

Abstract
Large macromolecular complexes such as the spliceosomal small nuclear ribonucleoproteins (snRNPs) play a variety of roles within the cell. Despite their biological importance, biochemical studies of snRNPs and other machines are often thwarted by practical difficulties in the isolation of sufficient amounts of material. Studies of the snRNPs as well as other macromolecular machines would be greatly facilitated by new approaches that enable their isolation and biochemical characterization. One such approach is single-molecule pull-down (SiMPull) that combines in situ immunopurification of complexes from cell lysates with subsequent single-molecule fluorescence microscopy experiments. We report the development of a new method, called SNAP-SiMPull, that can readily be applied to studies of splicing factors and snRNPs isolated from whole-cell lysates. SNAP-SiMPull overcomes many of the limitations imposed by conventional SiMPull strategies that rely on fluorescent proteins. We have used SNAP-SiMPull to study the yeast branchpoint bridging protein (BBP) as well as the U1 and U6 snRNPs. SNAP-SiMPull will likely find broad use for rapidly isolating complex cellular machines for single-molecule fluorescence colocalization experiments.
Keywords: snRNP, single-molecule, fluorescence, microscopy, splicing, spliceosome
INTRODUCTION
Dynamic multicomponent complexes are at the center of many cellular functions including pre-mRNA splicing. Recently, single-molecule colocalization and fluorescence resonance energy transfer (smFRET) experiments have provided many unique insights into the dynamics of spliceosome assembly and pre-mRNA conformational changes that occur during splicing (Hoskins et al. 2011a; Abelson et al. 2012; Shcherbakova et al. 2013; Krishnan et al. 2013). However, in all of these experiments spliceosome components are assembled onto pre-mRNA molecules in yeast whole-cell extract. The pre-mRNA molecules were either already tethered to a glass surface at the initiation of spliceosome assembly or subsequently immobilized after post-assembly via the pre-mRNA or by a TAP tag found on spliceosomal protein. This approach does not permit direct interrogation of the splicing machinery in the absence of pre-mRNA and is complicated by the presence of cellular extract, which possesses an unknown composition that may influence or interfere with the assay. As an alternate approach, spliceosomal components can be purified from cell extracts and studied in isolation. However, purification often requires days to complete during which many of these complexes may degrade or dissociate into individual components. Straightforward and rapid methods for obtaining spliceosome components for single-molecule assays are needed to address many fundamental questions in splicing such as heterogeneity in small nuclear ribonucleoprotein (snRNP) composition and substrate-independent interactions between snRNPs (Konarska and Sharp 1988; Stevens et al. 2002; Xu et al. 2004; Hoskins et al. 2011b; Abelson et al. 2012; Li et al. 2013; Shcherbakova et al. 2013).
Recently, a novel method called single-molecule pull-down (SiMPull) combined traditional coimmunoprecipitation (co-IP) assays with single-molecule fluorescence microscopy in order to isolate and study single molecules from lysates (Jain et al. 2011). To achieve this, the SiMPull method uses surface-immobilized antibodies to IP protein complexes tagged with spectrally distinct fluorescent proteins (FPs) such as YFP or mCherry. Importantly, SiMPull bridges the gap between biochemical purification and assays in cell extracts by purifying molecules in situ from lysates. Using the SiMPull approach, biomolecular complex heterogeneity, protein stoichiometry, and activity can be readily studied using single-molecule assays.
Despite their utility, SiMPull assays as currently described can be complicated by a number of factors. Incomplete chromophore maturation of the FP can confound analysis of complex stoichiometry and heterogeneity. For example, only 40% of mCherry FPs mature and fluoresce at 37°C leaving more than half of the FPs dark in the experiment (Ulbrich and Isacoff 2007, 2008). Additionally, many FPs are susceptible to blinking or very short lifetimes that can limit the duration of a single-molecule experiment (Dickson et al. 1997; Kubitscheck et al. 2000; Hendrix et al. 2008). Since many aspects of pre-mRNA splicing occur over tens of minutes in vitro, these features of FPs make their use in studying the spliceosome difficult.
We have developed SNAP-SiMPull as a tool for applying single-molecule pull-down techniques to the splicing machinery while avoiding the use of FPs. The SNAP-SiMPull approach uses a bifunctional genetic tag that allows for fluorescent derivitization and robust, biotin-dependent immobilization. This provides two distinct advantages over SiMPull: First, it simplifies immobilization by circumventing the need for antibodies, and second, it utilizes bright, organic fluorophores that have far superior photophysical properties over fluorescent proteins and do not require maturation (Shaner et al. 2005; Chen et al. 2013). We have used SNAP-SiMPull to isolate different spliceosome components including the branchpoint bridging protein (BBP) and the U1 and U6 snRNPs. This approach has allowed us to carry out functional assays of RNA-binding by BBP as well as elucidation of snRNA-dependent interactions between U6 snRNP proteins.
RESULTS
Design and testing of the SNAP-SiMPull tag
In creating a protein tag for SiMPull experiments, we sought to incorporate two features. First, the tag should be able to efficiently incorporate different types of fluorophores that emit light across a range of wavelengths to enable multicolor imaging. Second, the tag should facilitate stable surface immobilization under a range of conditions. We believed that the first feature could be accommodated by use of the SNAP tag. The SNAP tag is a derivative of human alkylguanine-S-transferase that forms covalent adducts with benzylguanine fluorophores (Juillerat et al. 2003). This has been widely used for a number of fluorescence microscopy applications including single-molecule imaging (Hoskins et al. 2011a) and a variety of benzylguanine fluorophores are available for the SNAP tag. Additionally, the SNAP tag exhibits a high degree of fluorophore incorporation and rapid labeling kinetics (Sun et al. 2011)—both beneficial attributes for SiMPull.
We next explored alternatives for stable surface immobilization of the SNAP tag. The very strong interaction between biotin and streptavidin has been routinely used for immobilizing molecules for a wide range of experiments and under many different conditions (Roy et al. 2008; Hoskins et al. 2011b). Based on this precedent, we created a derivative of the SNAP tag that could be biotinylated in vivo. While several options exist for biotinylation of proteins (Cronan 1990; Chen et al. 2007), we chose to utilize the well-characterized biotin acceptor peptide (AP) and Escherichia coli biotin ligase, BirA. BirA catalyzes robust biotinylation of the diminutive 15 amino acid AP when the enzyme is coexpressed in the cell (Schatz 1993; Beckett et al. 1999; van Werven and Timmers 2006). We predicted that fusion of the AP to the SNAP protein would permit biotinylation of SNAPAP-tagged proteins in the presence of BirA (Chen et al. 2007). The SNAPAP-tagged proteins could then be subsequently labeled with fluorophores, immobilized on a surface, and imaged (Fig. 1A).
FIGURE 1.

Overview and characterization of SNAP-SiMPull. (A) Schematized workflow of a SNAP-SiMPull experiment. A yeast strain is constructed containing a SNAPAP-derivitized protein. A whole-cell lysate is then prepared, labeled with a benzylguanine fluorophore, and passed through a gel filtration column before application to a glass slide. This results in immobilization of complexes containing the SNAP-tagged protein. (SA) Streptavidin, (B) biotin. (B) Streptavidin EMSA to determine the extent of biotinylation of the SNAPAP tag. The BBP protein was derivitized with a fluorophore prior to SDS-PAGE and fluorescence imaging. Addition of streptavidin causes a decrease in gel mobility indicative of biotinylation of the SNAPAP tag. Asterisk indicates the presence of higher-order streptavidin oligomers binding to fluorescent BBP-SNAPAP. (C) Cartoon representation of DY649-labeled BBP-SNAPAP pull-down on the surface of a passivated glass slide. (D) Representative microscopic fields of view of a SNAP-SiMPull assay with DY649 fluorophore-labeled BBP-SNAPAP. Pull-down is strongly dependent on the presence of streptavidin. Scale bar is 5 μm. (E) Quantification of the number of DY649 BBP-SNAPAP spots observed in the presence and absence of streptavidin coating from representative experiments. (F) Representative single-molecule trace highlighting single step photobleaching for immobilized BBP-SNAPAP molecules. (G) Quantification of the number of observed photobleaching steps for spots of BBP-SNAPAP fluorescence. The vast majority of spots photobleach in a single step consistent with the presence of single molecules of DY649 BBP-SNAPAP on the surface.
As an initial test, we generated an endogenously expressed fusion of the branchpoint bridging protein (BBP)/Msl5p to the SNAPAP tag in Saccharomyces cerevisiae by homologous recombination. BBP is an essential yeast protein and strains containing BBP-SNAPAP fusions exhibited wild-type growth (Supplemental Table S1). Plasmid-borne E. coli BirA was constitutively expressed in this strain in order to biotinylate the SNAPAP tag in vivo. Following extract preparation, fluorescent SNAP dye was added to the extract and specific labeling of the BBP-SNAPAP tag was observed (Supplemental Fig. S1). To test for biotinylation of the SNAPAP tag, we performed an electrophoretic mobility shift assay (EMSA) using streptavidin. Upon incubation of the labeled extract with streptavidin, a supershift was observed for the resulting BBP-SNAPAP/streptavidin complex (Fig 1B). The addition of streptavidin caused a quantitative shift in the fluorescence band indicating that the BBP-SNAPAP protein in the lysate was biotinylated and the biotin was accessible to streptavidin. We found that neither addition of excess biotin to the growth media nor addition of a nuclear localization signal to BirA were essential for biotin transfer to the nuclear BBP-SNAPAP protein (data not shown).
We next tested if the biotinylated BBP-SNAPAP protein could be pulled-down from cell lysate and captured on a streptavidin-coated glass surface (Fig. 1C). DY649 fluorophore-labeled BBP-SNAPAP cell extract (see Materials and Methods) was diluted to 40% under conditions typically used to carry out in vitro splicing assays (Hoskins et al. 2011a). The diluted extract was then flowed over a slide that had been previously passivated with a polyethylene glycol (PEG) and derivitized with streptavidin. Using a total internal reflection fluorescence (TIRF) microscope with laser excitation of DY649 at 633 nm, we observed rapid accumulation of red fluorescent spots on the surface. After spot accumulation, the slide was extensively washed with buffer containing oxygen scavengers and immediately imaged (Fig. 1D). Importantly, spot accumulation was dependent on the presence of streptavidin (Fig. 1D,E). The vast majority of spots (∼96%) that accumulated on the surface vanished in single steps after prolonged excitation, consistent with these fluorescent spots originating from single molecules of BBP-SNAPAP (Fig. 1F,G). Together these results indicate that yeast SNAPAP-tagged proteins can be efficiently biotinylated in vivo, immobilized on a streptavidin-coated slide directly from cell extract, and imaged at the single-molecule level.
SNAP-SiMPull analysis of RNA binding by BBP
We next tested if the isolated BBP-SNAPAP molecules were capable of binding RNA. It has previously been shown that BBP associates specifically with RNAs containing a UACUAAC sequence (Berglund et al. 1997). When a solution containing a 3′ Cy3-labeled UACUAAC RNA (MLR001) oligomer was added to the immobilized BBP-SNAPAP molecules, we observed Cy3 fluorescent spots appear at the surface upon excitation at 532 nm (Fig. 2A,B). These Cy3-labeled RNAs colocalized with a subset of the DY649-labeled BBP-SNAPAP molecules (on average 32%) and appeared transiently. BBP-SNAPAP molecules repeatedly bound and released the UACUAAC RNA during data collection (5 min) (Fig. 2B). When a control RNA (MLR002) oligomer that lacked the UACUAAC sequence was added, very few Cy3 spots appeared that could be colocalized with BBP-SNAPAP. Those spots that did colocalize departed very quickly from the surface—often lasting for just a single frame (dwell time ≤500 msec) (Fig. 2C,D). Likewise, very few interactions between the UACUAAC oligo and the surface were observed in the absence of BBP-SNAPAP pull-down (Fig. 2E) and unlabeled UACUAAC oligo competed with the Cy3-labeled oligo for BBP-SNAPAP binding (data not shown). Finally, we observed an increasing number of BBP-SNAPAP/UACUAAC oligo interactions as the concentration of the oligo in solution increased (Supplemental Fig. S3). These results are consistent with specific and reversible binding of UACUAAC-containing RNAs to immobilized BBP-SNAPAP molecules.
FIGURE 2.
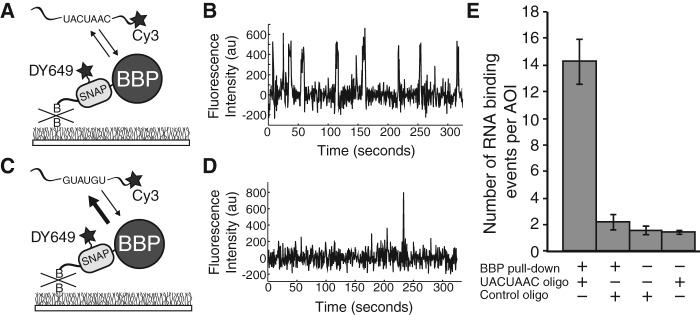
SNAP-SiMPull characterization of interactions between RNAs and immobilized BBP (A) Cartoon representation of the single-molecule assay for immobilized DY649 BBP-SNAPAP binding to a Cy3-labeled oligo containing the UACUAAC sequence. (B) Representative Cy3 fluorescence trace illustrating many binding events of Cy3-labeled RNAs to a single molecule of BBP-SNAPAP. (C) Cartoon representation of the single-molecule assay for immobilized DY649 BBP-SNAPAP binding to a Cy3-labeled oligo that does not contain a branchsite sequence (control oligo). (D) Representative Cy3 fluorescence trace illustrating few binding events of Cy3-labeled RNAs lacking a branchsite sequence to BBP-SNAPAP. (E) Quantification of the appearance of Cy3-RNA fluorescent spots in the presence and absence of immobilized BBP-SNAPAP. Error bars were calculated using the standard error of the mean. In the absence of BBP-SNAPAP pull-down, approximately sevenfold fewer Cy3-RNA spots appeared in randomly selected areas of interest (AOIs).
A detailed kinetic analysis of UACUAAC binding by BBP has not been carried out until now. By measuring the dwell times of the binding events for the UACUAAC oligo to immobilized BBP-SNAPAP, we were able to calculate an off rate for the interaction. The measured dwell times could be fit well to a single exponential distribution described by koff = 0.40 ± 0.02 sec−1 (Supplemental Fig. S2). This result indicates that the UACUAAC RNA oligo is dissociating by a single pathway from the BBP-SNAPAP molecules and that likely a single type of BBP-SNAPAP/RNA complex is being formed on these surface-immobilized molecules. These results provide an example of how SNAP-SiMPull can be used to characterize activity in vitro following in situ purification.
Isolation of the U1 snRNP using SNAP-SiMPull
Given our success at SNAP-SiMPull analysis of BBP, we next sought to immobilize larger, multicomponent complexes such as the spliceosomal snRNPs. We reasoned that these complexes could be identified and distinguished from individual proteins by colocalization of multiple fluorophores fused to different snRNP components by multiwavelength TIRF microscopy. To this end, we first sought to immobilize the yeast U1 snRNP. Since U1 has been previously isolated by a number of methods (Rigaut et al. 1999; Puig et al. 2001), we predicted that it should be amenable to SNAP-SiMPull.
We incorporated the SNAPAP tag onto the U1 protein Prp40 and an orthogonal CLIP tag onto the protein Snu71. The CLIP tag reacts with benzylcytosine fluorophores (Gautier et al. 2008), and thus could be simultaneously labeled in the presence of the SNAP tag without appearance of cross-labeling (Supplemental Fig. S1A). We confirmed fluorescent labeling efficiency of the SNAPAP and CLIP tags by a time course assay using unlabeled SNAP and CLIP substrates to quench the labeling reaction. We found that after 45 min of simultaneous labeling at room temperature, Prp40-SNAPAP was ∼99% labeled and Snu71-CLIP was ∼95% labeled (Supplemental Fig. S1B). Importantly, the doubly labeled U1 extract also retained in vitro splicing activity (Supplemental Fig. S1C).
Once labeled, U1 extract was flowed over a streptavidin-coated slide, Prp40-SNAPAP complexes were captured, and the complexes imaged. We observed both green (DY547) and red (DY649) fluorescent spots upon excitation indicating the presence of both Snu71-CLIP and immobilized Prp40-SNAPAP, respectively (Fig. 3A). To assess colocalization, a mapping file to relate the green and red images was created using fluorescent beads as fiducial markers. The locations of the Prp40-SNAPAP molecules were then translated to the Snu71-CLIP image and vice versa. As expected, we found Snu71 and Prp40 to be highly colocalized (Fig. 3B). Replicate experiments showed that 82% ± 3% of Snu71-CLIP molecules colocalized with Prp40-SNAPAP and 77% ± 1% of Prp40-SNAPAP molecules colocalized with Snu71-CLIP. These values are slightly lower than our maximum predicted extent of colocalization based on labeling efficiencies (∼94% colocalization being the product of the extent of labeling for each tag) (Supplemental Fig. S1B). The discrepancy could likely stem from the presence of dark fluorophores, protein complex heterogeneity, or a small population of incomplete U1 snRNPs.
FIGURE 3.

SNAP-SiMPull analysis of U1 snRNP components. (A) Representative microscopic fields of view of U1 snRNP components: Snu71-CLIP and Prp40-SNAPAP. The individual images were false-colored (Snu71-CLIP green and Prp40-SNAPAP red) and superimposed without correction for differences in magnification (overlay). Scale bars are 5 μm. (B) Quantification of colocalization of Snu71-CLIP spots with Prp40-SNAPAP spots (dark blue) and vice versa (light blue) after mapping of the spots between each field of view after correction for differences in magnification. Error was calculated using the standard error of the mean for three replicate experiments. (C) Representative microscopic fields of view between two additional U1 snRNP components, Snp1-CLIP and Prp40-SNAPAP. Scale bars are 5 μm. (D) Quantification of the colocalization between Snp1-CLIP and Prp40-SNAPAP. Error was calculated using the standard error of the mean for three replicate experiments. (E) Representative microscopic fields of view of SNAP-SiMPull of the U1 snRNP components, Snp1-CLIP and Prp40-SNAPAP following extract treatment with 50 µg of RNase A for 30 min at room temperature. (F) Quantification of the colocalization between Snu71/Prp40 and Snp1/Prp40 following RNase treatment of the extract.
We next analyzed the colocalization of Prp40 to another U1 snRNP protein, Snp1. As before Prp40-SNAPAP was immobilized on the slide surface and labeled with a red fluorophore. Snp1 was labeled with a green fluorophore using the CLIP tag. In contrast with Prp40 and Snu71, Prp40 and Snp1 showed a lower degree of colocalization (Fig. 3C,D). Only 41% ± 3% of Prp40 molecules colocalized with Snp1, suggesting that many Prp40 molecules were not fully incorporated into complete U1 snRNPs. These results are unlikely due to poor labeling kinetics since SDS-PAGE analysis showed >90% of proteins capable of incorporating a fluorophore were labeled in these experiments (data not shown). Instead these results suggest either the presence of a subpopulation of Prp40-SNAPAP or Snp1-CLIP-proteins that cannot be labeled (e.g., due to protein misfolding) or heterogeneous protein complexes.
While the results above are consistent with isolation of U1 snRNPs, they do not distinguish between protein-only complexes and those containing both the snRNA and protein. To test whether or not the snRNA impacted the observed colocalization, we carried out SNAP-SiMPull assays of both Prp40/Snu71 and Prp40/Snp1 after first degrading the snRNA by RNase treatment. RNase completely abolished our ability to pull-down Snp1 using Prp40-SNAPAP (Fig. 3E,F). This indicates that colocalization of these two proteins is dependent on the snRNA. Further, this likely means that the 35% of Snp1 molecules that did not colocalize with Prp40 when the snRNA was intact (Fig. 3D) were bound to either unlabeled or photobleached Prp40-SNAPAP/snRNA complexes.
RNase treatment did not impact our ability to colocalize Prp40 and Snu71 (Fig. 3F). This is consistent with pull-down of these two proteins being predominated by protein–protein interactions rather than the snRNA. Thus, while colocalization of Snp1 with Prp40 is indicative of the presence of the snRNA and (potentially) the intact U1 snRNP the same cannot be said for colocalized Prp40/Snu71 complexes. These results are in agreement with yeast two-hybrid assays showing an interaction between the FF domains of Prp40 and Snu71 (Ester and Uetz 2008). Our colocalization data further support the presence of a protein-only subcomplex of the U1 snRNP that contains at minimum Prp40 and Snu71 as been previously suggested (Ester and Uetz 2008).
SNAP-SiMPull analysis of the U6 snRNP and Prp24/Lsm complexes
During splicing several snRNPs undergo changes in abundance and composition due to formation and disassembly of spliceosomal complexes. This has best been documented with the U4/U6.U5 tri-snRNP that is disassembled during spliceosomal activation or in the presence of ATP (Raghunathan and Guthrie 1998b; Stevens et al. 2001). We sought to determine if SNAP-SiMPull could be used to detect changes in abundance of the free U6 snRNP in extract. U6 contains the fewest number of proteins of any spliceosomal snRNP including just the 112-nucleotide U6 snRNA, the Prp24 protein, and the heptameric Lsm ring (Shannon and Guthrie 1991; Stevens et al. 2001). Prp24 is thought to be specific only to the U6 snRNP (Stevens et al. 2001) while the Lsm2-8 ring is found in the U6 snRNP, the U4/U6 di-snRNP, the U4/U6.U5 tri-snRNP and fully assembled B-complex spliceosomes (Achsel et al. 1999; Gottschalk et al. 1999; Mayes et al. 1999; Stevens and Abelson 1999). Based on our results with U1, we predicted that U6 snRNPs could be specifically isolated and identified using a red-labeled SNAPAP tag on Prp24 and a green-labeled CLIP tag on the U6-specific Lsm protein, Lsm8 (Fig. 4A). To our knowledge, functional interaction between Lsm8 and the U6 snRNA in the absence of the other Lsm proteins has not been demonstrated and is unlikely based on the structure of the complex (Zhou et al. 2014). Thus, the presence of Lsm8 and the U6 snRNA likely indicates presence of a Lsm ring. As with the U1 snRNP, we determined efficiency of dual labeling to be ∼93% for Prp24-SNAPAP and ∼91% for Lsm8-CLIP after 45 min of labeling (Supplemental Fig. S1D). Additionally, we found that labeled U6 snRNP extract was active for in vitro splicing (Supplemental Fig. S1C).
FIGURE 4.
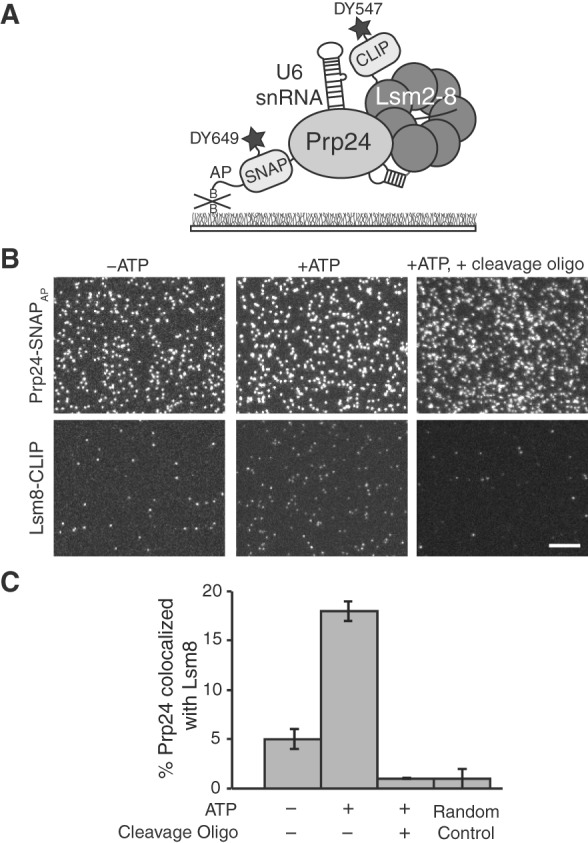
SNAP-SiMPull analysis of U6 snRNP components. (A) Illustration of a SNAP-SiMPull experiment involving colocalization of DY649-labeled Prp24-SNAPAP and DY549-labeled Lsm8-CLIP. (B) Representative microscopic fields of view from a U6 SNAP-SiMPull experiment. Lsm8-CLIP and Prp24-SNAPAP were imaged simultaneously using a dual-view apparatus either in the presence or absence of ATP (left and center) or in the presence of ATP and after RNase H-directed cleavage of the U6 snRNA (right). Scale bar is 10 μm and all images are of identical magnification. (C) Quantification of colocalization of Prp24-SNAPAP and Lsm8-CLIP from fields of view of identical dimensions. ATP causes a large increase in the extent of colocalization, while all colocalization is lost after cleavage of the U6 snRNA. The Random Control bar represents the probability of a Prp24-SNAPAP spot of fluorescence colocalizing with a Lsm8-CLIP spot of fluorescence in the absence of genuine complexes being present. Error bars represent the standard error of the mean derived from three independent experiments.
In the absence of ATP, pull-down of Prp24-SNAPAP revealed a large number of red Prp24-SNAPAP spots but only a few green spots of Lsm8-CLIP (Fig. 4B). Only 5% ± 1% of the Prp24-SNAPAP molecules were found to colocalize with Lsm8-CLIP spots. This value approached our lower limit of detection of complexes based on the random probability of two spots colocalizing (1% ± 1%). This result reveals that in yeast WCE in the absence of ATP most Prp24-SNAPAP proteins are not present in complexes with Lsm8-CLIP. It has previously been shown that addition of ATP to extract in the absence of splicing results in tri-snRNP disassembly and accumulation of U6 (Raghunathan and Guthrie 1998a,b). Upon incubation of the extract with 2 mM ATP, we observe a large increase in the number of Lsm8-CLIP spots that appear on the surface and approximately fourfold increase in the number of Prp24-SNAPAP spots that colocalize with Lsm8 (18% ± 1%) (Fig. 4B,C). These results are consistent with ATP-dependent accumulation of U6 containing both Prp24 and Lsm8 on the surface.
It is not known if stable interactions between Prp24 and the Lsm ring are dependent on the U6 snRNA. To test this, we carried out SNAP-SiMPull assays in the presence of ATP but after oligo-directed RNaseH ablation of the U6-snRNA. The oligo (MLR003) promoted cleavage of the central domain of the U6 snRNA (nucleotides 28–54; Fabrizio et al. 1989) and resulted in complete loss of splicing activity from the extract (Supplemental Fig. S4). Based on recent structures of Prp24 bound to a fragment of the U6 snRNA (Montemayor et al. 2014), we predicted that cleavage of the snRNA at this position would result in disruption of the Prp24/U6 complex. Therefore, if the snRNA is necessary for a stable interaction between Prp24 and the Lsm ring, SNAP-SiMPull would detect a decrease in the amount of Lsm8-CLIP isolated with Prp24-SNAPAP in the presence of ATP. In agreement with our prediction, we observed a complete loss in colocalization of Prp24-SNAPAP and Lsm8-CLIP after U6 snRNA cleavage (Fig. 4B,C). It should be noted that these experiments do not report on the fraction of Prp24-SNAPAP proteins bound to the U6 snRNA but not containing Lsm8. In control experiments, RNaseH ablation of the 5′ end of the U1 snRNA did not appear to disrupt the Snu71/Prp40 complex consistent with previous evidence for U1 snRNP complexes after ablation and/or the presence of protein-only complexes (Supplemental Fig. S5; Du and Rosbash 2001). We conclude that an intact U6 snRNA is required for SNAP-SiMPull of Lsm8 with Prp24 and that the complete U6 snRNP containing Prp24, the Lsm ring, and the snRNA is being isolated in these experiments.
DISCUSSION
Pull-down assays from unfractionated cell lysates are important biochemical tools for studying molecular interactions and activities. Typically these assays are carried out using antibodies to immunopurify biomolecules on a solid support or by engineering an affinity tag (e.g., a hexahistidine or GST tag) into the biomolecule to facilitate its isolation. The SiMPull approach developed by Jain et al. merges immunopurification with quantitative fluorescence imaging to provide a rapid and sensitive characterization of complexes isolated from cell lysates (Jain et al. 2011). Additionally, this can be used for in situ purification of molecules for functional assays using single-molecule methods.
SiMPull assays can be complicated by a number of factors. First, immunoprecipitation of biomolecules relies on maintaining the integrity of the antibody/antigen complex during the assay. This is particularly problematic if reducing agents such as dithiothreitol (DTT) are needed to maintain assembly or function of the biomolecule. These reducing agents can greatly interfere with antibody function by reduction of the disulfide bonds holding the heavy and light chains of the antibody together. Secondly, reliance on fluorescence proteins is not ideal for many types of experiments. Fluorescent proteins vary greatly in their extent of maturation, maturation time, brightness, and photostability (Shaner et al. 2005). For example, YFP and mCherry, which have previously been used for SiMPull, only mature to extents of 75% and 40%, respectively, at 37°C (Ulbrich and Isacoff 2007, 2008; Jain et al. 2011). The variable extent of maturation complicates analysis of protein stoichiometry and colocalization assays of protein complexes in SiMPull. The long, oxygen-dependent maturation times of many fluorescent proteins (Khmelinskii et al. 2012; Macdonald et al. 2012) also preclude their use in studying transiently expressed proteins or expression under anaerobic conditions since the FP may fail to mature and remain “dark” in the SiMPull assay. Finally, the photostability and brightness of the proteins themselves can also limit functional assays since blinking or rapid photobleaching of the FP (Dickson et al. 1997; Kubitscheck et al. 2000; Hendrix et al. 2008) may complicate analysis of complexes isolated by SiMPull.
As an alternative approach, the SNAP-SiMPull tag overcomes many of the limitations imposed by immunoprecipitation of the FPs. The SNAP tag is smaller than FPs (19.4 kDa versus 27 kDa for GFP) and has been used extensively as an alternative to FPs (Chen et al. 2013). In order to adapt the SNAP tag for SiMPull, we fused the SNAP gene to the biotin AP and showed that this tag is readily biotinylated in cells following coexpression with the E. coli biotin ligase. While other options for biotinylation of yeast proteins are available (Chen et al. 2007), the biotin AP has been used in many systems, is readily translated to other organisms, and is only 15 amino acids in length. Both the SNAP and AP tags are amenable to amino- or carboxy-terminal labeling highlighting the versatility of the combined SNAPAP tag. Additionally, fusion of the SNAPAP tag did not significantly interfere with protein function as all strains used in this study exhibited wild-type growth (Supplemental Table S1). The biotinylated SNAPAP protein can be easily pulled-down from cell lysate onto streptavidin-coated slides for imaging. Streptavidin immobilization is widely used for single-molecule experiments and is compatible with a range of buffers including those containing high concentrations of DTT or β-mercaptoethanol.
Most importantly, the SNAP-SiMPull tag allows single molecules to be imaged using organic fluorophores that overcome many of the drawbacks of fluorescent proteins. Organic fluorophores have superior photophysical properties than fluorescent proteins, do not require maturation, and easily permit long observations of biomolecules in cell extracts (Hoskins et al. 2011a). Moreover, the extents of labeling of the SNAP and CLIP tags are quite high, comparable with or exceeding the maturation extent of YFP (≥90%, Supplemental Fig. S1; Sun et al. 2011). Finally, SNAP-SiMPull tags allow the fluorescent properties of the tagged molecule (e.g., the wavelength of emission) to be altered by addition of differently colored fluorophores to the cell lysate rather than by expression of different protein-FP fusions. This is particularly advantageous in cases where different FP fusions have been shown to impact protein function in different ways such as mislocalization of RFP but not GFP-tagged proteins (Lee et al. 2013).
We have used the SNAP-SiMPull tag to isolate both BBP and U1 and U6 snRNPs from yeast cell lysate. In all cases, endogenous complexes were isolated by integration of the SNAP-SiMPull tag into the yeast genome and without protein overexpression. In the case of BBP, we were able to show that a subset of the molecules could interact specifically with UACUAAC-containing RNAs and determined a lifetime for this interaction. Biochemical studies of BBP have been limited since only a fragment of the protein has been previously isolated by recombinant expression and purification (Berglund et al. 1997). Using SNAP-SiMPull, we were able to rapidly isolate full-length BBP molecules from yeast cell lysate and provide the first kinetic description of RNA binding by BBP. Interestingly, only a subset of BBP molecules were capable of interacting with the RNA substrate. While this could be due to the impact of the SNAPAP tag or surface immobilization, it could also indicate novel regulatory features of BBP. For example, the presence of interacting partners or post-translational modification of BBP, such as at the seven predicted phosphorylation sites (Bodenmiller et al. 2010), could impact BBP's RNA-binding ability and result in mixed populations of BBP. These experiments clearly set the stage for more detailed investigations into RNA-binding mechanisms by BBP.
In our studies of the U1 and U6 snRNPs, we combined SNAP-SiMPull with multiwavelength colocalization experiments to identify complexes by the presence of differently colored fluorophores. Using SNAPAP and CLIP tags on U1 proteins, we demonstrated a very high degree of colocalization (∼80%) between Prp40 and Snu71. However, this colocalization was not dependent on the snRNA and could indicate a mixture of both U1 snRNPs and protein-only subcomplexes. In contrast, the proteins Prp40 and Snp1 showed a lower degree of colocalization (∼40%), but this was strictly dependent on the presence of the snRNA. This suggests that colocalization of Prp40 and Snp1 can be used to detect single molecules of the intact U1 snRNP. Because of the high amount of labeling, no significant corrective factors were needed to determine the preceding colocalization values. This is in contrast to use of YFP and mCherry FPs in which incomplete fluorophore maturation leads to a maximum possible colocalization of ∼30% for monomeric proteins.
Building upon the robustness and ease of SNAP-SiMPull analysis of U1 complexes, we next isolated the U6 snRNP. Importantly, we were able to demonstrate that SNAP-SiMPull could be used to isolate complexes that transiently accumulate in extract—in this case U6 snRNPs that formed due to destabilization of the tri-snRNP upon the addition of ATP but had not yet been incorporated back into U4/U6 di-snRNPs for tri-snRNP reassembly. To achieve this, we incorporated the SNAPAP tag onto a U6 snRNP-specific protein, Prp24, and identified snRNPs by colocalization with the Lsm8 protein. The colocalization of Lsm8 with Prp24 was dependent on the presence of an intact U6 snRNA, providing evidence that the snRNA can be required for stable Lsm8/Prp24 interactions. These data complement previous yeast two-hybrid assays that detected an interaction between the carboxy-terminal tail of Prp24 (the “SNFFL” box) and Lsm proteins (Fromont-Racine et al. 2000; Rader and Guthrie 2002). Our data suggest that these RNA-independent Prp24/Lsm8 interactions may be weak, which in turn could contribute to Prp24's absence from isolated U4/U6 di-snRNPs and tri-snRNPs (Gottschalk et al. 1999; Stevens and Abelson 1999; Stevens et al. 2001). Finally, these data support a model for U6 snRNP formation that does not involve binding of the snRNA to a preformed Prp24/Lsm ring complex since such complexes are rarely observed in the absence of ATP in our assay (Fig. 4B,C). It is likely that U6 snRNP formation can be driven by independent, high affinity (10−8–10−9 M) interactions (Kwan and Brow 2005) between the Lsm ring and Prp24 with the U6 snRNA. This is consistent with the SNFFL-box being nonessential in vivo (Rader and Guthrie 2002) and in agreement with a previously proposed mechanism for U6 snRNP assembly based upon U6 snRNA mutagenesis (Ryan et al. 2002). SNFFL-box/Lsm ring interactions may serve to optimize U6 snRNP formation (Ryan et al. 2002), facilitate nuclear localization of Prp24, or may have other roles in the cell.
In conclusion, the SNAP-SiMPull approach represents a significant advance in single-molecule pull-down methods since it makes use of bright organic fluorophores as well as high affinity interactions between streptavidin and in vivo biotinylated molecules. While we have concentrated these studies on dual-color experiments, this approach can likely be extended to three or more colors and components with other tagging methods (e.g., the Halotag) or by splitting the SNAPAP tag into its SNAP and AP components. We predict that in situ purification of molecules bearing bright, organic fluorophores by SNAP-SiMPull will present a viable and facile method for obtaining low abundance complexes for single-molecule studies. SNAP-SiMPull will prove particularly useful for studying processes, like spliceosome assembly, that occur over tens of minutes in vitro in which the use of long lifetime, nonblinking organic fluorophores is beneficial.
MATERIALS AND METHODS
Plasmids, yeast transformation and growth assays
A double stranded DNA fragment encoding the SNAPf (Juillerat et al. 2003; Sun et al. 2011) protein fused to the AP sequence (GLNDIFEAQKIEWHW) (van Werven and Timmers 2006) was purchased from GENEWIZ. The fragment was digested with BamHI and HindIII restriction enzymes and ligated into those same sites of the plasmid pAG32 (Euroscarf) that also encodes a hygromycin selectable marker to create plasmid pAAH0204. The E. coli birA gene was subcloned from pBTac2 (Chen et al. [2005]; courtesy of Alice Ting, Massachusetts Institute of Technology), digested with NheI and NcoI, and ligated into those same sites of the yeast expression vector pVTU260 (Melcher 2000) to generate plasmid pAAH0290. Plasmids have been deposited in Addgene.
SNAPAP and CLIP tagging of spliceosomal proteins were carried out by homologous recombination as described previously (Shcherbakova et al. 2013). Following SNAPAP and CLIP tagging, pAAH0290 was transformed into competent yeast prepared by the lithium acetate method (Gietz and Woods 2002) and selected for on –URA dropout media. DNA primers and yeast strains used for this study are summarized in Supplemental Tables S2 and S3, respectively.
To measure the growth rate of the five strains used in this study (Supplemental Table S1), three replicate 50 mL cultures were inoculated in YPD from a saturated 5 mL overnight culture to an optical density (OD600) of 0.1. Cultures of each strain were grown at 30°C over a period of 14 h. Hourly time points were taken to monitor growth by measuring the OD600. Growth data were plotted and fit to an exponential growth curve as described previously to calculate doubling times (Hoskins et al. 2011a). Doubling times are reported as an average of replicate growth experiments and the error was calculated using the standard error from these replicates (Supplemental Table S1).
Yeast extract preparation
Yeast extracts were prepared as described previously (Ansari and Schwer 1995; Crawford et al. 2008) except that strains transformed with the pAAH0290 were grown in –URA dropout media.
SNAP and CLIP fluorophore labeling
Aliquots of (1.2 mL) cell extract were thawed at room temperature and labeled using 1 μM SNAP dye and 2 μM CLIP dye for 45 min at room temperature protected from light. For each extract, the SNAP tag was labeled with SNAP-DY649 and the CLIP tag was labeled with CLIP-DY547 (New England Biolabs). Immediately following the labeling reaction, extract was applied to gel filtration column to remove excess dye as previously described (Hoskins et al. 2011a). Five fractions (0.5 mL) were taken and each assayed for fluorescent labeling by SDS-PAGE. Fractions containing the highest amount of fluorescently labeled protein, were aliquoted (40 µL), frozen in liquid N2, and stored at −80°C until use. Labeling efficiency of the SNAP or CLIP-proteins was determined as previously described (Hoskins et al. 2011a) except that CLIP-protein containing samples were quenched by addition of excess CLIP Cell Block (New England Biolabs).
Determination of the extent of Biotinylation of the SNAPAP tag
Streptavidin (50 µg in 5 µL; Prozyme) was incubated with 20–40 µL of labeled yeast whole-cell extract for 10 min at room temperature. Samples were then diluted 1:4 in H2O followed by addition of an equivalent volume of colorless Laemelli buffer prior to SDS-PAGE. The samples were not heat denatured prior to loading. Gels were subsequently imaged using a GE LAS4010 and SNAPAP tag fluorescence quantified with ImageQuant software.
Slide preparation for SNAP-SiMPull
SNAP-SiMPull assays were carried using either glass or quartz microscope slides constructed as previously described (Friedman et al. 2006; Roy et al. 2008). For passivation, all slides were treated with Vectabond (Vector Labs) for 10 min and washed thoroughly with ethanol. Activated slides were passivated overnight at room temperature with a 100:1 mixture of PEG/PEG-biotin (LaysanBio) to facilitate immobilization and reduce nonspecific binding.
SNAP-SiMPull colocalization experiments
Prior to sample application, the slides were flushed with buffer A (25 mM potassium phosphate pH 7.3, 3% PEG 8000, 1 mM DTT, 1.5 mM MgCl2, 10% glycerol, 25 mM HEPES KOH pH 7.9, 50 mM KCl) to remove unreacted PEG/PEG-biotin. Thereafter, flow chambers were incubated with 10 µg/mL streptavidin (Prozyme) for 2 min and then washed with buffer A. Experiments were carried out by first diluting the extract to 40% (v/v) in buffer B (40 mM potassium phosphate pH 7.3, 5% PEG 8000, 2 mM DTT, 3 mM MgCl2) to recapitulate in vitro splicing conditions. In all experiments, extract was allowed to equilibrate in buffer B for 5–10 min at room temperature while protected from light. For U1 snRNA digestion experiments, 50 μg RNaseA was added to 40 µL undiluted extract and allowed to incubate for 30 min at room temperature. In experiments containing ATP, ATP (2 mM) was added to the diluted extract before incubation at room temperature (10 min). For ablation experiments, the ablation oligos (Supplemental Table S5) were added (to final concentration of 3 μM for MLR003; Fabrizio et al. 1989 and 75 μg of MLR004; Du and Rosbash 2001) to the diluted extract and allowed to incubate at 30°C for 30 min prior to application to the slide. Ablation was confirmed by primer extension analysis. Diluted extract (20–40 µL) was applied to the slide and immediately washed with 200 µL buffer C (buffer A + 450 µg/µL glucose, 40 units glucose oxidase, 2 units catalase, and 2 mM Trolox). Typical fluorescent spot densities ranged from 0.07 to 0.12 spots/µm2, and data were only analyzed if individual spots could be resolved and were well separated from one another. For unknown reasons, some extracts prepared from strain yAAH0327 showed increased numbers of immobilized fluorescent spots if the extract was first briefly incubated with ATP.
Images were collected continuously using a 500 msec or 1 sec exposures. Images collected from samples applied to glass slides were collected on a micromirror (mm) TIRF microscope with excitation from 532 to 633 nm lasers at powers of 100–300 µW. Images collected from samples applied to quartz slides were collected on a prism TIRF microscope with excitation from 532 to 640 nm lasers at powers of 1–10 mW.
Single-molecule RNA-binding experiments
RNA oligomers containing a 3′ Cy3 were purchased from IDT (Supplemental Table S5; MLR001 and MLR002). All single-molecule binding experiments were performed on the mmTIRF microscope with glass slides. Slide preparation was the same except lanes were treated with yeast tRNA (10 µg/mL; Sigma) following streptavidin coating to reduce nonspecific surface binding of the RNA. Extract containing labeled BBP-SNAPAP was applied to the slide and immediately washed with buffer C. Lanes were then imaged to confirm efficient protein immobilization. Buffer C containing 1 nM fluorescent oligomer was then flowed into the imaging chamber. Images were then collected continuously with an exposure time of 500 msec for ∼5 min. BBP-SNAPAP spots were imaged periodically (∼50 sec) to correct for any microscope drift.
Single-molecule data analysis
All images were analyzed using custom software implemented in MATLAB (The Mathworks). A mapping function that mathematically relates each field-of-view in the microscope images was created for each experiment by determining the locations of surface-immobilized fluorescent beads that appeared in both emission channels (< and >635 nm). Using this mapping function, single molecule locations were selected individually in one emission channel, mapped onto the other channel, and then analyzed for colocalization with any fluorescent spots that were present.
For off-rate analysis, RNA-binding events to individual BBP molecules were selected as previously described (Hoskins et al. 2011a). Dwell times for RNA-binding events were binned and a probability density plot was constructed by dividing by the bin width and total number of observations. Error in the number of events per bin was calculated from the variance of a binomial distribution as previously described (Hoskins et al. 2011a). To determine an off-rate for the BBP/RNA interaction, the unbinned data were fit with a maximum likelihood algorithm to a single exponential function in which the probability of an event with length t is given by Equation 1 where tmin represents the time between consecutive frames, tmax is the maximum detectable dwell time, τ (the fitted parameter) is the off-rate constant, and t is the measured dwell times. Error in the fit was determined by bootstrapping 1000 random samples of the data and determining the standard deviation of the resultant values.
| (1) |
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge support from startup funding from the University of Wisconsin-Madison, Wisconsin Alumni Research Foundation (WARF), and the Department of Biochemistry. A.A.H., M.L.R., and J.P. are also supported by a K99/R00 award from the National Institutes of Health (R00 GM086471), the Arnold and Mabel Beckman Foundation, and the Shaw Scientist Program of the Greater Milwaukee Foundation. M.L.R. is supported by the Molecular Biophysics Training Program (NIH T32-GM08293). We thank members of the Hoskins laboratory as well as David Brow, Samuel Butcher, Allison Didychuk, and Eric Montemayor for helpful discussions.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.047845.114.
REFERENCES
- Abelson J, Blanco M, Ditzler MA, Fuller F, Aravamudhan P, Wood M, Villa T, Ryan DE, Pleiss JA, Maeder C, et al. 2012. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat Struct Mol Biol 17: 504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achsel T, Brahms H, Kastner B, Bachi A, Wilm M, Lührmann R 1999. A doughnut-shaped heteromer of human Sm-like proteins binds to the 3′-end of U6 snRNA, thereby facilitating U4/U6 duplex formation in vitro. EMBO J 18: 5789–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A, Schwer B 1995. SLU7 and a novel activity, SSF1, act during the PRP16-dependent step of yeast pre-mRNA splicing. EMBO J 14: 4001–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett D, Kovaleva E, Schatz PJ 1999. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci 8: 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund JA, Chua K, Abovich N, Reed R, Rosbash M 1997. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 89: 781–787. [DOI] [PubMed] [Google Scholar]
- Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O, et al. 2010. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal 3: rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Howarth M, Lin W, Ting AY 2005. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat Methods 2: 99–104. [DOI] [PubMed] [Google Scholar]
- Chen I, Choi YA, Ting AY 2007. Phage display evolution of a peptide substrate for yeast biotin ligase and application to two-color quantum dot labeling of cell surface proteins. J Am Chem Soc 129: 6619–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Cornish VW, Min W 2013. Chemical tags: inspiration for advanced imaging techniques. Curr Opin Chem Biol 17: 637–643. [DOI] [PubMed] [Google Scholar]
- Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ 2008. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA 14: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE 1990. Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J Biol Chem 265: 10327–10333. [PubMed] [Google Scholar]
- Dickson RM, Cubitt AB, Tsien RY, Moerner WE 1997. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 388: 355–358. [DOI] [PubMed] [Google Scholar]
- Du H, Rosbash M 2001. Yeast U1 snRNP–pre-mRNA complex formation without U1snRNA–pre-mRNA base pairing. RNA 7: 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester C, Uetz P 2008. The FF domains of yeast U1 snRNP protein Prp40 mediate interactions with Luc7 and Snu71. BMC Biochem 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, McPheeters DS, Abelson J 1989. In vitro assembly of yeast U6 snRNP: a functional assay. Genes Dev 3: 2137–2150. [DOI] [PubMed] [Google Scholar]
- Friedman LJ, Chung J, Gelles J 2006. Viewing dynamic assembly of molecular complexes by multi-wavelength single-molecule fluorescence. Biophys J 91: 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromont-Racine MM, Mayes AE, Brunet-Simon A, Rain JC, Colley A, Dix I, Decourty L, Joly N, Ricard F, Beggs JD, et al. 2000. Genome-wide protein interaction screens reveal functional networks involving Sm-like proteins. Yeast 17: 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Corrêa IR Jr, Kindermann M, Beaufils F, Johnsson K 2008. An engineered protein tag for multiprotein labeling in living cells. Chem Biol 15: 128–136. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350: 87–96. [DOI] [PubMed] [Google Scholar]
- Gottschalk A, Neubauer G, Banroques J, Mann M, Lührmann R, Fabrizio P 1999. Identification by mass spectrometry and functional analysis of novel proteins of the yeast [U4/U6.U5] tri-snRNP. EMBO J 18: 4535–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix J, Flors C, Dedecker P, Hofkens J, Engelborghs Y 2008. Dark states in monomeric red fluorescent proteins studied by fluorescence correlation and single molecule spectroscopy. Biophys J 94: 4103–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ 2011a. Ordered and dynamic assembly of single spliceosomes. Science 331: 1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Gelles J, Moore MJ 2011b. New insights into the spliceosome by single molecule fluorescence microscopy. Curr Opin Chem Biol 15: 864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Liu R, Ramani B, Arauz E, Ishitsuka Y, Ragunathan K, Park J, Chen J, Xiang YK, Ha T 2011. Probing cellular protein complexes using single-molecule pull-down. Nature 473: 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, Johnsson K 2003. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem Biol 10: 313–317. [DOI] [PubMed] [Google Scholar]
- Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, Kaufmann A, Trautmann S, Wachsmuth M, Pereira G, et al. 2012. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol 30: 708–714. [DOI] [PubMed] [Google Scholar]
- Konarska MM, Sharp PA 1988. Association of U2, U4, U5, and U6 small nuclear ribonucleoproteins in a spliceosome-type complex in absence of precursor RNA. Proc Natl Acad Sci 85: 5459–5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R, Blanco MR, Kahlscheuer ML, Abelson J, Guthrie C, Walter NG 2013. Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nat Struct Mol Biol 20: 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitscheck U, Kückmann O, Kues T, Peters R 2000. Imaging and tracking of single GFP molecules in solution. Biophys J 78: 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan SS, Brow DA 2005. The N- and C-terminal RNA recognition motifs of splicing factor Prp24 have distinct functions in U6 RNA binding. RNA 11: 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lim WA, Thorn KS 2013. Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One 8: e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang W, Xu T, Ramsey J, Zhang L, Hill R, Hansen KC, Hesselberth JR, Zhao R 2013. Comprehensive in vivo RNA-binding site analyses reveal a role of Prp8 in spliceosomal assembly. Nucleic Acids Res 41: 3805–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald PJ, Chen Y, Mueller JD 2012. Chromophore maturation and fluorescence fluctuation spectroscopy of fluorescent proteins in a cell-free expression system. Anal Biochem 421: 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes AE, Verdone L, Legrain P, Beggs JD 1999. Characterization of Sm-like proteins in yeast and their association with U6 snRNA. EMBO J 18: 4321–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher K 2000. A modular set of prokaryotic and eukaryotic expression vectors. Anal Biochem 277: 109–120. [DOI] [PubMed] [Google Scholar]
- Montemayor EJ, Curran EC, Liao HH, Andrews KL, Treba CN, Butcher SE, Brow DA 2014. Core structure of the U6 small nuclear ribonucleoprotein at 1.7-Å resolution. Nat Struct Mol Biol 21: 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24: 218–229. [DOI] [PubMed] [Google Scholar]
- Rader SD, Guthrie C 2002. A conserved Lsm-interaction motif in Prp24 required for efficient U4/U6 di-snRNP formation. RNA 8: 1378–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C 1998a. A spliceosomal recycling factor that reanneals U4 and U6 small nuclear ribonucleoprotein particles. Science 279: 857–860. [DOI] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C 1998b. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol 8: 847–855. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032. [DOI] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T 2008. A practical guide to single-molecule FRET. Nat Methods 5: 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DE, Stevens SW, Abelson J 2002. The 5′ and 3′ domains of yeast U6 snRNA: Lsm proteins facilitate binding of Prp24 protein to the U6 telestem region. RNA 8: 1011–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz PJ 1993. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology (NY) 11: 1138–1143. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY 2005. A guide to choosing fluorescent proteins. Nat Methods 2: 905–909. [DOI] [PubMed] [Google Scholar]
- Shannon KW, Guthrie C 1991. Suppressors of a U4 snRNA mutation define a novel U6 snRNP protein with RNA-binding motifs. Genes Dev 5: 773–785. [DOI] [PubMed] [Google Scholar]
- Shcherbakova I, Hoskins AA, Friedman LJ, Serebrov V, Corrêa IR, Xu M-Q, Gelles J, Moore MJ 2013. Alternative spliceosome assembly pathways revealed by single-molecule fluorescence microscopy. Cell Rep 5: 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SW, Abelson J 1999. Purification of the yeast U4/U6.U5 small nuclear ribonucleoprotein particle and identification of its proteins. Proc Natl Acad Sci 96: 7226–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SW, Barta I, Ge HY, Moore RE, Young MK, Lee TD, Abelson J 2001. Biochemical and genetic analyses of the U5, U6, and U4/U6 × U5 small nuclear ribonucleoproteins from Saccharomyces cerevisiae. RNA 7: 1543–1553. [PMC free article] [PubMed] [Google Scholar]
- Stevens SW, Ryan DE, Ge HY, Moore RE, Young MK, Lee TD, Abelson J 2002. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol Cell 9: 31–44. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhang A, Baker B, Sun L, Howard A, Buswell J, Maurel D, Masharina A, Johnsson K, Noren CJ, et al. 2011. Development of SNAP-tag fluorogenic probes for wash-free fluorescence imaging. ChemBioChem 12: 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY 2007. Subunit counting in membrane-bound proteins. Nat Methods 4: 319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY 2008. Rules of engagement for NMDA receptor subunits. Proc Natl Acad Sci 105: 14163–14168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Werven FJ, Timmers HTM 2006. The use of biotin tagging in Saccharomyces cerevisiae improves the sensitivity of chromatin immunoprecipitation. Nucleic Acids Res 34: e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, Newnham CM, Kameoka S, Huang T, Konarska MM, Query CC 2004. Prp5 bridges U1 and U2 snRNPs and enables stable U2 snRNP association with intron RNA. EMBO J 23: 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Hang J, Zhou Y, Wan R, Lu G, Yin P, Yan C, Shi Y 2014. Crystal structures of the Lsm complex bound to the 3′ end sequence of U6 small nuclear RNA. Nature 506: 116–120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.