

Abstract
Nucleosome positioning is critical for gene expression and of major biological interest. The high cost of experimentally mapping nucleosomal arrangement signifies the need for computational approaches to predict nucleosome positions at high resolution. Here, we present a web-based application to fulfill this need by implementing two models, YR and W/S schemes, for the translational and rotational positioning of nucleosomes, respectively. Our methods are based on sequence-dependent anisotropic bending that dictates how DNA is wrapped around a histone octamer. This application allows users to specify a number of options such as schemes and parameters for threading calculation and provides multiple layout formats. The nuMap is implemented in Java/Perl/MySQL and is freely available for public use at http://numap.rit.edu. The user manual, implementation notes, description of the methodology and examples are available at the site.
Keywords: Nucleosome rotational positioning, Sequence-dependent DNA anisotropy, Prediction of nucleosome positioning, Sequence patterns, Web server
Introduction
The basic repeating unit of eukaryotic chromatin is the nucleosome, which contains a 145–147 bp DNA fragment wound around a histone core. The positioning of nucleosomes on a DNA fragment is critical for gene expression [1]. Along with other factors such as DNA methylation and histone modifications, DNA sequence is one of the most important factors guiding nucleosome positioning in vitro and in vivo [2]. Accurate determination of nucleosome positions is extremely important to study gene regulatory mechanisms. Unfortunately, micrococcal nuclease, which is commonly used for mapping nucleosome positions experimentally, exhibits high AT-specificity [3,4] and lacks the resolution to identify exact nucleosome positions, especially for nucleosomes with the GC-rich boundaries [5]. Therefore, prediction of nucleosome positioning on genomic sequence at high resolution is of great biological interest.
Nucleosome positioning is usually characterized by two parameters: rotational positioning, which describes the side of the DNA helix that faces the histones, and translational positioning, which determines the nucleosome midpoint (or dyad) with regard to the DNA sequence [6]. We have developed two simple models, the YR scheme [7] and W/S scheme [8], for the prediction of translational and rotational positioning of nucleosomes, respectively. Both methods successfully predict ∼80% of nucleosome positions in vitro with 2-bp precision (see Supplementary materials at http://numap.rit.edu/app/dna/suppMaterial.xhtml). Here, we present a web-based application that implements both models for prediction of nucleosome positioning patterns.
Methods
Both YR and W/S schemes are based on the sequence-dependent DNA anisotropy [9,10], which dictates how DNA is wrapped around a histone octamer. One of the best established sequence patterns consistent with this anisotropy is the periodic occurrence of AT-containing dinucleotides (WW) and GC-containing dinucleotides (SS) in the nucleosomal locations where DNA is bent in the minor and major grooves, respectively [11]. The minor and major groove bending sites are defined by the base-pair step Roll values observed in nucleosome crystal structures, in which a DNA fragment of 147 bp or 146 bp in length is co-crystallized with histones [12]. Based on the Roll values, 14 minor-groove DNA bending sites and 12 major-groove DNA bending sites were selected from the 147-bp or 146-bp nucleosomal DNA template; each site is 4 bp in length (see the Methods section on the server website for details). The W/S scheme implements the aforementioned periodic WW and SS patterns. The W/S score SW/S(n) of the threaded sequence with the center at position n can be calculated as
| (1) |
where CWW and CSS are the total occurrences of WW and SS dinucleotides at a given minor-groove or major-groove DNA bending site.
In addition to the WW and SS motifs, the YR scheme incorporates GC, pyrimidine–purine (YR), YYRR and RYRY motifs to take into account their anisotropic bending into DNA grooves (see Methods at the site for details). The YR score S(n) of the threaded sequence with the center at position n can be calculated as
| (2) |
where Cx is the occurrence of a designated motif x at a given site and wx is the weight of the motif at this site. A total of 28 DNA sequence patterns are used in the YR scheme [7]. If a motif occurs at the site where DNA is severely bent, its occurrence is given a higher weight than at other sites (see Methods section on the website for details).
Implementation
The nuMap web application takes a DNA sequence as input and returns both numeric scores and corresponding profiles. We use the model-view-controller (MVC) design, in which the communication between the client and the database is mediated by the web application server (Figure 1). The server is implemented using a combination of extensible hypertext markup language pages (XHTML), and JavaServer Faces (JSF) as a rich component-based user interface. Nucleosome positioning scores can be computed by the YR and W/S schemes implemented at the backend of the server as Perl scripts. An open-source reporting engine, JasperReports library, is used with the combination of Java codes for reporting the results in graphical output in multiple formats including PDF, Excel and CSV (see Implementation Notes at the server site for details).
Figure 1.
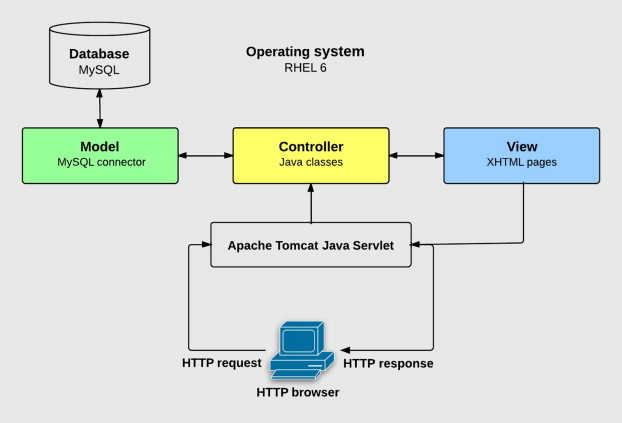
System infrastructure of the nuMap server
The MVC model describes an application in terms of three different design modules, the Model, the View and the Controller. The Model describes how the application represents the data model, including data files and a database management system (e.g., MySQL). The View describes what the user interface looks like and how the information is displayed at the user end (e.g., XHTML). The Controller part represents how the user interface is managed and controlled by the application code. MVC, model-view-controller.
Prediction options and features
Figure 2 shows the general layout for nuMap user interface. The user can select from four different output layouts. The “Single Layout” is used to generate single graph for each of the input sequences, whereas the “Superimpose Layout” overlays more than one graph, if multiple sequences are used as input. The “Average Layout (Asymmetric)” is used to calculate the average scores for multiple sequences, while the “Average Layout (Symmetric)” produces a symmetrical graph by generating the reverse complement of all the input sequences and then calculating the average scores at each base pair position. The setting panel allows the user to choose from two prediction schemes, YR and W/S. By default, a chosen scheme will calculate a score for the first nucleosome starting at the position 1 of the input sequence, and assign that score to the dyad position 74. The user can also give a specific value to the start of the input sequence (if not 1) by changing the value in the “Starting Position” box. The “Weight Set” option is specific to the YR scheme, the user can choose from three different sets used initially to establish this scheme with each set evaluating the occurrence of specific pattern at specific site differently (see Table SIII in [7]).
Figure 2.
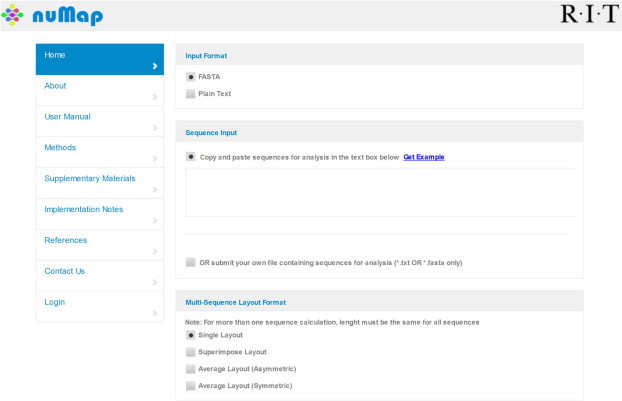
A screen shot of the nuMap server interface
.
The nuMap provides numerous reporting formats such as PDF (Figure 3), Word and Excel. Moreover, the server offers many features that allow the user to interact with output data in different ways such as zooming in a large graph, which is useful to investigate a certain range of values, and exploring dynamic data features, which show the corresponding score/base pair position when a user places the mouse over any point in the graph (Figure 4).
Figure 3.
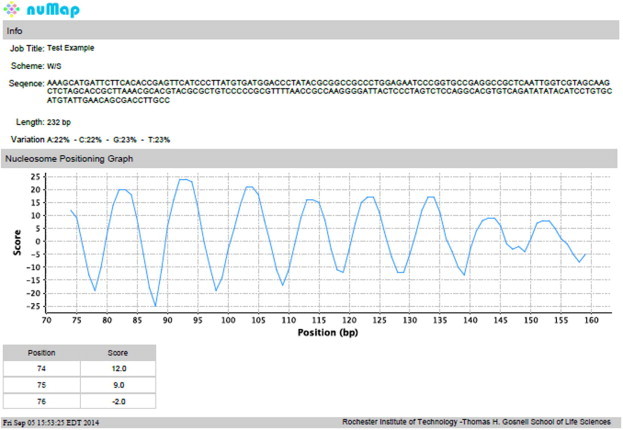
Example of nuMap output in PDF format
A sample out of the nuMap server is shown. The Information Panel is shown on the top, containing job-related information such as sequence name and user-selected options including prediction scheme, sequence, its length and composition. A profile of the nucleosome positioning score of the ‘601’ sequence, in this case, predicted by the W/S scheme is shown underneath. The physical meaning of the score is straightforward – the lower the DNA deformation energy for particular sequence motifs placed at the sites of DNA distortion, the higher the score. The score is calculated for each 147-bp window and is assigned to the center position of a given window. The actual score at each position is given in a tabular form.
Figure 4.
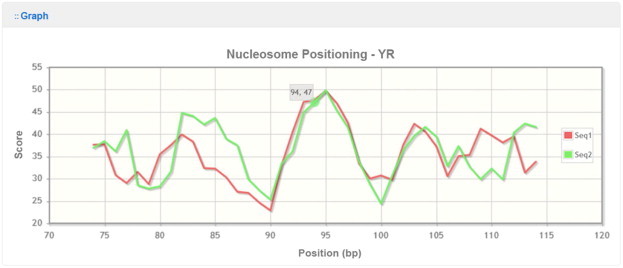
A screen shot of nuMap output of superimposing profiles for two sequences
A screen shot of the nuMap output for two sequences superimposed together is shown. The score value on a particular position can be shown on the screen by mouse. In this case, a mouse hovering over position 94 shows the score, 47, at this position. This example illustrates dynamic data features of the server. The YR (pyrimidine–purine) scheme was used to calculate the scores. The physical meaning of the scores is given in the legend in Figure 3.
Future developments
Other models [13,14] work well in predicting nucleosome occupancy in vivo by introducing a position-independent component, PL, to represent sequences that are generally favored or disfavored regardless of their position within the nucleosome (most notably, poly(dA:dT) tracts, which are strongly disfavored by nucleosomes) [14]. Detailed comparisons in prediction accuracy between these models and ours have been made and will be published separately. The incorporation of this component into the YR or W/S scheme has a potential to improve both the rotational and translational positioning predictions of nucleosomes in vivo.
Various models have been developed for nucleosome positioning predictions [15–19] and for gene regulatory analysis [20,21]. Nonetheless, one has to browse through different servers, which often have different formats and representations, making the comparison of the results extremely difficult. We will overcome this obstacle by reprogramming these methods, incorporating them into the nuMap server and presenting the results in the same format. Comparison of all the methods in this way will allow detailed analyses of the strengths and weaknesses of each approach, facilitating our understanding of the biophysical principles of nucleosome positioning.
Authors’ contributions
BAA and THA developed the server, performed the analyses and drafted the paper. NLF set up the server. VBZ and FC contributed to data analysis and paper writing. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
This study was supported by the scholarship of the King Abdullah International Medical Research Center (KAIMRC) in Saudi Arabia to BA, the start-up fund, Faculty of Development (FEAD) fund and Dean’s Research Initiation Grant (D-RIG) fund of Rochester Institute of Technology in the USA awarded to FC and the Intramural Research Program of National Cancer Institute, NIH of the USA to VBZ.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
References
- 1.Kornberg R.D., Lorch Y. Twenty-five years of the nucleosomes, fundamental particle of the eukaryotic chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 2.Struhl K., Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dingwall C., Lomonossoff G.P., Laskey R.A. High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 1981;9:2659–2673. doi: 10.1093/nar/9.12.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hörz W., Altenburger W. Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res. 1981;9:2643–2658. doi: 10.1093/nar/9.12.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikitina T., Wang D., Gomberg M., Grigoryev S.A., Zhurkin V.B. Combined micrococcal nuclease and exonuclease III reveals precise positions of the nucleosome core/linker junctions: implications for high-resolution nucleosome mapping. J Mol Biol. 2013;425:1146–1160. doi: 10.1016/j.jmb.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Travers A.A., Klug A. The bending of DNA in nucleosomes and its wider implications. Philos Trans R Soc Lond B Biol Sci. 1987;317:537–561. doi: 10.1098/rstb.1987.0080. [DOI] [PubMed] [Google Scholar]
- 7.Cui F., Zhurkin V.B. Structure-based analysis of DNA sequence patterns guiding nucleosome positioning in vitro. J Biomol Struct Dyn. 2010;27:821–841. doi: 10.1080/073911010010524947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui F., Zhurkin V.B. Rotational positioning of nucleosomes facilitates selective binding of p53 to response elements associated with cell cycle arrest. Nucleic Acids Res. 2014;42:836–847. doi: 10.1093/nar/gkt943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhurkin V.B. Anisotropic flexibility of DNA and the nucleosomal structure. Nucleic Acids Res. 1979;6:1081–1096. doi: 10.1093/nar/6.3.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trifonov E.N. Sequence-dependent deformational anisotropy of chromatin DNA. Nucleic Acids Res. 1980;8:4041–4053. doi: 10.1093/nar/8.17.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satchwell S.C., Drew H.R., Travers A.A. Sequence periodicities in chicken nucleosome core DNA. J Mol Biol. 1986;191:659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- 12.Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 13.Segal E., Fondufe-Mittendorf Y., Chen L., Thåström A., Field Y., Moore I.K. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaplan N., Moore I.K., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tolstorukov M.Y., Choudhary V., Olson W.K., Zhurkin V.B., Park P.J. nuScore: a web-interface for nucleosome positioning prediction. Bioinformatics. 2008;24:1456–1458. doi: 10.1093/bioinformatics/btn212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo S.H., Deng E.Z., Xu L.Q., Ding H., Lin H., Chen W. iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics. 2014;30:1522–1529. doi: 10.1093/bioinformatics/btu083. [DOI] [PubMed] [Google Scholar]
- 17.Xi L., Fondufe-Mittendor Y., Xia L., Flatow J., Widom J., Wang J.P. Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinformatics. 2010;11:346. doi: 10.1186/1471-2105-11-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stolz R.C., Bishop T.C. ICM web: the interactive chromatin modeling web server. Nucleic Acids Res. 2010;38:W254–W261. doi: 10.1093/nar/gkq496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabdank I., Barash D., Trifonov E.N. FineStr: a web server for single-base resolution nucleosome positioning. Bioinformatics. 2010;26:845–846. doi: 10.1093/bioinformatics/btq030. [DOI] [PubMed] [Google Scholar]
- 20.Guan D., Shao J., Zhao Z., Wang P., Qin J., Deng Y. PTHGRN: unraveling post-translational hierarchical gene regulatory networks using PPI, ChIP-seq and gene expression data. Nucleic Acid Res. 2014;12:W130–W136. doi: 10.1093/nar/gku471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guan D., Shao J., Deng Y., Wang P., Zhao Z., Liang Y. CMGRN: a web server for constructing multilevel gene regulatory networks using ChIP-seq and gene expression data. Bioinformatics. 2014;30:1190–1192. doi: 10.1093/bioinformatics/btt761. [DOI] [PubMed] [Google Scholar]