional expression of α2β4 and α2β2 nAChR subtypes, containing or not containing α2-Ile297Phe.

(A) Representative current traces elicited by the indicated concentration of nicotine in HEK cells expressing either the α2β4 (WT) or the α2/α2Ile297Pheβ4 (heterozygous) nAChR isoform. Vm was −70 mV. The continuous lines above the current traces mark the time of nicotine application. Panels display typical currents recorded from WT and heterozygous nAChRs, as indicated. (B) No measurable current was elicited when cells were transfected with the homozygous (α2Ile297Pheβ4) nAChRs. Experimental conditions were as in panel A. (C) Bars indicate the average peak current density for WT and heterozygous receptors in either α2β4 or α2β2 condition, as indicated. Currents were elicited at −70 mV, by 100 μM (for the α2β4 subtype) or 300 μM nicotine (for the α2β2 isoform). In particular, the average current values were (pA/pF): 22.97 ± 3.73 for WT α2β4 (n = 21), and 9.72 ± 1.85 for the corresponding heterozygote (n = 27, p < 0.01). In these experiments, the current densities obtained with 10 μM nicotine were (pA/pF): 11.33 ± 2.47 for WT (n = 21), and 3.49 ± 0.74 for the heterozygote (n = 27). No measurable current was ever detected in the homozygous mutant form (n = 30). Data summarize the results obtained in 4 runs of transfection. For the α2β2 subtype, the peak current density (pA/pF) was 2.45 ± 0.35 (n = 9) for the WT, and 0.55 ± 0.15 for the heterozygote (n = 5, p < 0.05). These data summarize the results obtained in 2 runs of transfection. (D) Representative current traces for the α2β2 subtype, in WT and heterozygous condition, as indicated. Currents were elicited by 300 μM nicotine, at −70 mV. Continuous lines mark the time of nicotine application. Once again, no current was ever observed in the homozygous condition (not shown). HEK = human embryonic kidney; nAChR = nicotinic acetylcholine receptor; WT = wild-type.
The functional expression of the α2β2 subtype generally produces lower current amplitudes than other subtypes such as α2β4 and α4β2.12,21–23 Therefore, to obtain measurable currents from the α2β2 subtype, before patch-clamp experiments, we incubated the transfected cells at lower temperature for 24 hours14 (see the methods section). As shown in figure 3D, the results we obtained with α2β2 were qualitatively similar to those obtained with α2β4, in that α2-Ile297Phe strongly decreased nAChR currents. These results show that the p.Ile297Phe substitution decreases the functional expression of the heteromeric nAChR isoforms containing the mutant α2 subunit.
We subsequently performed a fuller characterization of the properties conferred to nAChRs by α2-Ile297Phe, conducting a functional study in the α2β4 nAChR subtype. To assess whether the mutant subunit significantly altered ion selectivity, we measured Vrev from current/voltage (I/V) relations obtained by applying voltage ramps from −60 to +10 mV. Representative I/V curves from cells expressing WT or heterozygote nAChRs are shown in figure 4A. Both types of receptors showed the inward rectification typical of neuronal nAChRs.15 We observed no significant difference in the average Vrev for WT and heterozygous receptors in a series of similar experiments (figure 4B). These results indicate that α2-Ile297Phe is unlikely to produce major alterations in channel permeability, thus not modifying the depolarizing nature of nicotinic currents in CNS neurons.
Figure 4. Functional features of α2β4 nAChRs containing or not containing α2-Ile297Phe.

(A) I/V relationships for WT and heterozygous receptors, from 2 cells expressing whole-cell currents of similar amplitude. Currents were elicited by applying voltage ramps from −60 to +10 mV (duration was 1 second). The current flowing through nAChRs was isolated by subtracting the background current from the current recorded in the presence of 100 μM nicotine. The illustrated currents are averages of 3 trials, applied consecutively (interval between trials was 1 second). Notice the typical inward rectification displayed by neuronal nAChRs, and the Vrev between −10 mV and 0 mV. (B) Bars give the average Vrev calculated from a series of experiments in the same run of transfection, for WT and heterozygous receptors. Experimental procedure was as illustrated in panel A. The average Vrev was −1.1 ± 3 mV for the WT (n = 9) and −4.2 ± 2.1 for the heterozygote (n = 5, not statistically different from the WT). (C) Concentration-response relations obtained by applying different nicotine concentrations to WT (squares) and heterozygous (circles) receptors. The applied nicotine concentrations were 1, 3, 10, and 100 μM. Vm was −70 mV. Data points are average peak currents normalized to the maximal WT value, and plotted as a function of ligand concentration. Each point represents at least 6 determinations in different cells. Continuous lines were obtained by fitting the data with equation 1. The EC50 was 10.6 ± 0.03 μM (nH = 1.52 ± 0.06) for WT, and 23.9 ± 2.4 μM (nH = 1.56 ± 0.53) for heterozygous nAChRs. Data summarize the results obtained from 48 cells, in 4 runs of transfection. EC50 = 50% effective concentration; nAChR = nicotinic acetylcholine receptor; WT = wild-type.
Through experiments such as those illustrated in figure 3, we generated concentration-response relations to nicotine for WTs and heterozygotes. The peak currents measured at different agonist concentrations were normalized to the maximal value and plotted as a function of ligand concentration (figure 4C). Data points were fitted with a properly scaled Hill-type equation (equation 1). The half-effective nicotine concentrations (EC50) for the 2 components were approximately 11 μM for WT receptors and 22 μM for the heterozygote, with a Hill coefficient of about 1.5. Statistics are given in the figure legend.
DISCUSSION
ADNFLE is genetically heterogeneous, with only a few disease-causing mutations having been reported so far in 6 different genes.2 DEPDC5 was recently identified as a major ADNFLE-causing gene, with about 13% of affected families exhibiting mutations of this gene.10 DEPDC5-related ADNFLE is often drug-resistant and associated with diurnal seizures.10 Families carrying DEPDC5 mutations are smaller than those related to other causative genes,10 possibly as a consequence of some degree of reproductive disadvantage resulting from severe epilepsy. Mutations in the 5 remaining ADNFLE-related genes (CHRNA4, CHRNB2, CHRNA2, KCNT1, and CRH) collectively account for about 10% to 15% of familial cases.2,10 Among these genes, CHRNA2 has until now revealed the lower mutation rate, with only one reported mutation in one family.12
The second ADNFLE point mutation in CHRNA2 we are reporting here confirms the causative role of this subunit for this syndrome. Although CHRNA2 only accounts for 1.2% of the ADNFLE families we screened after reporting the first mutation,12 neither CHRNA4 nor CHRNB2 were ever mutated in the same cohort.
The previously reported CHRNA2 mutation (p.Ile279Asn) resulted in a marked increase of the receptor sensitivity to the agonist, suggesting gain of function.12,21,24 Conversely, the p.Ile297Phe mutation has scarce effect on the receptor sensitivity to nicotine, but causes complete loss of current expression in homozygosity and a decrease to about 40% in heterozygosity, thus pointing to loss of receptor function. These functional differences might be due to either the position of the mutated amino acid in the protein or the physicochemical alterations introduced by the substitution, or both. The p.Ile279Asn mutation resulted in the nonconservative substitution of an amino acid with a hydrophobic side chain with another having a polar uncharged side chain.12 The affected amino acid is located in the first transmembrane domain and, apparently, does not directly contribute to the structure of the channel pore, but is probably implicated in the conformational transition operated by agonist binding.25 The p.Ile297Phe mutation affects an amino acid located in the second transmembrane domain, which is more directly implicated in the gating structure.26 The decreased current expression caused by p.Ile297Phe might depend on either impaired channel expression onto the cell surface or on a drastic decrease in the channel open probability. Although both Ile and Phe have a hydrophobic side chain, the substitution cannot be considered conservative, because of the different sizes and chemical properties of the aliphatic and aromatic benzyl side chain.27 As a consequence of these differences, p.Ile297Phe might cause a steric hindrance that disrupts the tight association of amino acids in the ion gate, impairing folding and trafficking of the mutated receptor, or the gating transition, or both.
Observing similar phenotypes in relation to nAChR mutations exhibiting opposite cellular effects in vitro is not entirely surprising. This phenomenon has been demonstrated, for example, in congenital myasthenia in which most alterations in the CHRNE gene result in loss of function, but a small fraction cause gain of function, in either case ultimately resulting in reduced efficiency of the synaptic transmission.28 Loss of receptor function as a consequence of disease-causing mutations of nAChR has also been reported in ADNFLE. Analysis of the Ser248Phe (S248F) CHRNA4 mutant revealed that these receptors exhibit a marked desensitization.29–31 Based on these data, it was hypothesized that seizures were somehow caused by loss of receptor function, although a gain-of-function mechanism seems to be operating in most functionally characterized mutations. Heteromeric nAChRs control both excitatory and inhibitory transmission in the frontal cortex and elsewhere, and the normal firing activity is generated by a delicate balance of excitation and inhibition. For example, a gain-of-function nAChR expressed in GABAergic cells can produce hyperexcitability because abnormally strong bouts of γ-aminobutyric acid (GABA) release can synchronize pyramidal cells.32 However, a loss-of-function nAChR expressed in the same GABAergic cells can also produce hyperexcitability by decreasing feedback inhibition of pyramidal cells.33
Focusing on the p.Ile297Phe mutation, one might assume that because of its strong effect on channel expression, it would cause an abnormal reorganization of the subunits participating in the heteromeric nAChRs comprising or not α2, resulting in a global alteration of the nicotinic function in vivo.
Clinical manifestations occurring in the family we are describing were conclusively diagnosed as ADNFLE only after video-EEG recordings. ADNFLE is indeed often misdiagnosed as parasomnias and may consequently be underdiagnosed. When ADNFLE is suspected, mutation screening of CHRNA2 should be considered along with CHRNA4, CHRNB2, and KCNT1. Since ADNFLE caused by nAChR mutations carries an overall good long-term seizure outlook and is likely responsive to medication, assigning it to its specific genetic etiology may have implications for management.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families for participating in the research.
GLOSSARY
- ADNFLE
autosomal dominant nocturnal frontal lobe epilepsy
- HEK
human embryonic kidney
- nAChR
nicotinic acetylcholine receptor
- NFLE
nocturnal frontal lobe epilepsy
- WT
wild-type
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Study concept and design: V. Conti, A. Becchetti, R. Guerrini. Patient collection, mutation screening, and data analysis: V. Conti, L. Chiti, F. Mari, C. Marini, A. Romigi, M. Albanese, A. Marchi, C. Liguori, F. Placidi, R. Guerrini. Functional studies: P. Aracri, S. Brusco, A. Becchetti. Drafting of the manuscript: V. Conti, F. Mari, A. Becchetti, R. Guerrini. Critical revision of the manuscript for important intellectual content: A. Becchetti, C. Marini, R. Guerrini. Obtained funding: A. Becchetti, R. Guerrini.
STUDY FUNDING
This research was supported by Telethon Italia (GGP12147) to A.B.; University of Milano-Bicocca (FAR 2013) to A.B.; and the European Union Seventh Framework Programme FP7/2007–2013 under the project DESIRE (grant agreement 602531) to R.G.
DISCLOSURE
V. Conti serves on the editorial board of Epilepsia. P. Aracri, L. Chiti, S. Brusco, and F. Mari report no disclosures relevant to the manuscript. C. Marini served on the editorial board of Epilepsia (2007–2012). She received research support from the Italian Ministry of Health, Research Program Section (RF-2009-1525669) and from CARIPLO Foundation, Italy. M. Albanese, A. Marchi, C. Liguori, F. Placidi, and A. Romigi report no disclosures relevant to the manuscript. A. Becchetti received research support from Telethon Italia (GGP12147) and University of Milano-Bicocca (FAR 2013). R. Guerrini received honoraria from Biocodex, UCB, Eisai Inc., ValueBox, and ViroPharma, and research support from the Italian Ministry of Health, the European Community Sixth and Seventh Framework Thematic Priority Health, the Italian Ministry of Education, University and Research, the Tuscany Region, the Telethon Foundation, and the Mariani Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant nocturnal frontal lobe epilepsy: a distinctive clinical disorder. Brain 1995;118:61–73. [DOI] [PubMed] [Google Scholar]
- 2.Nobili L, Proserpio P, Combi R, et al. Nocturnal frontal lobe epilepsy. Curr Neurol Neurosci Rep 2014;14:424. [DOI] [PubMed] [Google Scholar]
- 3.Oldani A, Manconi M, Zucconi M, Martinelli C, Ferini-Strambi L. Topiramate treatment for nocturnal frontal lobe epilepsy. Seizure 2006;15:649–652. [DOI] [PubMed] [Google Scholar]
- 4.Provini F, Plazzi G, Tinuper P, Vandi S, Lugaresi E, Montagna P. Nocturnal frontal lobe epilepsy: a clinical and polygraphic overview of 100 consecutive cases. Brain 1999;122:1017–1031. [DOI] [PubMed] [Google Scholar]
- 5.Raju GP, Sarco DP, Poduri A, Riviello JJ, Bergin AMR, Takeoka M. Oxcarbazepine in children with nocturnal frontal-lobe epilepsy. Pediatr Neurol 2007;37:345–349. [DOI] [PubMed] [Google Scholar]
- 6.Romigi A, Marciani MG, Placidi F, et al. Oxcarbazepine in nocturnal frontal-lobe epilepsy: a further interesting report. Pediatr Neurol 2008;39:298. [DOI] [PubMed] [Google Scholar]
- 7.Combi R, Ferini-Strambi L, Tenchini ML. CHRNA2 mutations are rare in the NFLE population: evaluation of a large cohort of Italian patients. Sleep Med 2009;10:139–142. [DOI] [PubMed] [Google Scholar]
- 8.Ferini-Strambi L, Sansoni V, Combi R. Nocturnal frontal lobe epilepsy and the acetylcholine receptor. Neurologist 2012;18:343–349. [DOI] [PubMed] [Google Scholar]
- 9.Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45:552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 2014;82:2101–2106. [DOI] [PubMed] [Google Scholar]
- 11.Sansoni V, Forcella M, Mozzi A, et al. Functional characterization of a CRH missense mutation identified in an ADNFLE family. PLoS One 2013;8:e61306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aridon P, Marini C, Di Resta C, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet 2006;79:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC. Channelopathies in idiopathic epilepsy. Neurotherapeutics 2007;4:295–304. [DOI] [PubMed] [Google Scholar]
- 14.Cooper ST, Harkness PC, Baker ER, Millar NS. Up-regulation of cell-surface alpha4beta2 neuronal nicotinic receptors by lower temperature and expression of chimeric subunits. J Biol Chem 1999;274:27145–27152. [DOI] [PubMed] [Google Scholar]
- 15.Haghighi AP, Cooper E. A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine alpha3beta4 and alpha4beta2 receptors. J Neurosci 2000;20:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cherepanova NS, Leslie E, Ferguson PJ, Bamshad MJ, Bassuk AG. Presence of epilepsy-associated variants in large exome databases. J Neurogenet 2013;27:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009;25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han ZY, Le Novère N, Zoli M, Hill JA, Jr, Champtiaux N, Changeux JP. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur J Neurosci 2000;12:3664–3674. [DOI] [PubMed] [Google Scholar]
- 19.Quik M, Polonskaya Y, Gillespie A, Jakowec M, Lloyd GK, Langston JW. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol 2000;425:58–69. [DOI] [PubMed] [Google Scholar]
- 20.Zwart R, Vijverberg HP. Potentiation and inhibition of neuronal nicotinic receptors by atropine: competitive and noncompetitive effects. Mol Pharmacol 1997;52:886–895. [DOI] [PubMed] [Google Scholar]
- 21.Di Resta C, Ambrosi P, Curia G, Becchetti A. Effect of carbamazepine and oxcarbazepine on wild-type and mutant neuronal nicotinic acetylcholine receptors linked to nocturnal frontal lobe epilepsy. Eur J Pharmacol 2010;643:13–20. [DOI] [PubMed] [Google Scholar]
- 22.Groot-Kormelink PJ, Broadbent SD, Boorman JP, Sivilotti LG. Incomplete incorporation of tandem subunits in recombinant neuronal nicotinic receptors. J Gen Physiol 2004;123:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao Y, Kellar K. The comparative pharmacology and up-regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. J Pharmacol Exp Ther 2004;310:98–107. [DOI] [PubMed] [Google Scholar]
- 24.Hoda JC, Wanischeck M, Bertrand D, Steinlein OK. Pleiotropic functional effects of the first epilepsy-associated mutation in the human CHRNA2 gene. FEBS Lett 2009;583:1599–1604. [DOI] [PubMed] [Google Scholar]
- 25.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature 2003;423:949–955. [DOI] [PubMed] [Google Scholar]
- 26.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 2009;89:73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Fan F, Xiao X, Sun Y. Constitutive activation of the thyroid-stimulating hormone receptor (TSHR) by mutating Ile691 in the cytoplasmic tail segment. PLoS One 2011;6:e16335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinlein OK, Bertrand D. Neuronal nicotinic acetylcholine receptors: from the genetic analysis to neurological diseases. Biochem Pharmacol 2008;76:1175–1183. [DOI] [PubMed] [Google Scholar]
- 29.Weiland S, Witzemann V, Villarroel A, Propping P, Steinlein O. An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett 1996;398:91–96. [DOI] [PubMed] [Google Scholar]
- 30.Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human alpha4beta2 nicotinic acetylcholine receptors. J Neurosci 1997;17:9035–9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertrand S, Weiland S, Berkovic SF, Steinlein OK, Bertrand D. Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol 1998;125:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci USA 2006;103:19152–19157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aracri P, Consonni S, Morini R, et al. Tonic modulation of GABA release by nicotinic acetylcholine receptors in layer V of the murine prefrontal cortex. Cereb Cortex 2010;20:1539–1555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.