

Background: Selective inhibitors of the chemokine receptor CCR5, a co-receptor for HIV, can inhibit HIV infection.
Results: Inhibitory activity of the selective CCR5 inhibitor Schering C can be conferred to CCR2b by a single site mutation, R206I, in CCR2b.
Conclusion: Exploration of the inhibitor binding site reveals that targeting Ile198 contributes to drug selectivity for CCR5.
Significance: Understanding receptor/drug interactions facilitates drug design.
Keywords: CC Chemokine Receptor Type 5 (CCR5), Drug Design, Human Immunodeficiency Virus (HIV), Molecular Modeling, Site-directed Mutagenesis, CC Chemokine Receptor Type 2b (CCR2b)
Abstract
The chemokine receptors CCR5 and CCR2b share 89% amino acid homology. CCR5 is a co-receptor for HIV and CCR5 antagonists have been investigated as inhibitors of HIV infection. We describe the use of two CCR5 antagonists, Schering-C (SCH-C), which is specific for CCR5, and TAK-779, a dual inhibitor of CCR5 and CCR2b, to probe the CCR5 inhibitor binding site using CCR5/CCR2b chimeric receptors. Compound inhibition in the different chimeras was assessed by inhibition of chemokine-induced calcium flux. SCH-C inhibited RANTES (regulated on activation, normal T cell expressed and secreted) (CCL5)-mediated calcium flux on CCR5 with an IC50 of 22.8 nm but was inactive against monocyte chemoattractant protein-1 (CCL2)-mediated calcium flux on CCR2b. However, SCH-C inhibited CCL2-induced calcium flux against a CCR5/CCR2b chimera consisting of transmembrane domains IV–VI of CCR5 with an IC50 of 55 nm. A sequence comparison of CCR5 and CCR2b identified a divergent amino acid sequence located at the junction of transmembrane domain V and second extracellular loop. Transfer of the CCR5 sequence KNFQTLKIV into CCR2b conferred SCH-C inhibition (IC50 of 122 nm) into the predominantly CCR2b chimera. Furthermore, a single substitution, R206I, conferred partial but significant inhibition (IC50 of 1023 nm) by SCH-C. These results show that a limited amino acid sequence is responsible for SCH-C specificity to CCR5, and we propose a model showing the interaction with CCR5 Ile198.
Introduction
The chemokine receptor family is the largest subfamily of peptide-binding G-protein-coupled receptors. Chemokines, the ligands for these receptors, are 8–10-kilodalton proteins defined by the number and relative spacing of cysteine residues at the N-terminal end of the protein. The two major families are CC and CXC in which there are two cysteine residues that are either adjacent (CC) or separated by one amino acid residue (CXC). They are mediators of hematopoiesis and inflammation, regulating lymphocyte development, homing, and trafficking (1–3). The chemokine receptors CXCR42 and CCR5 are of pharmacological importance because they are used by HIV-1 as co-receptors to enter CD4+ cells (4, 5). Specifically, interaction of CCR5 with the CD4-activated gp120 subunit of HIV-1 envelope glycoproteins is essential for the viral entry of CCR5-using or R5 virus. CCR5 is the principal co-receptor for viral transmission in the early, clinically latent stage of the disease, whereas CXCR4 usage emerges in the later stages and is associated with a decrease in CD4 cell count and accelerated disease progression (6).
The inhibition of viral entry by inhibition of the viral gp120/chemokine co-receptor interaction is an attractive target for the design of novel anti-HIV drugs. Inhibition of CXCR4 was validated as a target in a Phase II clinical trial using the CXCR4 antagonist plerixafor (AMD3100) (7). Inhibition of CXCR4 by plerixafor also leads to leukocytosis and mobilization of hematopoietic stem cells from the bone marrow, and plerixafor was subsequently approved for hematopoietic stem cell mobilization for autologous transplantation in patients with non-Hodgkin lymphoma and multiple myeloma (8–10). A second generation oral CXCR4 antagonist, AMD11070 (11), subsequently entered clinical trial in HIV-infected patients harboring X4 virus (12).
Several CCR5 inhibitors have entered clinical trials including maraviroc, aplaviroc, vicriviroc, Schering C (SCH-C), and TAK-779 (5). Most of these compounds encountered hurdles during clinical development; however, maraviroc was successfully approved by the United States Food and Drug Administration for use in treatment-experienced patients in August 2007 and for use in treatment-naïve patients with CCR5-using virus in November 2009 (13–15).
The molecular pharmacology and mechanism of these chemokine receptor inhibitors have been extensively studied. Plerixafor and AMD11070 have been shown to interact with three positively charged amino acids, Asp171, Asp262, and Glu288, in transmembrane domain (TM)-IV and TM-VI of CXCR4 (16–18), whereas the binding motif for CCR5 inhibitors has been largely defined by two hydrophobic pockets defined by TM-I, TM-II (Leu33, Tyr37, Phe79, and Trp86), TM-III, TM-V, and TM-VI regions (Thr105, Tyr108, Phe109, Ile198, and Tyr251), which flank a polar center Glu283. Specific interactions for each individual molecule have been further defined by a combination of site-directed mutagenesis and molecular modeling (19–25). There is debate over the role of certain of these interactions. For example, Ile198 in CCR5 has been implicated in the binding of aplaviroc, TAK-779, SCH-C, and maraviroc to CCR5. Earlier studies suggested that this amino acid had an indirect effect on compound binding and was too distant from the compound to be within the binding domain but acted to stabilize the receptor structure (24–26). However, more recent studies including the determination of the crystal structure of maraviroc binding to CCR5 have suggested that Ile198 can directly interact with a variety of inhibitors and play an integral role in compound binding (21, 23, 27–29). We have used SCH-C, a selective antagonist of CCR5, and TAK-779 (30), a dual antagonist of CCR5 and CCR2b (Fig. 1), to further probe the role of Ile198.
FIGURE 1.
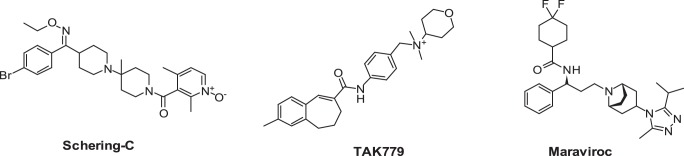
Structures of the CCR5 inhibitors maraviroc and Schering C and the CCR5/CCR2b dual inhibitor TAK-779.
SCH-C was chosen to investigate the role of Ile198 as this compound specifically inhibits human CCR5 but not macaque CCR5 (26). The sequence of the latter is highly conserved compared with human CCR5 with only a difference of nine residues, one of which is the substitution of methionine for Ile198 (Fig. 2A). Interestingly, the replacement of methionine at position 198 in macaque CCR5 by the natural human CCR5 residue isoleucine conferred inhibition by SCH-C to the macaque CCR5 (26).
FIGURE 2.

Amino acid sequence alignment of the chemokine receptors CCR5 and CCR2b. A, alignment of human and macaque CCR5. The amino acids that differ between the two species are shown in bold. Ile198 (human) and Arg206 (macaque) are circled. B, alignment of human CCR5 and CCR2b.TM-I–TM-VII are shown in boxes.
CCR5 and CCR2b are the two most closely related receptors in the CC chemokine receptor family with an overall homology of 89% amino acid homology, in the transmembrane region (Fig. 2B). CCR2b is expressed in monocytes and recognizes the ligands CCL2 (MCP-1), CCL8 (MCP-2), CCL7 (MCP-3), and CCL13 (MCP-4) (31, 32), whereas CCR5 is expressed on T cells and recognizes the ligands CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) (33–35). The homology between CCR5 and CCR2b has been exploited to explore the regions of CCR5 necessary for chemokine specificity and binding and for HIV-1 infectivity by engineering chimeras of the two receptors using a “centaur” approach in which the N terminus of the chimera is derived from one receptor and the C-terminus is derived from the second receptor (36–38). Recently a “Trojan horse” domain-related approach was described in which the intracellular domains were derived from one receptor and the extracellular domains were derived from the second receptor (39). These CCR2/CCR5 chimeras were used to explore allosteric agonist and antagonist binding sites on CCR5.
Despite the high homology between CCR5 and CCR2b, SCH-C is a potent inhibitor of CCR5 but is completely inactive toward CCR2b. We therefore adopted a combination of the two approaches described above to investigate the structure of the binding domain conferring specificity of SCH-C to CCR5. We used chimeras of decreasing size to create a “minidomain chimera” which allowed us to create and identify a nine-amino acid sequence (KNFQTLKIV) in the near extracellular region of TM-V of CCR5. Upon transfer of this domain into a CCR2b background, SCH-C was able to inhibit the response to the CCR2 ligand CCL2. The sequence of the CCR5 domain in the “minichimera” contains Ile198. In CCR2b, this position is occupied by arginine (Arg206). We hence created a single site mutation, R206I, and we were subsequently able to demonstrate inhibition of this mutant CCR2b by SCH-C. With these data obtained using this novel chimera approach with the CCR5 receptor expressed in a natural environment and utilizing the published crystal structure of maraviroc bound to an engineered CCR5 construct, we confirmed the importance of the direct interaction of Ile198 for CCR5 inhibition and selectivity over CCR2b and propose a model of SCH-C binding to CCR5.
EXPERIMENTAL PROCEDURES
Construction of Plasmids Encoding Wild-type Human and Macaque CCR5, Mutant CCR5, and CCR5/CCR2b Chimeric Receptors
The coding regions of human CCR5 and CCR2b were amplified by polymerase chain reaction (PCR) using genomic DNA and cloned into the pcDNA3.1 TOPO directional vector (Invitrogen). The following primers were used: R5F1 (5′-CACCATGGATTATCAAGTGTCA-3′) and R5R1 (5′-TCCGTGTCACAAGCCCACAGA-3′) for CCR5 and R2bF1 (5′-CACCATGCTGTCCACATCTCGTTC-3′) and R2bR1 (5′-GTTTTATAAACCAGCCGAGAC-3′) for CCR2b. Macaque CCR5 was PCR-cloned from genomic DNA isolated from cynomolgus buffy coat using primers 5′-CACCATGGATTATCAAGTGTCAAGTCCAACCTATGAC-3′) and 5′-TCACAAGCCCACAGATATTTCCTGCTC-3′ into the pcDNA3.1 TOPO directional vector as described above.
Three CCR5/CCR2b chimeric receptors were produced: 2252, 2255, and 2b199–207R5 (Fig. 3A). The 2252 and 2255 chimeric constructs were prepared based on published procedures (36) adopting a restriction-enzyme based approach that makes use of the conserved restriction sites in CCR5 and CCR2b. Appropriate restriction fragments (BamHI and ClaI for 2252 and EcoRI and ClaI for 2255) of CCR5 and CCR2b were ligated to create the desired constructs. Chimera 2b199–207R5 was created using a PCR-based strategy. Briefly, the two desired CCR2b fragments (I and II) were amplified by Pfu Turbo DNA polymerase (Stratagene) using the pcDNA3.1.CCR2b construct as the template such that each fragment contained an overlapping CCR5 sequence that codes for a stretch of eight amino acids at the beginning of TM-V. Primers used were: CCR2b fragment I, R2bF1 (5′-CACCATGCTGTCCACATCTCGTTC-3′) and 2b199–207R5-R1 (5′-GACTATCTTTAATGTCTGGAAATTCTTCCATCCTCGTGGAAAATAAG-3′); CCR2b fragment II, 2b199–207R5-F1 (5′-GACTATCTTTAATGTCTGGAAATTCTTCCATCCTCGTGGAAAATAAG-3′) and R2bR1 (5′-GTTTTATAAACCAGCCGAGAC-3′). The gel-purified fragments were then combined and subjected to one PCR cycle (denaturation at 94 °C for 4 min, annealing at 50 °C for 2 min, and extension at 72 °C for 10 min), allowing the hybridization between the top strand of fragment I and bottom strand of fragment II. Extension of 3′-ends from both strands yielded the recombinant product, which was then used as a template for the final PCR of the chimeric receptor. The PCR product was gel-purified and cloned into the pcDNA3.1 TOPO directional vector (Invitrogen). The point mutation R206I in the CCR2b receptor was prepared using the Stratagene QuikChange site-directed mutagenesis kit (Stratagene, Inc.) according to the manufacturer's instructions. Sequences of receptor constructs were confirmed by DNA sequence analysis.
FIGURE 3.

Generation of CCR5/CCR2b receptor chimeras. A, schematic structure of chimeras 2252, 2255, and 2b199–207R5; the CCR5 component of the chimera is shown in black. B, expression of the chimeric receptor 2252 in HEK293F cells by flow cytometry. The chimera is recognized by antibodies specific for ECL2 of CCR5 (2D7) and CCR2b but not by the antibody to the N terminus of CCR5 (3A9).
Expression in Cell Lines
Human embryonic kidney (HEK) 293F cells (3 × 107 cells plated on a 15-cm dish the day prior to the day of transfection) were grown in Dulbecco's modified Eagle's medium (HyClone Inc.) supplemented with 10% fetal bovine serum (Invitrogen), 1 mm sodium pyruvate (HyClone Inc.), 1 mm non-essential amino acids (HyClone Inc.), and 4 mm l-glutamine (HyClone Inc.). The CCR5, CCR2b, and CCR2b/CCR5 chimeric and mutant constructs (60 μg) were transfected transiently into HEK293F cells with Gqi5-HA.pcDNA (30 μg) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Cells were split 1:2 the next day, and cell surface expression levels were assessed 48 h after transfection by flow cytometry using the phycoerythrin-conjugated 2D7 monoclonal antibody to CCR5 that recognizes the second extracellular loop and 3A9, which recognizes the N terminus of CCR5 (Pharmingen) and phycoerythrin-conjugated anti-human CCR2 antibody (R&D Systems).
Chemokine Receptor-mediated Intracellular Ca2+ Flux
HEK293F transient transfectants of chimeric and mutant receptors were assayed for intracellular Ca2+ changes in response to chemokine induction. Cells were washed and resuspended (5 × 106 cells/ml) in serum-reduced medium (DMEM containing 2% fetal calf serum) and loaded with the fluorescence probe Fluo-4/AM (4 μm) (Molecular Probes, Inc., Eugene, OR) for 30 min at 37 °C in the dark. The dye-loaded cells were subsequently washed twice with Hanks' balanced salt solution in 20 mm HEPES, 0.2% BSA, 2.5 mm probenecid, pH 7.4. The cells were resuspended in the same buffer (4 × 106 cells/ml) followed by a 20-min incubation in the dark at room temperature. Cells were pre-equilibrated in a tissue culture 96-well clear bottom plate (BD Biosciences) with either Hanks' balanced salt solution or varying concentrations of chemokine receptor inhibitors for 15 min at 37 °C. Changes in intracellular calcium concentration upon chemokine addition (RANTES (CCL5) for CCR5 and MCP-1 (CCL2) for CCR2b) were monitored using the FLEXstationTM (Molecular Devices, Sunnyvale, CA) at 525 nm (excitation λ, 485 nm). Data were analyzed using the programs Softmax® PRO 4.3.1 (Molecular Devices) and GraphPad Prism® 3.0 Software (San Diego, CA).
125I-RANTES Ligand Binding Assay
The 125I-RANTES binding assay was performed with membranes prepared from HEK293F cells expressing wild-type CCR5 or mutant CCR5 as described previously (22). Briefly, membranes were incubated with 125I-RANTES (50 μl of 50 pm; specific activity, 2,200 Ci/mmol) and compound over a concentration range of 1 × 10−5–6.4 × 10−10 m (final assay volume, 150 μl) for 45 min at room temperature. The plates were washed with ice-cold 50 mm HEPES, 0.5 m NaCl, pH 7.4; dried; and counted on a Wallac 1450 Microbeta Jet liquid scintillation counter. Ligand binding dose-response curves were analyzed, and IC50 values were calculated using Prism 3.0.
CCR5/gp120 Cell Fusion Assay
A cell-cell fusion assay was used to mimic the first stage of the HIV infection process. The principle of the assay is the fusion of one cell line expressing the HIV-1 JRFL viral envelope protein and the Tat transcription factor (CHO-Tat) with a second cell line (HeLa-CD4-LTR-β-gal) expressing CD4, CCR5, and the LacZ gene under the control of the HIV-1 LTR promoter (22). Briefly, HeLa-CD4-LTR-β-gal cells were transiently transfected with either wild-type or mutant CCR5 for 24 or 48 h. After transfection, the CCR5-expressing cells were incubated with CHO-Tat cells in the presence of test compound for 20 h at 37 °C in 5% CO2 and then assayed for β-galactosidase activity using the Gal-Screen homogenous chemiluminescent reporter gene assay (Applied Biosystems). The plates were read on a Victor 2 plate reader. IC50 values were calculated using non-linear regression curve fitting (ExcelFit 3.0).
Molecular Modeling
For this work, Schrödinger's small molecule drug discovery suite was used (Schrödinger, LLC, New York, NY, 2014: (a) Maestro, version 9.8; (b) Prime, version 3.6; (c) Macromodel, version 10.4; and (d) Glide, version 6.3. The crystal structure of CCR5 with the small molecule antagonist maraviroc was downloaded from the Research Collaboratory for Structural Bioinformatics (Protein Data Bank code 4MBS) and prepared within Maestro as recommended for use with Glide (40–42). All water molecules were removed. For all docking calculations, the standard precision mode of Glide was used. If not mentioned otherwise the receptor structure was kept rigid during docking calculations. In a first step to assess the docking algorithm, the binding mode of maraviroc was reproduced using an arbitrarily generated ligand conformation (Fig. 4A). After successful method evaluation, next SCH-C was docked into the binding site of maraviroc. An example how one of its docking poses aligns with that of maraviroc is shown in Fig. 4B. Similar mutagenesis data for both compounds and the generally accepted view that small molecules bind beneath the ECL2 of chemokine receptors support this assumption (19–27). The docking of TAK-779 was less straightforward. Because Thr284 is sterically not accessible for ligand binding, the double mutant Y37A/Q280A was used first for modeling efforts. Based on mutagenesis results, TAK-779 was manually placed into the binding site by allowing the oxygen of tetrahydropyran to form a hydrogen bond interaction with Thr284 and geometry-optimized while keeping the receptor atoms rigid. The optimized conformation of TAK-779 was placed into the wild-type CCR5, and steric clashes were removed by first visual inspection and then geometry optimization of the small molecule in the context of a rigid receptor. In a final step, the ligand-receptor complex was refined by using Prime. This protocol allows for conformational changes of side chain atoms and to a minor degree of back bone atoms. The Ramachandran plot of this somewhat geometrically modified receptor shows no dihedral violations (data not shown).
FIGURE 4.
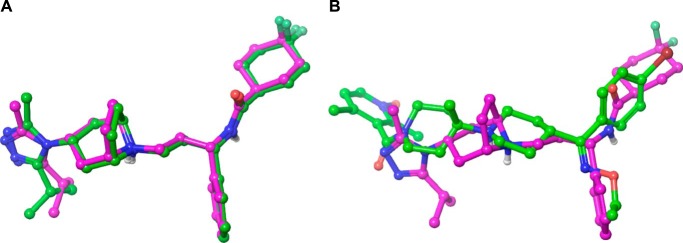
Automatic docking of small molecules to CCR5. Computationally derived docking modes are shown with carbons in green; the binding mode obtained by crystallography is shown with carbons in magenta. A, successful method evaluation as self-docking reproduces the crystal binding mode of maraviroc to CCR5. For clarity, only the small molecules are shown as the receptor was kept rigid in the docking process. B, aligned docking poses of maraviroc and SCH-C.
RESULTS
Schering C Is a Potent Inhibitor of Human CCR5
SCH-C was developed as a specific inhibitor of human CCR5. We confirmed the specific inhibition of human CCR5 in human CCR5-expressing HEK293F cells. SCH-C inhibited RANTES-mediated calcium flux with an IC50 of 4.96 ± 0.61 nm, whereas it was unable to inhibit MCP-1-mediated calcium flux in HEK293F cells expressing CCR2b (Table 1). By comparison, the CCR2b/CCR5 dual inhibitor TAK-779 was able to inhibit calcium flux in both CCR5- and CCR2b-expressing cells (IC50 values of 5.9 ± 1.0 and <1.6 nm, respectively). We further confirmed SCH-C selectivity for the human CCR5 as SCH-C was able to inhibit the human CCR5/gp120 interaction but was unable to inhibit the interaction between macaque CCR5 and gp120 in a cell fusion assay, whereas TAK-779 was able to inhibit both (Table 2).
TABLE 1.
Inhibition of chemokine receptor-mediated calcium flux by SCH-C and TAK-779
Results are expressed as IC50 (nm) ± S.E.
| CCR5 | CCR2b | |
|---|---|---|
| SCH-C | 4.96 ± 0.61 (n = 9) | >25,000 (n = 3) |
| TAK-779 | 5.9 ± 1.0 (n = 4) | <1.6 (n = 3) |
TABLE 2.
Inhibition of CCR5/gp120-mediated cell fusion by SCH-C and TAK-779
Cells were transiently transfected with either human or simian CCR5 for 24 h. Results are expressed as IC50 (nm).
| Human | Macaque | |
|---|---|---|
| SCH-C | 15.6 | >10,000 |
| TAK-779 | 59 | 341.5 |
Although CCR5 is highly conserved between human and macaque, the substitution of Ile198 in human CCR5 for methionine in macaque CCR5 has been shown to be an important determinant for lack of SCH-C inhibition against the simian receptor. The significance of this residue was confirmed by the attenuation of both the inhibition of RANTES binding to human CCR5 and CCR5/gp120-mediated cell fusion by both I198A and I198M single site mutations. Neither of these mutations affected inhibition by TAK-779 (Table 3). We hence adopted a chimeric receptor approach to further investigate the role of Ile198 in the CCR5 binding of the SCH-C antagonist.
TABLE 3.
Effect of mutating Ile198 of human CCR5 on inhibition of RANTES ligand binding and CCR5/gp120 cell fusion by SCH-C and TAK-779
Results are expressed as IC50 (nm) ± S.E.
| RANTES binding |
Cell fusion |
|||||
|---|---|---|---|---|---|---|
| CCR5 WT | I198A | I198M | CCR5 WT | I198A | I198M | |
| SCH-C | 43.0 ± 2.7 (n = 9) | 396.7 ± 121.7 (n = 3) | 911.3 ± 61.5 (n = 34) | 3.7 ± 0.2 (n = 71) | 350.0 ± 64.0 (n = 9) | 1277.4 ± 102.2 (n = 44) |
| TAK-779 | 6.7 | 6.9 (n = 1) | 9.2 ± 3.67 (n = 3) | 20.8 ± 2.7 (n = 9) | 15.2 | 13.4 ± 2.1 (n = 14) |
Expression of Wild-type and Chimeric Receptors
To initially define the major regions of inhibitor/receptor interaction on CCR5 required to confer inhibition, CCR2b/CCR5 chimeras were prepared. The construction of two of the chimeras made use of common EcoRI and ClaI restriction endonuclease sites in regions conserved between CCR2b and CCR5 (36). One of these, the 2252 chimera, has a CCR2b background construct with the second intracellular loop to TM-III region substituted with the corresponding CCR5 sequence. The second construct, the 2255 chimera, is made up primarily of CCR5 with the N terminus to the end of TM-III replaced with the corresponding region of CCR2b (Fig. 3A).
These constructs were transiently expressed in HEK293F cells, and their surface expression was confirmed by flow cytometry (an example flow cytometry analysis for 2252 is shown in Fig. 3B). As predicted, expression of both 2252 and 2255 chimeras could only be recognized by antibodies specific for ECL2 (2D7) but not the N terminus (3A9) of CCR5. Both chimeras were recognized by the CCR2 antibody.
Response of the CCR2b/CCR5 2252 and 2255 Chimeras to Chemokine Agonist Stimulation and SCH-C Inhibition
The functionality of these chimeras was investigated by their ability to mobilize intracellular calcium in response to RANTES and MCP-1. The chimeric and wild-type receptors were transiently expressed in HEK293F cells together with the chimeric G protein (Gqi5) to enhance the calcium response (43–45). The wild-type CCR5 responded to RANTES with an EC50 of 2.16 ± 1.21 nm, whereas wild-type CCR2b responded to MCP-1 with an EC50 of 2.73 ± 0.69 nm (Table 4). The chimera 2255 responded only to RANTES (EC50 of 1.4 ± 0.5 nm), whereas the 2252 chimera responded to both RANTES and MCP-1 (EC50 values of 3.44 ± 1.54 and 9.22 ± 1.88 nm, respectively) (Fig. 5, A and B).
TABLE 4.
Activity of CCR5, CCR2b, and receptor chimeras; agonist response to chemokines MCP-1/CCL2 and RANTES/CCL5 (EC50); and inhibition by SCH-C (IC50)
NR, no response; NT, not tested.
| Receptor | Chemokine EC50 |
SCH-C IC50 |
||
|---|---|---|---|---|
| MCP-1/CCL2 | RANTES/CCL5 | MCP-1/CCL2 | RANTES/CCL5 | |
| nm | nm | |||
| CCR5 | NR | 2.16 ± 1.21 | NT | 20.57 ± 4.68 |
| 2255 | NR | 1.4 ± 0.5 | NT | 248 ± 37 |
| 2252 | 9.22 ± 1.88 | 3.44 ± 1.54 | 55.33 ± 18.37 | 113.67 ± 31.75 |
| 2b199–207R5 | 3.62 ± 0.85 | NR | 122 ± 15.01 | NT |
| CCR2b R206I | 3.75 ± 1.68 | NR | 1023.00 ± 167.03 | NT |
| CCR2b | 2.73 ± 0.69 | NR | >25,000 | NT |
FIGURE 5.

Response of the CCR5/CCR2b receptor chimeras to chemokines and inhibition by SCH-C. A, chemokine-mediated calcium flux response to the CCR5 chemokine RANTES by 2252, 2255, and wild-type CCR5. B, chemokine-mediated calcium flux response to the CCR2b chemokine MCP-1 by the chimera 2252 and wild-type CCR2b. C, inhibition of the receptor chimeras 2252 and 2255 and wild-type receptors CCR2b and CCR5 by the CCR5 antagonist SCH-C. The chimeras and CCR5 were stimulated with RANTES, and CCR2b was stimulated with MCP-1. Error bars represent S.E. RFU, relative fluorescence units.
Next we examined the inhibition of MCP-1- and RANTES-induced calcium mobilization in the 2252 chimera by SCH-C (Fig. 5C). Interestingly, we found that SCH-C inhibited the calcium flux induced by MCP-1 and RANTES with very similar IC50 values of 92 and 86 nm, respectively, indicating that SCH-C can bind to and antagonize the 2252 signaling induced by either ligand. Thus, one or more specific features included in the region spanning from TM-IV to TM-VI must be required for SCH-C activity.
TM-V of CCR5 Gives Specificity of SCH-C Antagonism on CCR5
Because we succeeded in obtaining significant SCH-C inhibition by inserting the CCR5 sequence spanning from intracellular loop 2 to TM-VI into a CCR2b background, we proceeded to focus further on the features associated with this gain of function. Comparison of the amino acid sequences reveals that most of the TM-IV to TM-VI regions of CCR5 and CCR2b are well conserved except for a stretch of nine amino acids (KNFQTLKIV) in the near extracellular section of TM-V (Fig. 2B). Moreover, two reports have shown that within this region conservation of Ile198 is essential for the anti-HIV-1 activity of SCH-C (23, 25). To investigate further whether this region could be important for the action of SCH-C on the CCR5 receptor, a chimera on the CCR2b background bearing a short region of CCR5 (KNFQTLKIV) was created by a PCR overlap strategy (Fig. 6A).
FIGURE 6.

A short N-terminal portion of TM-V confers inhibition to CCR5 antagonist SCH-C. A, amino acid sequence of the TM-V portion of the 2b199–207R5 receptor chimera. The 191–199 portion of CCR5 (KNFQTLKIV) is inserted at the 199–207 region of CCR2b. B, chemokine-mediated calcium flux response to the CCR2b chemokine MCP-1 by the 2b199–207R5 receptor chimera and the single site R206I CCR2b mutant. C, inhibition of MCP-1-mediated calcium flux of the 2b199–207R5 receptor chimera and the single site R206I CCR2b mutant by the CCR5 antagonist SCH-C. D, inhibition of CCR2b, the 2b199–207R5 receptor chimera, and the single site R206I CCR2b mutant by TAK-779. Error bars represent S.E. RFU, relative fluorescence units.
Using FACS analysis with an anti-CCR2b antibody, this chimera (2b199–207R5) was found to be well expressed, and its functionality was assessed by stimulation of intracellular calcium flux with MCP-1. Fig. 6B showed that 2b199–207R5 responded well to MCP-1 in the calcium flux assay with an EC50 of 4 ± 1 nm, a value similar to that of the wild-type CCR2b control (1.8 ± 0.4 nm). As expected, RANTES did not induce calcium mobilization by 2b199–207R5 (Table 4). Importantly these results showed that this short CCR5 region can replace the corresponding region of CCR2b while retaining specific MCP-1 agonism. Thus, this region seems to bear some essential features that are shared between the two receptors even though their primary sequences are divergent.
Next we tested SCH-C antagonism in 2b199–207R5. Strikingly, the CCR5 antagonist was found to inhibit MCP-1-induced calcium flux with an IC50 of 151 nm, which is only about a 7-fold decrease in potency compared with the effect on wild-type CCR5 (Fig. 6C). This result showed that substitution of this short stretch of nine amino acids in TM-V is able to confer SCH-C inhibition in a CCR2b background. As expected, TAK-779 remained a potent inhibitor of 2b199–207R5 with an IC50 of <1.6 nm with activity comparable with that against the native CCR2b (Fig. 6D). The inhibition of MCP-1-stimulated calcium flux by TAK-779 further confirms the integrity of the 2b199–207R5 chimera.
CCR2b Single Mutant R206I Is Inhibited by SCH-C
The next step was to identify specific residues within the extracellular TM-V region responsible for specificity of SCH-C activity. Based upon the cumulative data from the 2b199–207R5 chimera, CCR5 mutational analysis, and comparison of the CCR5 and CCR2b sequences and the human and macaque CCR5 sequences, we decided to specifically explore the role of Ile198. We therefore converted the corresponding arginine (Arg206) residue of CCR2b into an isoleucine by site-directed mutagenesis. This CCR2b R206I mutant was transiently expressed in the HEK293F cell line and characterized. The mutant was found to be functional by its calcium flux response to MCP-1, generating a dose-response EC50 similar to that of wild-type CCR2b (Table 4 and Fig. 6B). Most significantly, SCH-C inhibited MCP-1-induced calcium flux in CCR2b R206I with an IC50 of 1023 nm, which is about 40-fold above the IC50 of SCH-C for wild-type CCR5 and ∼8-fold higher than the IC50 for the 2b199–207R5 chimera (Fig. 6C). Therefore this single site mutation is able to confer SCH-C inhibition of CCR2b. Again, TAK-779 remained cross-reactive in the R206I mutant with an IC50 of <1.6 nm (Fig. 6D). The inhibition of MCP-1-stimulated calcium flux by TAK-779 further confirms the integrity of the R206I mutant.
Role of Ile198 in the Binding of SCH-C to CCR5
Molecular modeling studies were used to provide rationale on the molecular level for interaction with Ile198. Binding models of TAK-779 and SCH-C with CCR5 (Figs. 7 and 8) were generated taking into account experimental data including crystallographic and mutagenesis data (19–27).
FIGURE 7.
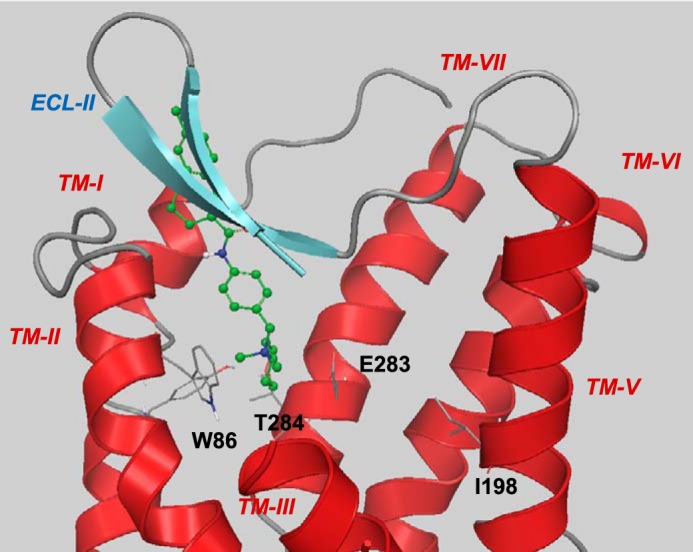
Suggested binding mode of TAK-779. For clarity, TM-IV (residues 158–166) and the extracellular part of TM-III (residues 98–106) are not shown. The tetrahydropyran interacts with Thr284, and the ionic nitrogen interacts with Glu283. The non-polar tail of TAK-779 extends into the extracellular domains of CCR5. Ile198 does not participate in any interaction with TAK-779.
FIGURE 8.

Two docked binding modes of SCH-C in the maraviroc binding site of CCR5. A, the ethyl oxime group points toward the extracellular region, and the bromophenyl group points into the center of the transmembrane domain. B, the oxime and the bromophenyl group are rotated by 180°. In both potential binding modes, SCH-C is able to form a non-polar interaction with Ile198. For clarity, TM-IV and the extracellular part of TM-III (residues 98–106) are not shown.
The I198A and I198M mutations on CCR5 demonstrate the importance of Ile198 for the SCH-C inhibition. Likewise the chimera data and the R206I mutation in CCR2b indicate the essential role of Ile198 in the SCH-C/CCR5 interaction. Conversely neither the I198A and I198M mutations, the minichimera, nor the R206I mutation had any effect on the dual inhibitory action of TAK-779, implying a potentially different mechanism of inhibition and different binding mode (21, 22, 26). Although standard docking would place TAK-779 (data not provided) in the same binding site of maraviroc and SCH-C, the mutagenesis data suggest a binding mode in which TAK-779 does not interact with Ile198 (Fig. 7). The activity loss of TAK-779 upon mutation of Thr284 to alanine as well as the activity loss with the E283A mutation, albeit less pronounced compared with maraviroc and SCH-C, suggests that the oxygen atom of the tetrahydropyran ring forms a hydrogen bond interaction with Thr284 and that the quaternary sterically shielded ammonium ion forms an ionic interaction with Glu283 (46). The hydrophobic tail of TAK-779 reaches toward ECL2 in this model.
All generated docking poses of SCH-C bound to CCR5 are characterized by an ionic interaction of its protonated piperidyl ring with Glu283 as demonstrated by multiple studies. Otherwise the mostly hydrophobic nature of the small molecule interactions with CCR5 allows for several docking solutions. However, in every case, the molecular model supports a direct hydrophobic interaction between Ile198 and SCH-C (Fig. 8, A and B).
DISCUSSION
The chemokine receptor CCR5 is expressed on multiple cell types including Th1 effector and memory T cells, natural killer cells, monocytes, macrophages, immature dendritic cells, and cells of the CNS including neurons, astrocytes, and microglial cells (33–35). CCR5 has thus been implicated in the pathophysiology of numerous inflammatory diseases including rheumatoid arthritis, multiple sclerosis, psoriasis, atherosclerosis, and hepatitis (35, 47, 48). Several studies attempted to link CCR5 to several of these diseases by looking for an association with the CCR5Δ32 deletion, which results in a truncated receptor that does not get expressed on the cell surface (49). Results from these studies are inconsistent and generally have failed to show a relationship with this CCR5 polymorphism. However, CCR5Δ32 individuals are resistant to HIV infection, and following the discovery that CCR5 is one of two chemokine co-receptors involved in viral infection, this has become the therapeutic area that has attracted the most attention (33).
Several laboratories have worked on the development of small molecule antagonists, a number of which have entered clinical trial, culminating in the approval of maraviroc in 2007 (13–15). Structural biology and molecular modeling have played a role in elucidating the mechanism of action and inhibitory binding of these molecules. Several laboratories including our own have shown that the binding site for CCR5 inhibitors generally consists of two hydrophobic binding pockets (21–24). We have further demonstrated how this approach can be used to successfully design novel CCR5 inhibitors with improved inhibitory and absorption, distribution, metabolism, and excretion properties (50). One early prototypical CCR5 inhibitor was SCH-C, and we have used this as a tool molecule to further refine our understanding of the CCR5 inhibitor binding site.
SCH-C is a potent selective inhibitor of CCR5 and has no inhibitory activity against CCR2b, a structurally closely related chemokine receptor (51). In control studies, we confirmed the selective inhibition of CCR5 by SCH-C compared with a lack of activity on CCR2b in contrast to the dual CCR5/CCR2b inhibitor TAK-779 (30). We further confirmed the selectivity of SCH-C for human over simian CCR5. One difference between human and simian CCR5 is the substitution of a methionine for an isoleucine in TM-V in simian CCR5. Using site-directed mutagenesis, we created two human CCR5 mutants, I198A and I198M. In both cases, we showed that the mutations attenuated CCR5 inhibition by SCH-C in a RANTES ligand binding assay and in a cell fusion assay that models the binding of HIV to a host cell, thus confirming the importance of this residue for inhibitor/receptor interaction. Neither mutation had an effect on CCR5 inhibition by TAK-779. On the basis of these data, we further explored the role of TM-V in inhibitor binding.
We adopted a chimeric receptor approach to interrogate the structural determinants of TM-V of CCR5. Initially we created two CCR5/CCR2b chimeric receptors, 2255 and 2252. Whereas 2252 responded to both RANTES (a ligand for CCR5) and MCP-1 (a ligand for CCR2b) as assessed by receptor-stimulated calcium flux, 2255 was only stimulated by RANTES. SCH-C was able to inhibit RANTES-stimulated calcium flux of both chimeric receptors but in addition was able to inhibit MCP-1-stimulated calcium flux of 2252, the chimera incorporating TM-III–TM-V of CCR5 on the CCR2b backbone. These data demonstrated the importance of this region in ligand and inhibitor interaction with CCR5.
The chimeras such as 2252 and 2255 have been described as centaur or sequence-related chimeras. An alternative chimera, the Trojan horse chimera, a domain-related chimera, has been described for CCR2 and CCR5 in which the extracellular domains of CCR5 were replaced by the extracellular domains of CCR2. The Trojan horse chimera retained binding affinity and response to CCR2 chemokines. Interestingly SCH-C was able to inhibit the response to the CCR2 chemokine CCL7 (MCP-3) in the Trojan horse chimera, confirming that the binding site for SCH-C resided in the transmembrane domains of CCR5.
Comparison of the amino acid sequences of CCR5 and CCR2b within the TM-III–TM-V region reveals that most of the TM-IV–TM-VI regions of CCR5 and CCR2b are well conserved except for a stretch of nine amino acids (KNFQTLKIV) in the near extracellular section of TM-V, which also contains the amino acid Ile198. To focus on specific regions within the CCR5 TM-V region, we created a novel minichimera on the CCR2b backbone by substituting the residues 199–207 with the CCR5 sequence KNFQTLKIV. This chimera, 2b199–207R5, consisting of primarily CCR2b responded only to MCP-1 in the calcium flux assay and not RANTES. Interestingly this response could now be inhibited by the CCR5-selective inhibitor SCH-C with an IC50 only 2-fold higher than the inhibition of 2252 calcium flux and 6-fold higher than the inhibition of RANTES-induced calcium flux in wild-type CCR5.
Based on this result, we decided to create a single site change in CCR2b, this time substituting Arg206 for an isoleucine to mimic the Ile198 of human CCR5. Gratifyingly SCH-C was still able to inhibit MCP-1-stimulated calcium flux in this CCR2b R206I mutant albeit with an IC50 ∼8-fold higher than that for the 2b199–207R5 chimera. These data therefore not only demonstrated the necessity of the TM-V region of CCR5 for inhibitor/receptor interaction but clearly showed that a single amino acid residue could confer sensitivity to the CCR5-selective inhibitor SCH-C.
The modeling data suggest that SCH-C can directly interact with Ile198.This is compatible with the binding mode for maraviroc, which like SCH-C interacts with Ile198 as demonstrated by mutagenesis data and confirmed by the crystal structure of CCR5 with bound maraviroc. The chimera data described here provide additional evidence that SCH-C interacts directly with Ile198 in TM-V of CCR5 rather than this amino acid residue acting indirectly by conferring stability to the CCR5 structure. Furthermore the above observations indicate that Ile198 is in part responsible for conferring CCR5 selectivity upon SCH-C. The observation that the IC50 for the 2b199–207R5 chimera is 8-fold lower than that for the R206I CCR2b mutant suggests that other residues within TM-V, specifically within the 191–199 region of the CCR5 sequence, contribute both to inhibitor binding and selectivity.
We have previously shown how a structural biology approach can aid the medicinal chemistry process and compound design (50, 52) specifically for the design of novel inhibitors of CCR5. We have identified two hydrophobic binding regions within TM-I/TM-II and TM-III/TM-V/TM-VI, respectively, and important ionic interactions with Gln283 and Lys26. The data presented herein further define the TM-III/TM-V/TM-VI hydrophobic binding region, demonstrating the role for a direct interaction of CCR5 inhibitors with Ile198. These data, which further refine our understanding of the interaction between CCR5 and selective inhibitors, will prove valuable in the design of novel inhibitors of CCR5.
Acknowledgment
The authors would like to thank Alison Schroeer, Principal Medical Illustrator in Genzyme Biomedical Media Services, for her help in preparing the figures.
Footnotes
- CXCR4
- CXC chemokine receptor type 4
- CCR5
- CC chemokine receptor type 5
- CCR2b
- CC chemokine receptor type 2b
- MCP
- monocyte chemoattractant protein
- RANTES
- regulated on activation, normal T cell expressed and secreted
- SCH-C
- Schering-C
- TM
- transmembrane domain
- ECL2
- second extracellular loop
- HIV-1
- human immunodeficiency virus, type 1
- LTR
- long terminal repeat.
REFERENCES
- 1. Allen S. J., Crown S. E., Handel T. M. (2007) Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820 [DOI] [PubMed] [Google Scholar]
- 2. Viola A., Luster A. D. (2008) Chemokines and their receptors: drug targets in immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 48, 171–197 [DOI] [PubMed] [Google Scholar]
- 3. Zlotnik A., Yoshie O. (2000) Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 [DOI] [PubMed] [Google Scholar]
- 4. Moore J. P., Doms R. W. (2003) The entry of entry inhibitors: a fusion of science and medicine. Proc. Natl. Acad. Sci. U.S.A. 100, 10598–10602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tilton J. C., Doms R. W. (2010) Entry inhibitors in the treatment of HIV-1 infection. Antiviral Res. 85, 91–100 [DOI] [PubMed] [Google Scholar]
- 6. Doranz B. J., Berson J. F., Rucker J., Doms R. W. (1997) Chemokine receptors as fusion cofactors for human immunodeficiency virus type 1 (HIV-1). Immunol. Res. 16, 15–28 [DOI] [PubMed] [Google Scholar]
- 7. Hendrix C. W., Collier A. C., Lederman M. M., Schols D., Pollard R. B., Brown S., Jackson J. B., Coombs R. W., Glesby M. J., Flexner C. W., Bridger G. J., Badel K., MacFarland R. T., Henson G. W., Calandra G. (2004) Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J. Acquir. Immune Defic. Syndr. 37, 1253–1262 [DOI] [PubMed] [Google Scholar]
- 8. Calandra G., Bridger G., Fricker S. (2010) CXCR4 in clinical hematology. Curr. Top. Microbiol. Immunol. 341, 173–191 [DOI] [PubMed] [Google Scholar]
- 9. DiPersio J. F., Micallef I. N., Stiff P. J., Bolwell B. J., Maziarz R. T., Jacobsen E., Nademanee A., McCarty J., Bridger G., Calandra G. (2009) Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin's lymphoma. J. Clin. Oncol. 27, 4767–4773 [DOI] [PubMed] [Google Scholar]
- 10. DiPersio J. F., Stadtmauer E. A., Nademanee A., Micallef I. N., Stiff P. J., Kaufman J. L., Maziarz R. T., Hosing C., Früehauf S., Horwitz M., Cooper D., Bridger G., Calandra G., and 3102 Investigators (2009) Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood 113, 5720–5726 [DOI] [PubMed] [Google Scholar]
- 11. Skerlj R. T., Bridger G. J., Kaller A., McEachern E. J., Crawford J. B., Zhou Y., Atsma B., Langille J., Nan S., Veale D., Wilson T., Harwig C., Hatse S., Princen K., De Clercq E., Schols D. (2010) Discovery of novel small molecule orally bioavailable C-X-C chemokine receptor 4 antagonists that are potent inhibitors of T-tropic (X4) HIV-1 replication. J. Med. Chem. 53, 3376–3388 [DOI] [PubMed] [Google Scholar]
- 12. Moyle G., DeJesus E., Boffito M., Wong R. S., Gibney C., Badel K., MacFarland R., Calandra G., Bridger G., Becker S., and X4 Antagonist Concept Trial Study Team (2009) Proof of activity with AMD11070, an orally bioavailable inhibitor of CXCR4-tropic HIV type 1. Clin. Infect. Dis. 48, 798–805 [DOI] [PubMed] [Google Scholar]
- 13. Kromdijk W., Huitema A. D., Mulder J. W. (2010) Treatment of HIV infection with the CCR5 antagonist maraviroc. Expert Opin. Pharmacother. 11, 1215–1223 [DOI] [PubMed] [Google Scholar]
- 14. Perry C. M. (2010) Maraviroc: a review of its use in the management of CCR5-tropic HIV-1 infection. Drugs 70, 1189–1213 [DOI] [PubMed] [Google Scholar]
- 15. Sayana S., Khanlou H. (2009) Maraviroc: a new CCR5 antagonist. Expert Rev. Anti Infect. Ther. 7, 9–19 [DOI] [PubMed] [Google Scholar]
- 16. Gerlach L. O., Skerlj R. T., Bridger G. J., Schwartz T. W. (2001) Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J. Biol. Chem. 276, 14153–14160 [DOI] [PubMed] [Google Scholar]
- 17. Rosenkilde M. M., Gerlach L. O., Hatse S., Skerlj R. T., Schols D., Bridger G. J., Schwartz T. W. (2007) Molecular mechanism of action of monocyclam versus bicyclam non-peptide antagonists in the CXCR4 chemokine receptor. J. Biol. Chem. 282, 27354–27365 [DOI] [PubMed] [Google Scholar]
- 18. Wong R. S., Bodart V., Metz M., Labrecque J., Bridger G., Fricker S. P. (2008) Comparison of the potential multiple binding modes of bicyclam, monocylam, and noncyclam small-molecule CXC chemokine receptor 4 inhibitors. Mol. Pharmacol. 74, 1485–1495 [DOI] [PubMed] [Google Scholar]
- 19. Dragic T., Trkola A., Thompson D. A., Cormier E. G., Kajumo F. A., Maxwell E., Lin S. W., Ying W., Smith S. O., Sakmar T. P., Moore J. P. (2000) A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl. Acad. Sci. U.S.A. 97, 5639–5644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garcia-Perez J., Rueda P., Alcami J., Rognan D., Arenzana-Seisdedos F., Lagane B., Kellenberger E. (2011) Allosteric model of maraviroc binding to CC chemokine receptor 5 (CCR5). J. Biol. Chem. 286, 33409–33421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kondru R., Zhang J., Ji C., Mirzadegan T., Rotstein D., Sankuratri S., Dioszegi M. (2008) Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 73, 789–800 [DOI] [PubMed] [Google Scholar]
- 22. Labrecque J., Metz M., Lau G., Darkes M. C., Wong R. S., Bogucki D., Carpenter B., Chen G., Li T., Nan S., Schols D., Bridger G. J., Fricker S. P., Skerlj R. T. (2011) HIV-1 entry inhibition by small-molecule CCR5 antagonists: a combined molecular modeling and mutant study using a high-throughput assay. Virology 413, 231–243 [DOI] [PubMed] [Google Scholar]
- 23. Maeda K., Das D., Ogata-Aoki H., Nakata H., Miyakawa T., Tojo Y., Norman R., Takaoka Y., Ding J., Arnold G. F., Arnold E., Mitsuya H. (2006) Structural and molecular interactions of CCR5 inhibitors with CCR5. J. Biol. Chem. 281, 12688–12698 [DOI] [PubMed] [Google Scholar]
- 24. Seibert C., Ying W., Gavrilov S., Tsamis F., Kuhmann S. E., Palani A., Tagat J. R., Clader J. W., McCombie S. W., Baroudy B. M., Smith S. O., Dragic T., Moore J. P., Sakmar T. P. (2006) Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology 349, 41–54 [DOI] [PubMed] [Google Scholar]
- 25. Tsamis F., Gavrilov S., Kajumo F., Seibert C., Kuhmann S., Ketas T., Trkola A., Palani A., Clader J. W., Tagat J. R., McCombie S., Baroudy B., Moore J. P., Sakmar T. P., Dragic T. (2003) Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J. Virol. 77, 5201–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Billick E., Seibert C., Pugach P., Ketas T., Trkola A., Endres M. J., Murgolo N. J., Coates E., Reyes G. R., Baroudy B. M., Sakmar T. P., Moore J. P., Kuhmann S. E. (2004) The differential sensitivity of human and rhesus macaque CCR5 to small-molecule inhibitors of human immunodeficiency virus type 1 entry is explained by a single amino acid difference and suggests a mechanism of action for these inhibitors. J. Virol. 78, 4134–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nishikawa M., Takashima K., Nishi T., Furuta R. A., Kanzaki N., Yamamoto Y., Fujisawa J. (2005) Analysis of binding sites for the new small-molecule CCR5 antagonist TAK-220 on human CCR5. Antimicrob. Agents Chemother. 49, 4708–4715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tan Q., Zhu Y., Li J., Chen Z., Han G. W., Kufareva I., Li T., Ma L., Fenalti G., Li J., Zhang W., Xie X., Yang H., Jiang H., Cherezov V., Liu H., Stevens R. C., Zhao Q., Wu B. (2013) Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341, 1387–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kothandan G., Gadhe C. G., Cho S. J. (2012) Structural insights from binding poses of CCR2 and CCR5 with clinically important antagonists: a combined in silico study. PLoS One 7, e32864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shiraishi M., Aramaki Y., Seto M., Imoto H., Nishikawa Y., Kanzaki N., Okamoto M., Sawada H., Nishimura O., Baba M., Fujino M. (2000) Discovery of novel, potent, and selective small-molecule CCR5 antagonists as anti-HIV-1 agents: synthesis and biological evaluation of anilide derivatives with a quaternary ammonium moiety. J. Med. Chem. 43, 2049–2063 [DOI] [PubMed] [Google Scholar]
- 31. Charo I. F., Ransohoff R. M. (2006) The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621 [DOI] [PubMed] [Google Scholar]
- 32. Yamasaki R., Liu L., Lin J., Ransohoff R. M. (2012) Role of CCR2 in immunobiology and neurobiology. Clin. Exp. Neuroimmunol. 3, 16–29 [Google Scholar]
- 33. Lederman M. M., Penn-Nicholson A., Cho M., Mosier D. (2006) Biology of CCR5 and its role in HIV infection and treatment. JAMA 296, 815–826 [DOI] [PubMed] [Google Scholar]
- 34. Mueller A., Strange P. G. (2004) The chemokine receptor, CCR5. Int. J. Biochem. Cell Biol. 36, 35–38 [DOI] [PubMed] [Google Scholar]
- 35. Sorce S., Myburgh R., Krause K. H. (2011) The chemokine receptor CCR5 in the central nervous system. Prog. Neurobiol. 93, 297–311 [DOI] [PubMed] [Google Scholar]
- 36. Rucker J., Samson M., Doranz B. J., Libert F., Berson J. F., Yi Y., Smyth R. J., Collman R. G., Broder C. C., Vassart G., Doms R. W., Parmentier M. (1996) Regions in β-chemokine receptors CCR5 and CCR2b that determine HIV-1 cofactor specificity. Cell 87, 437–446 [DOI] [PubMed] [Google Scholar]
- 37. Wu L., LaRosa G., Kassam N., Gordon C. J., Heath H., Ruffing N., Chen H., Humblias J., Samson M., Parmentier M., Moore J. P., Mackay C. R. (1997) Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J. Exp. Med. 186, 1373–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Samson M., LaRosa G., Libert F., Paindavoine P., Detheux M., Vassart G., Parmentier M. (1997) The second extracellular loop of CCR5 is the major determinant of ligand specificity. J. Biol. Chem. 272, 24934–24941 [DOI] [PubMed] [Google Scholar]
- 39. Thiele S., Steen A., Jensen P. C., Mokrosinski J., Frimurer T. M., Rosenkilde M. M. (2011) Allosteric and orthosteric sites in CC chemokine receptor (CCR5), a chimeric receptor approach. J. Biol. Chem. 286, 37543–37554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 41. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L. (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 [DOI] [PubMed] [Google Scholar]
- 42. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 [DOI] [PubMed] [Google Scholar]
- 43. Coward P., Chan S. D., Wada H. G., Humphries G. M., Conklin B. R. (1999) Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal. Biochem. 270, 242–248 [DOI] [PubMed] [Google Scholar]
- 44. Fricker S. P., Anastassov V., Cox J., Darkes M. C., Grujic O., Idzan S. R., Labrecque J., Lau G., Mosi R. M., Nelson K. L., Qin L., Santucci Z., Wong R. S. (2006) Characterization of the molecular pharmacology of AMD3100: a specific antagonist of the G-protein coupled chemokine receptor, CXCR4. Biochem. Pharmacol. 72, 588–596 [DOI] [PubMed] [Google Scholar]
- 45. Skerlj R., Bridger G., Zhou Y., Bourque E., McEachern E., Metz M., Harwig C., Li T. S., Yang W., Bogucki D., Zhu Y., Langille J., Veale D., Ba T., Bey M., Baird I., Kaller A., Krumpak M., Leitch D., Satori M., Vocadlo K., Guay D., Nan S., Yee H., Crawford J., Chen G., Wilson T., Carpenter B., Gauthier D., Macfarland R., Mosi R., Bodart V., Wong R., Fricker S., Schols D. (2013) Design of substituted imidazolidinylpiperidinylbenzoic acids as chemokine receptor 5 antagonists: potent inhibitors of R5 HIV-1 replication. J. Med. Chem. 56, 8049–8065 [DOI] [PubMed] [Google Scholar]
- 46. Berkhout T. A., Blaney F. E., Bridges A. M., Cooper D. G., Forbes I. T., Gribble A. D., Groot P. H., Hardy A., Ife R. J., Kaur R., Moores K. E., Shillito H., Willetts J., Witherington J. (2003) CCR2: characterization of the antagonist binding site from a combined receptor modeling/mutagenesis approach. J. Med. Chem. 46, 4070–4086 [DOI] [PubMed] [Google Scholar]
- 47. Filer A. D., Burman A. C., Haworth G., Parsonage G., Salmon M., Buckley C. D. (2004) Chemokines and persistent inflammation in rheumatoid arthritis: hunting for therapeutic targets. Curr. Med. Chem. Anti Inflamm. Anti Allergy Agents 3, 103–117 [Google Scholar]
- 48. Jones K. L., Maguire J. J., Davenport A. P. (2011) Chemokine receptor CCR5: from AIDS to atherosclerosis. Br. J. Pharmacol. 162, 1453–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghorban K., Dadmanesh M., Hassanshahi G., Momeni M., Zare-Bidaki M., Arababadi M. K., Kennedy D. (2013) Is the CCR5Δ32 mutation associated with immune system-related diseases? Inflammation 36, 633–642 [DOI] [PubMed] [Google Scholar]
- 50. Metz M., Bourque E., Labrecque J., Danthi S. J., Langille J., Harwig C., Yang W., Darkes M. C., Lau G., Santucci Z., Bridger G. J., Schols D., Fricker S. P., Skerlj R. T. (2011) Prospective CCR5 small molecule antagonist compound design using a combined mutagenesis/modeling approach. J. Am. Chem. Soc. 133, 16477–16485 [DOI] [PubMed] [Google Scholar]
- 51. Strizki J. M., Xu S., Wagner N. E., Wojcik L., Liu J., Hou Y., Endres M., Palani A., Shapiro S., Clader J. W., Greenlee W. J., Tagat J. R., McCombie S., Cox K., Fawzi A. B., Chou C. C., Pugliese-Sivo C., Davies L., Moreno M. E., Ho D. D., Trkola A., Stoddart C. A., Moore J. P., Reyes G. R., Baroudy B. M. (2001) SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 12718–12723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fricker S. P., Metz M. (2014) Chemokine receptor modeling: an interdisciplinary approach to drug design. Future Med. Chem. 6, 91–114 [DOI] [PubMed] [Google Scholar]