

Abstract
Background
The coronavirus 3 chymotrypsin-like protease (3CLpro) is a validated target in the design of potential anticoronavirus inhibitors. The high degree of homology within the protease’s active site and substrate conservation supports the identification of broad spectrum lead compounds. A previous study identified the compound ML188, also termed 16R, as an inhibitor of the Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) 3CLpro. This study will detail the generation of a homology model of the 3CLpro of the human coronavirus OC43 and determine the potential of 16R to form a broad-spectrum lead compound. MODELLER was used to generate a suitable three-dimensional model of the OC43 3CLpro and the Prime module of Schrӧdinger predicted the binding conformation and free energy of binding of 16R within the 3CLpro active site. Molecular dynamics further confirmed ligand stability and hydrogen bonding networks.
Results
A high quality homology model of the OC43 3CLpro was successfully generated in an active conformation. Further studies reproduced the binding pose of 16R within the active site of the generated model, where its free energy of binding was shown to equal that of the 3CLpro of SARS-CoV, a receptor it is experimentally proven to inhibit. The stability of the ligand was subsequently confirmed by molecular dynamics.
Conclusion
The lead compound 16R may represent a broad-spectrum inhibitor of the 3CLpro of OC43 and potentially other coronaviruses. This study provides an atomistic structure of the 3CLpro of OC43 and supports further experimental validation of the inhibitory effects of 16R. These findings further confirm that the 3CLpro of coronaviruses can be inhibited by broad spectrum lead compounds.
Keywords: Human coronavirus, OC43, 3CLpro, Homology modelling, Molecular dynamics
Background
Human coronaviruses have a worldwide distribution and are frequently associated with self-limiting upper respiratory tract disease or “the common cold”. They can, however, also present with high morbidity outcomes of the lower respiratory tract including bronchiolitis, pneumonia, [1-3], asthmatic exacerbations [4] and acute exacerbations of chronic obstructive pulmonary disease (COPD) [5], where human coronavirus OC43 has been shown to be the prevalent strain in several regions [6]. Given the high burden of coronaviruses to human health, and their potential for genetic recombination to give rise to the emergence of completely novel strains, the presence of antiviral strategies is paramount. However, there is currently no commercially available molecular entity which is capable of inhibiting infection with human coronaviruses [7]. The 3CLpro has been validated as an effective drug target in several studies and has even been termed “the Achilles’ heel of coronaviruses” [8] making it an ideal target for the identification of novel lead compounds.
During coronavirus replication the large open reading frame (ORF) 1a/1ab genes, located at the 5’ end of the genome, are responsible for expressing two large replicase polyproteins (pp). These are co- or post-translationally cleaved by the virally encoded 3CLpro and Papain-like protease to yield 16 non-structural proteins responsible for viral replication [9]. The 3CLpro is also termed the main protease as it cleaves a total of 11 cleavage sites within pp1a and pp1ab [10], in comparison to only three cleavage sites predicted for the papain-like protease [11].
The 3CLpro has three distinct domains. Domains I and II are largely composed of several anti-parallel β barrels [12] and is connected to domain III by a long loop region. Domain III is composed of several globular α-helices and plays a role in protein dimerization, along with the N-terminal N-finger (residues 1–7). The active site is located in a chymotrypsin-like fold between domains I and II and contains a catalytic dyad of His41, which acts as a proton acceptor, and Cys144/5, which undergoes nucleophilic attack on the carbonyl carbon of the substrate [8,13]. The 3CLpro is so named for its close structural and sequence homology to the 3 chymotrypsin protease (3Cpro) of rhinoviruses, which contains a catalytic triad composed of His, Cys and Glu or Asp. Superimposition of the structures of 3CLpro and 3Cpro shows that the His and Cys of both proteases is almost perfectly aligned, which may explain the similar substrate specificity and catalytic mechanism [14]. The position of the Glu/Asp in 3Cpro is, however, replaced by a water molecule in 3CLpro, which shares three hydrogen bonds with surrounding residues including the catalytic dyad member His41. It is assumed that this water molecule is responsible for stabilization of the histidine in the intermediate state during enzymatic cleavage [8].
All coronavirus 3CLpro share a high sequence homology, as well as main chain architecture and substrate conservation [15,16]. The substrate binding site of the 3CLpro has two deeply buried S1 and S2 subsites, with shallow S1’, S3 and S4 subsites with varying degrees of solvent exposure. Substrate specificity of coronavirus 3CLpro is mainly determined by the P1, P2 and P1’ positions [16]. The P1 position has an absolute specificity for glutamine which stabilizes the S1 subsite via a hydrogen bond with the imidazole Nε2 of His162/3 and van der Waals interactions with surrounding residues of the S1 pocket. The P2 site has a preference for leucine or methionine to fill the hydrophobic S2 pocket. The sidechains of the S3 site are solvent exposed and therefore this site is expected to tolerate a wide range of functionality, but shows a preference for basic residues [17]. Sidechains and backbones of residues surrounding the S4 site create a highly congested pocket which favors a small, hydrophobic residue in the P4 position, either Ser, Thr, Val or Pro [17-19]. The S1’ and S2’ subsites also accommodate small residues in the P1’ and P2’ positions, which may include Ser, Ala or Gly [18,20]. A typical cleavage recognition site is therefore (Ser, Ala)-(Val, Thr)-Leu-Glu ↓ (Ser, Ala, Gly), which is conserved among all coronavirus 3CLpro [21]. Theoretically, since these viruses share a highly homologous binding site and substrate conservation, it is reasonable to anticipate that broad spectrum inhibitor strategies might be successful. [13].
A noncovalent, furyl amide ligand, 16R, which was identified by a high throughput screen, with subsequent lead optimization based on structure-activity relationships within the S1, S1’ and S2 binding pockets was shown to inhibit the 3CLpro of SARS-CoV at an IC50 of 1.5 ± 0.3 μM [12]. This study will detail the generation of a theoretical model of the OC43 3CLpro based on a high sequence homology with the solved 3CLpro structure of HKU1 and assess the potential of the inhibitor, 16R, to inhibit this complex in a broad-spectrum manner. This study will therefore provide further evidence to support the identification of broad spectrum 3CLpro inhibitors.
Results and discussion
Homology modelling of the OC43 3CLpro
The 3CLpro structure of HKU1 [PDB: 3D23] displayed a high sequence identity of 82.3% to the 3CLpro of OC43, with an e-value of 0.0. The high degree of identity can be partly expected as both OC43 and HKU1 are human coronaviruses from the Betacoronavirus genus. The exceptionally high degree of identity may even further suggest a recent common ancestor, which has yet to be identified. The active site residues are also highly conserved between both sequences indicating that 3D23 forms a highly suitable template for model generation (Figure 1).
Figure 1.

Pairwise sequence alignment of OC43 3CLpro with the template structure of 3D23. Sequence alignment revealed a high identity of 82.3%. Asterisks indicate conserved residues between target and template. Conserved active site residues are highlighted in red. Important residues within the oxyanion loop (yellow), S1 pocket (blue) and S2 pocket (black) are also highlighted to display high degree of conservation within the active site.
Homology models were built with MODELLER (9v10) [22,23] where the lowest discrete optimized protein energy (DOPE) score corresponded to model five with a GA341 score of 1, indicating that the model quality corresponded with low resolution crystallographic structures. The DOPE score profile of target and template (Figure 2) were nearly perfectly overlaid, indicating that the model was close to its native state. A peak in DOPE score for HKU1 3CLpro (3D23) was observed at approximately residue 50, where OC43 3CLpro showed a moderate conservation in DOPE score. Colouring the HKU1 3CLpro (3D23) structure by B-factor indicates the presence of a highly variable loop region from Ser46 to Asp53 (Figure 3). The presence of this highly variable loop structure could explain the increase in the DOPE score profile in this region and may suggest that the homology model has assumed a more stable conformation than the template. Structural alignments where the root mean square deviation (RMSD) is below 2 Å between target and template indicates that the positions of all backbone elements are correct [24,25]. Superimposition of the 3D23 template and modelled OC43 3CLpro structure displayed an RMSD of 0.327 Å suggesting a highly accurate prediction of the position of all backbone elements (Figure 4). Analysis of the overall model quality of target and template by ProSA Z-score indicated that both fall within an acceptable range for crystallographic structures with a Z-score for 3D23 of −7.04 and −7.34 for the homology model of OC43 3CLpro (Figure 5). Stereochemical analysis of phi-psi dihedral angles indicated that 91.8% of residues were in the most favoured regions with none in the disallowed regions (Figure 6).
Figure 2.
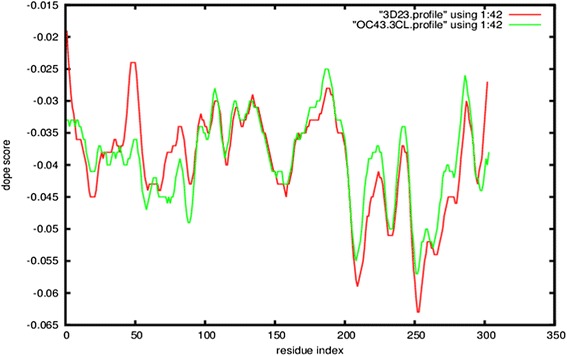
DOPE score profiles of template, 3D23, and homology model of OC43 3CLpro . General overlay of profiles indicates the generated model is close to its native structure. The spike at residue 50 corresponds to a variable loop structure for which OC43 3CLpro has assumed a more stable conformation.
Figure 3.
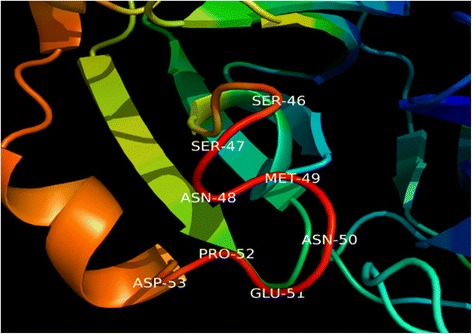
Location of highly variable loop region in the 3CLpro of HKU1 (3D23). Colouring of backbone elements by B-factor indicates the presence of a highly variable loop from residues Ser46 to Asp53. This variable loop region may be responsible for the spike in DOPE score for 3D23, for which OC43 3CLpro has assumed a more stable conformation.
Figure 4.
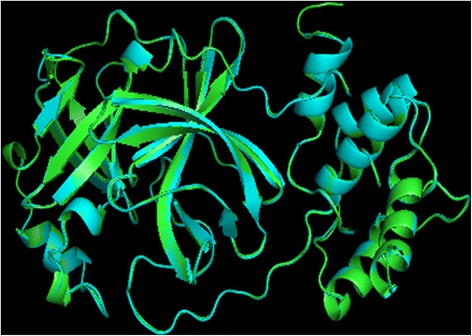
Superimposition of 3D23 and homology model of OC43 3CLpro. Blue ribbon represents the template structure of 3D23 with green representing the homology model of OC43 3CLpro. Superimposition shows a high degree of structural homology with a low RMSD of 0.327 Å.
Figure 5.
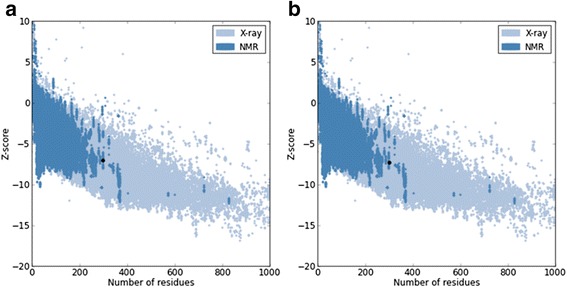
Overall quality of the model as assessed by Z-score. (a) Z-score for crystallographic model of 3D23. (b) Z-score for the homology model of OC43 3CLpro. A strong correlation between template and target structures indicates an accurate model.
Figure 6.
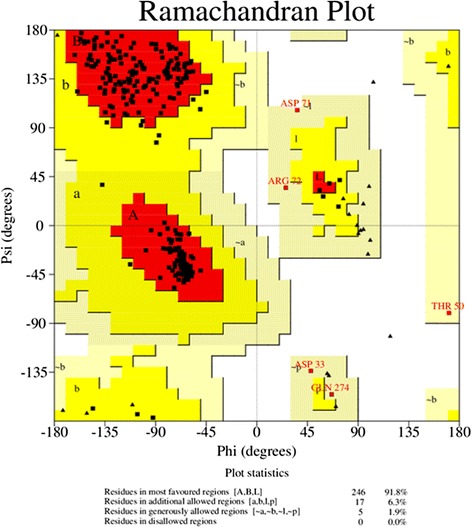
Stereochemical analysis of phi-psi dihedral angle of the OC43 3CLpro. Ramachandran plot generated by PROCHECK indicates that 91.8% of residues are located in the most favoured regions with none in the disallowed regions. Based on analysis of 118 solved structures, a good model is expected to have above 90% of residues in regions A, B and I [35].
For the latter purposes of this model, in characterization of the inhibitory potential of 16R, it is essential to confirm that the receptor in its active conformation. To ensure the modelled 3CLpro of OC43 is in its active conformation, several features can be used to differentiate between the active and inactive states. These features include the maintenance of a loop conformation of the loop connecting domain II and III and the oxyanion loop. If these features assume an alpha helical conformation it directly or indirectly leads to the collapse of the oxyanion hole and thereby forms an inactive state [26]. An alpha helix conformation of residues 186–190 in the loop region between domain II and III causes a further collapse of the S2 and S4 subsites [19]. The orientation of the His163 sidechain in the S1 pocket is also vital for substrate binding [27]. This residue is largely kept in place by a stacking interaction with the benzene ring of Phe140 and imidazole ring of His163 [26]. The orientation of His172 is also stabilized by a hydrogen bond with the sidechain of Glu166. This prevents steric interactions between His172 and His163 allowing it to maintain its orientation in the S1 pocket. A hydrogen bond between His163 and Tyr161 has also been implicated in stabilizing the sidechain of His163 [27], however we could not observe the formation of this bond in the crystal structure of SARS-CoV 3CLpro (3V3M), raising the question of its importance to maintain the orientation of His162/3.
Utilising these previously mentioned parameters it is possible to ascertain if the generated homology model of OC43 3CLpro is in an active conformation. With the exception of the hydrogen bond between Tyr161 and His163, which may form in a dynamic system as the Tyr161 hydroxyl is in close proximity to the imidazole nitrogen of His163 but with incorrect geometry to form a hydrogen bond, all other interactions and conformations were maintained indicating that the homology model generated using the 3CLpro structure of HKU1 (3D23) as a template is representative of the active conformation of the enzyme (Figure 7). With this and previously mentioned results, indicating the generation of an appropriate homology model of the OC43 3CLpro, suggests the generated structure is suitable for further structure-based drug design techniques.
Figure 7.
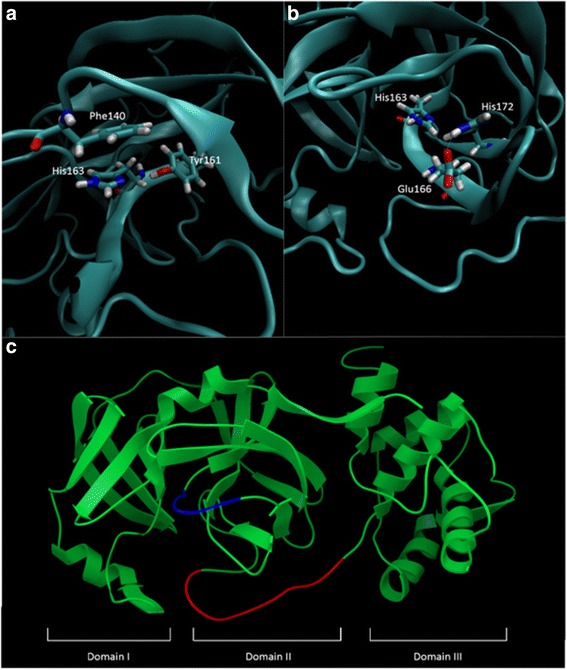
Features present in the homology model of OC43 3CLpro which represent of an active state of the enzyme. (a) Orientation of His163 is essential for substrate binding. This orientation is maintained by stacking interactions with Phe140 and a hydrogen bond with Tyr161. The importance of this bond is questionable as it is not observed in all crystallographic models. (b) Steric interactions between His172 and His163 disrupt the active conformation of His163. To prevent this, His172 is stabilized by a hydrogen bond with Glu166. (c) Maintenance of loop structures of the oxyanion loop (blue) and the loop connecting domain II and III (red) are essential in stabilizing the oxyanion hole. The general three domain structure of all 3CLpro is also depicted.
Assessing the binding conformation and free energy of binding of 16R
Analysis of the crystallographic structure of the SARS-CoV 3CLpro, with the bound inhibitor 16R, illustrates a pyridine occupying the S1 pocket and forming a hydrogen bond with the imidazole Nε2 of His163. A further two hydrogen bonds are formed between ligand carbonyls and Gly143 and Glu166 with three methyl groups inserting into the deep hydrophobic S2 pocket [12]. The conformation of 16R is almost completely conserved when bound to the 3CLpro of OC43. Notable exceptions include the shifting of the hydrogen bond formed with Gly143 to a furan ring oxygen as opposed to the carbonyl oxygen seen in SARS-CoV 3CLpro. The hydrogen bond formed with Glu166 is also absent in OC43 3CLpro, however the distance between the ligand carbonyl and backbone hydrogen is 2.75 Å and therefore this may be capable of mediating hydrogen bond formation in a dynamic system (Figure 8). Free energy of binding between 16R and SARS-CoV and OC43 3CLpro, as assessed by MM-GBSA, is -85 kcal/mol for both receptors. These results suggest that the inhibitor 16R may be capable of inhibiting both complexes.
Figure 8.
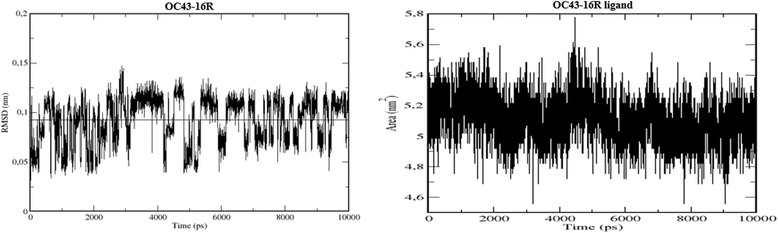
Binding conformation of 16R within the active pocket of the 3CLpro. (a) Pose of 16R within the active site of the SARS-CoV 3CLpro. Hydrogen bonds (yellow dashes) are formed with Gly143, His163 and Glu166. (b) 16R assumes a similar pose when bound to the OC43 3CLpro. A notable difference is the loss a hydrogen bond with Glu166. Hydrogen bonds with Gly143 and His163 are however maintained.
Molecular dynamic simulation of OC43 and 16R
To further confirm the findings described in the previous section, and to assess the potential of Glu166 to form a hydrogen bond in a dynamic system, molecular dynamic simulations were used. Molecular dynamics of the SARS-CoV 3CLpro-16R complex are in strong agreement with the crystal data of 3V3M [12]. The simulation predicts an average of 3.17 hydrogen bonds formed over the 10 ns trajectory, with these bonds predominantly forming with Glu166, His163 and Gly143. The bond formed at Gly143 does however display the potential to form a bifurcated interaction with Asn142. As predicted by the previous results, detailed in the section describing the binding conformation, the 3CLpro-16R complex of OC43 displayed a high occupancy of 100% and 85% at both His163 and Gly143 positions respectively. These results however suggested that the bond formed with Gly143 was mediated via the furan ring oxygen, where molecular dynamic simulations indicated that this bond was formed between the carbonyl oxygen and Gly143, as seen with the crystal structure of SARS-CoV. Molecular dynamics also confirmed that the hydrogen bond at the Glu166 does not stably form in a dynamic environment and only displays the potential to form at 0-2 ns and 4-5 ns, with an overall occupancy of 21.62%.
The stability of the bound ligand within the active pocket was also assessed via RMSD, radius of gyration and changes in solvent accessible surface area. A RMS deviation below 2 Å is an indication of a ligand which is stably bound to its receptor [28], where an increase in radius of gyration from the starting reference would suggest ligand instability. An increase in solvent accessible surface area of the ligand may indicate the ligand is dissociating into surrounding solvent [29]. Ligand RMSD within the active site was approximately 0.75 Å and radius of gyration and solvent accessible surface area remained within a stable range of 3.9 Å to 4.0 Å and 4.8 nm2 to 5.4 nm2 respectively (Figure 9). These parameters suggest that the ligand is highly stable within the active site.
Figure 9.
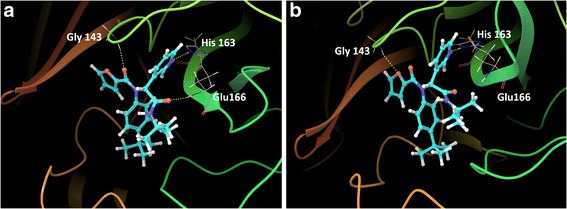
RMSD and solvent accessible surface area of 16R when bound to the OC43 3CLpro. Analysis of ligand RMSD and solvent accessible surface area depicts a ligand which is stably bound to its receptor.
Conclusion
The three-dimensional structure of the OC43 3CLpro was successfully solved by homology modelling with MODELLER. Analysis of various side chains and loop conformations within and surrounding the substrate-binding site is indicative of an active conformation of the enzyme. The solved structure has been made publicly available in the Protein Model Database (PMDB; https://bioinformatics.cineca.it/PMDB/) [PMDB ID: PM0079872]. Further analysis of a previously identified lead compound, 16R, suggests that it makes extensive and stable interactions with the 3CLpro binding site of OC43. Additional analysis of the free energy of binding by MM-GBSA suggests that the ligand binds with the same affinity to the 3CLpro of OC43 as it does to SARS-CoV, a receptor it is experimentally proven to inhibit. These results are strongly suggestive that the lead compound, 16R, not only displays the potential to inhibit the 3CLpro of SARS-CoV but also that of the highly homologous OC43. In additional studies (data not shown) 16R was shown to remain stably bound to the 3CLpro of 229E and NL63 in molecular dynamic simulations and may therefore even display the potential to inhibit the 3CLpro of additional coronaviruses. 16R is therefore an excellent lead compound in the pursuit of true broad spectrum inhibitors of all coronaviruses.
Methods
Homology modelling of the OC43 3CLpro
The amino acid sequence of the OC43 3CLpro was obtained from the NCBI database [GenBank: AEN19363]. Fold assignment with the profile.build module of MODELLER (9v10) identified the most suitable template structure from PDB with an 82.3% sequence identity and e-value of 0. Template-target alignment was conducted with the align.2d module, which also incorporates structural information from the template. Models of the OC43 3CLpro were built by satisfaction of homology derived spatial restraints on distances and dihedral angles [23], stereochemical restraints obtained from the CHARMM22 force field [30] and statistical preferences on dihedral angles and non-bonded distances obtained from known protein structures [22,31]. Models were built by the automodel class by minimizing the violations on all restraints and united-atom PDB models were generated.
DOPE scores are assigned by considering the positions of all non-hydrogen atoms, where the lowest DOPE score corresponds to the model that is predicted to be most accurate [31]. The evaluate_model.py script was further utilised to calculate per residue DOPE scores, which were superimposed over the template structure DOPE scores. A general conservation in the pattern of respective DOPE scores was used as an indication of an accurate model. A low RMSD, as assessed by PyMOL [32], indicated structural homology between solved structure and template. The GA341 score was used to analyse fold-assignment. Z-scores, obtained from ProSA-web [33,34] were used to analyse overall model quality and to assure template and query models were in a range acceptable for structures of their respective size. A Z-score outside a range characteristic for native proteins will suggest the production of an erroneous model [33]. Stereochemical analysis of dihedral angles was then further conducted using PROCHECK [35,36] on models corresponding to the lowest DOPE score with GA341 scores amounting to 1.0. Stereochemical analysis by PROCHECK assessed residue geometry, where more than 90% of residues should be located in the “most favoured region” to indicate a stereochemically favourable structure.
Once an appropriate model accuracy was achieved based on these previous observations, the active site of the enzyme was analysed to ensure it was in its active state. Several features are able to differentiate an active from inactive state of the 3CLpro. These include the conformation of the oxyanion loop and loop connecting domain II and III [19,26] and the sidechain orientation of residues, including His163, Phe140, His172, Tyr161 and Glu166, within the S1 pocket [26,27].
Assessing the binding conformation and free energy of binding of 16R
Protein structures of 3V3M (3CLpro of SARS-CoV) and the homology modelling derived structure of OC43 3CLpro were prepared with the Schrӧdinger Protein Preparation Wizard. Histidine protonation was assigned to the epsilon nitrogen in accordance with crystal data. A restrained minimization was then performed using the OPLS2005 force field [37,38], during which heavy atoms were restrained to remain within 0.3 Å of the original structure. To assess the conformation of the known inhibitor, 16R, when binding to the OC43 3CLpro, the experimental structure of 3V3M and OC43 3CLpro were aligned in Maestro, the interface for all Schrӧdinger’s software. The inhibitor, 16R, was subsequently superimposed over the OC43 3CLpro active site using coordinates present in the 3V3M crystal structure. Side chains of residues in the active pocket assuming orientations that resulted in a steric clash with the ligand were refined and the ligand was minimized with Prime [39,40] to better fit the pocket. Prime utilizes both empirically and theoretically derived constraints to predict and replicate the dynamic motion of protein sidechains, allowing for conformational changes that arise through the “induced-fit” phenomenon, which is often neglected in molecular docking screens.
The Prime/MM-GBSA method was used to calculate the binding free energy of 16R within the respective receptor. The free energy of binding is the calculated difference between the minimized receptor-inhibitor complex and the sum of the energies of the minimized unbound inhibitor and receptor. Ligand poses were minimized using the local optimization feature in Prime, where energies of the complex were calculated with the OPLS-2005 force field and GBSA continuum solvent model.
Molecular dynamic simulation of the 3CLpro-16R complex
The CHARMM27 all atom force field was assigned to receptor structures using the three point TIP3P water model. All input hydrogens in the coordinate file were ignored and were assigned according to the force field. Histidine protonation states were assigned to the epsilon nitrogen in the neutral form in accordance with crystallographic data. The ligand, in the previously predicted pose, was prepared by SwisParam [41]. The protein-ligand complex was then placed in the centre of a solvated, cubic box, 1 nm from the box edge. The system was solvated with the spc216 water model and sodium counter ions were added to neutralize the system. The system was energy minimized by a brief dynamics simulation using Steepest Descent Minimization until steepest descent converged to Fmax < 1000. Long range electrostatic interactions were calculated with the Particle Mesh Ewald (PME) method [42] with a cutoff of 1 nm and periodic boundary conditions were applied. Both NVT and NPT equilibrations were run for 50 000 steps or 100 ps using a 2 fs time step and a leap-frog integrator. All bonds were constrained by the lincs algorithm. PME was again used for long range electrostatics with a 0.16 fourier spacing. Short range electrostatic and van der Waals cutoffs were 1.0 nm and 1.4 nm respectively. Coordinates, velocities and energies were saved every 0.2 ps. During NVT equilibration temperature coupling was achieved by V-rescale algorithm with a target temperature of 300 K. Pressure coupling during NPT equilibration was achieved via the Parrinello-Rahman algorithm and is incorporated in the NPT equilibration once temperature is stable to ensure the proper density is reached (approximately 1000 kg/m3). Following NVT and NPT system equilibration an unrestrained, 10 ns simulation (5 000 000 steps) was run with the leap frog integrator, saving coordinates, velocities and energies every 2 ps. All other parameters were maintained from the NVT and NPT equilibration including both temperature and pressure coupling.
Acknowledgements
This research was supported by grants provided by the Council for Scientific and Industrial Research (CSIR, South Africa), Poliomyelitis Research Foundation (PRF) and Ernst and Ethel Erikson Trust. The authors thank Novartis (Basel, Switzerland) and H3D (University of the Cape Town) for significant intellectual input and access to resources.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MB conducted all experimental and bioinformatics analysis detailed in the manuscript and drafted the manuscript. JG participated in the design of the study and editing of the manuscript. BF edited the manuscript and contributed towards study rationale. All authors read and approved the final manuscript.
Contributor Information
Michael Berry, Email: michael@sanbi.ac.za.
Burtram Fielding, Email: bfielding@uwc.ac.za.
Junaid Gamieldien, Email: junaid@sanbi.ac.za.
References
- 1.Pene F, Merlat A, Vabret A, Rozenberg F, Buzyn A, Dreyfus F, et al. Coronavirus 229E-related pneumonia in immunocompromised patients. Clin Infect Dis. 2003;37(7):929–932. doi: 10.1086/377612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woo PC, Lau SK, Tsoi H-w, Huang Y, Poon RW, Chu C-m, et al. Clinical and molecular epidemiological features of coronavirus HKU1–associated community-acquired pneumonia. J Infect Dis. 2005;192(11):1898–1907. doi: 10.1086/497151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vabret A, Mourez T, Gouarin S, Petitjean J, Freymuth F. An outbreak of coronavirus OC43 respiratory infection in Normandy, France. Clin Infect Dis. 2003;36(8):985–989. doi: 10.1086/374222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau SKP, Woo PCY, Yip CCY, Tse H, Tsoi H, Cheng VCC, et al. Coronavirus HKU1 and Other Coronavirus Infections in Hong Kong. J Clin Microbiol. 2006;44(6):2063–2071. doi: 10.1128/JCM.02614-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorse GJ, O’Connor TZ, Hall SL, Vitale JN, Nichol KL. Human coronavirus and acute respiratory illness in older adults with chronic obstructive pulmonary disease. J Infect Dis. 2009;199(6):847–857. doi: 10.1086/597122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jean A, Quach C, Yung A, Semret M. Severity and outcome associated with human coronavirus OC43 infections among children. Pediatr Infect Dis J. 2013;32(4):325–329. doi: 10.1097/INF.0b013e3182812787. [DOI] [PubMed] [Google Scholar]
- 7.Ramajayam R, Tan KP, Liu HG, Liang PH. Synthesis, docking studies, and evaluation of pyrimidines as inhibitors of SARS-CoV 3CL protease. Bioorg Med Chem Lett. 2010;20(12):3569–3572. doi: 10.1016/j.bmcl.2010.04.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H, Bartlam M, Rao Z. Drug Design targeting the main protease, the Achilles heel of Coronaviruses. Curr Pharm Des. 2006;12(35):4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 9.Pyrc K, Berkhout B, van der Hoek L. The novel human coronaviruses NL63 and HKU1. J Virol. 2007;81(7):3051–3057. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziebuhr J: The coronavirus replicase. Coronavirus replication and reverse genetics. Springer Berlin Heidelberg, 2005. 57-94.
- 11.Gao F, Ou H-Y, Chen L-L, Zheng W-X, Zhang C-T. Prediction of proteinase cleavage sites in polyproteins of coronaviruses and its applications in analyzing SARS-CoV genomes. FEBS Lett. 2003;553(3):451–456. doi: 10.1016/S0014-5793(03)01091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs J, Grum-Tokars V, Zhou Y, Turlington M, Saldanha SA, Chase P, et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J Med Chem. 2013;56(2):534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3(10) doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CC, Kuo CJ, Ko TP, Hsu MF, Tsui YC, Chang SC, et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J Biol Chem. 2009;284(12):7646–7655. doi: 10.1074/jbc.M807947200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Q, Li S, Xue F, Zou Y, Chen C, Bartlam M, et al. Structure of the main protease from a global infectious human coronavirus, HCoV-HKU1. J Virol. 2008;82(17):8647–8655. doi: 10.1128/JVI.00298-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hegyi A, Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J Gen Virol. 2002;83(3):595–599. doi: 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- 17.Chuck C-P, Chow H-F, Wan DC-C, Wong K-B. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS One. 2011;6(11) doi: 10.1371/journal.pone.0027228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 19.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu MF, Kuo CJ, Chang KT, Chang HC, Chou CC, Ko TP, et al. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem. 2005;280(35):31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81(4):853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 22.Šali A, Overington JP. Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci. 1994;3(9):1582–1596. doi: 10.1002/pro.5560030923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 24.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 25.Costanzi S. On the applicability of GPCR homology models to computer-aided drug discovery: a comparison between in silico and crystal structures of the β2-adrenergic receptor. J Med Chem. 2008;51(10):2907–2914. doi: 10.1021/jm800044k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CG, Cheng SC, Chen SC, Li JY, Fang YH, Chen YH, et al. Mechanism for controlling the monomer-dimer conversion of SARS coronavirus main protease. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 5):747–755. doi: 10.1107/S0907444913001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng K, Ma G, Zhou J, Zen M, Zhao W, Jiang Y, et al. Insight into the activity of SARS main protease: Molecular dynamics study of dimeric and monomeric form of enzyme. Proteins. 2007;66(2):467–479. doi: 10.1002/prot.21160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni H, Sotriffer CA, McCammon JA. Ordered water and ligand mobility in the HIV-1 integrase-5CITEP complex: a molecular dynamics study. J Med Chem. 2001;44(19):3043–3047. doi: 10.1021/jm010205y. [DOI] [PubMed] [Google Scholar]
- 29.Eisenhaber F, Lijnzaad P, Argos P, Sander C, Scharf M. The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J Comput Chem. 1995;16(3):273–284. doi: 10.1002/jcc.540160303. [DOI] [Google Scholar]
- 30.MacKerell AD, Bashford D, Bellott M, Dunbrack R, Evanseck J, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 31.My S, Šali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15(11):2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLano WL. The PyMOL molecular graphics system. 2002. [Google Scholar]
- 33.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(suppl 2):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sippl MJ. Recognition of errors in three‐dimensional structures of proteins. Proteins: Struct, Funct, Bioinf. 1993;17(4):355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 36.Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK: A program to produce both detailed and schematic plots of proteins. J Appl Crystallogr. 1993;24:946–956. [Google Scholar]
- 37.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- 38.Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J Chem Theory Computation. 2010;6(5):1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 39.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320(3):597–608. doi: 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 40.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, et al. A hierarchical approach to all‐atom protein loop prediction. Proteins: Struct, Funct, Bioinf. 2004;55(2):351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 41.Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 42.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Physics. 1995;103(19):8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]