

Abstract
IMPORTANCE
Currently, fewer than 40% of patients treated for major depressive disorder achieve remission with initial treatment. Identification of a biological marker that might improve these odds could have significant health and economic impact.
OBJECTIVE
To identify a candidate neuroimaging “treatment-specific biomarker” that predicts differential outcome to either medication or psychotherapy.
DESIGN
Brain glucose metabolism was measured with positron emission tomography prior to treatment randomization to either escitalopram oxalate or cognitive behavior therapy for 12 weeks. Patients who did not remit on completion of their phase 1 treatment were offered enrollment in phase 2 comprising an additional 12 weeks of treatment with combination escitalopram and cognitive behavior therapy.
SETTING
Mood and anxiety disorders research program at an academic medical center.
PARTICIPANTS
Men and women aged 18 to 60 years with currently untreated major depressive disorder.
INTERVENTION
Randomized assignment to 12 weeks of treatment with either escitalopram oxalate (10–20 mg/d) or 16 sessions of manual-based cognitive behavior therapy.
MAIN OUTCOME AND MEASURE
Remission, defined as a 17-item Hamilton Depression Rating Scale score of 7 or less at both weeks 10 and 12, as assessed by raters blinded to treatment.
RESULTS
Positive and negative predictors of remission were identified with a 2-way analysis of variance treatment (escitalopram or cognitive behavior therapy) × outcome (remission or nonresponse) interaction. Of 65 protocol completers, 38 patients with clear outcomes and usable positron emission tomography scans were included in the primary analysis: 12 remitters to cognitive behavior therapy, 11 remitters to escitalopram, 9 nonresponders to cognitive behavior therapy, and 6 nonresponders to escitalopram. Six limbic and cortical regions were identified, with the right anterior insula showing the most robust discriminant properties across groups (effect size = 1.43). Insula hypometabolism (relative to whole-brain mean) was associated with remission to cognitive behavior therapy and poor response to escitalopram, while insula hypermetabolism was associated with remission to escitalopram and poor response to cognitive behavior therapy.
CONCLUSIONS AND RELEVANCE
If verified with prospective testing, the insula metabolism-based treatment-specific biomarker defined in this study provides the first objective marker, to our knowledge, to guide initial treatment selection for depression.
TRIAL REGISTRATION
Registered at clinicaltrials.gov (NCT00367341)
Major depressive disorder (MDD) is a highly prevalent, disabling, and costly illness.1–3 For a patient presenting with MDD, an antidepressant medication or evidence-based psychotherapy is currently recommended as first-line treatment.4–7 However, fewer than 40% of patients achieve remission with initial treatment,8,9 and choosing the “wrong” initial treatment has significant individual and societal costs due to continued distress, risk of suicide, loss of productivity, and wasted resources associated with 2 to 3 months of an ineffective treatment.10,11 Given the public health consequences of inadequately treated depression, a clinical or biological marker to guide initial treatment selection for MDD could have major health and economic impact.12
In other areas of medicine, identification of markers to guide treatment has significantly improved clinical outcome. For example, in cancer13 and heart disease,14 biomarkers are currently used to optimize initial treatment selection as well as guide treatment modifications with disease progression. Over the past several decades, a number of potential markers to guide antidepressant treatment have been investigated including clinical,15 immune,16 inflammatory,17 endocrine,18 genetic,19–21 and imaging/electroencephalography22–27 measures. Despite this extensive research, to our knowledge, no clinically useful marker to guide treatment selection has emerged.
In the process of developing a marker to guide antidepressant treatment selection, it is important to consider what qualities such a marker should have. Toward this goal, a nonspecific biomarker that predicts improvement regardless of treatment is not useful. Rather, a clinically meaningful and treatment-specific biomarker (TSB) should (1) predict an individual’s improvement to a specific treatment and (2) predict nonresponse to an alternative treatment. Such a biomarker can only be identified in a study that assesses outcome to 2 or more different treatments.
Previous neuroimaging studies have suggested that pre-treatment brain activity patterns can predict efficacy, but those studies have generally focused on a particular treatment.26,27 For example, higher rostral cingulate and/or subgenual cingulate activity has been associated with greater improvement with antidepressant medications,28,29 sleep deprivation,30 and cingulotomy.31 Comparisons of different treatments have thus far identified markers of response and nonresponse but not patterns that differentiate among the treatments tested.22,32,33 Further, imaging studies demonstrate that medications and psychotherapy have differential effects on distinct brain regions,23,34 suggesting that baseline activity may indicate response to one treatment vs the other. Although, to our knowledge, no prior imaging study has directly assessed the association of pretreatment brain activity patterns with differential response to different treatments (eg, medication vs psychotherapy), these past studies strongly suggest that a neuroimaging-based TSB can be defined.
In this study, we measured pretreatment brain glucose metabolism in patients with MDD randomized to receive a selective serotonin reuptake inhibitor (escitalopram oxalate) or cognitive behavior therapy (CBT).35,36 Positron emission tomography (PET) scan measurement of glucose metabolism was selected based on its high reliability and availability combined with its established use for studies of baseline scan patterns in depression and effects of various antidepressant treatments.22,23,28,34,37–43 Our aim was to define an imaging TSB for these 2 potential first-line treatments, ie, a brain activity pattern that distinguishes escitalopram remitters from both escitalopram nonresponders and CBT remitters while concurrently distinguishing CBT remitters from both CBT nonresponders and escitalopram remitters.
Methods
Patient Selection
Written informed consent was obtained from all participants, with the protocol conducted as approved by the Emory institutional review board and registered at clinicaltrials.gov (NCT00367341). Eligible participants were adult outpatients with a primary diagnosis of MDD as assessed by the Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition With Psychotic Screen44 and confirmed through a psychiatric evaluation conducted by a study psychiatrist. Patients aged 18 to 60 years were recruited through the Mood and Anxiety Disorders Program at Emory University via advertisements and clinician referrals.45 Patients were required to have moderate to severe symptoms of depression, defined as a 17-item Hamilton Depression Rating Scale (HDRS)46 score of 18 or more at screening and 15 or more at the baseline randomization visit. Exclusion criteria included a current diagnosis of a primary psychiatric disorder other than MDD; a medical or neurological condition that could contribute to depression or that might interfere with response to treatment such as chronic pain syndromes and irritable bowel syndrome; current suicidal ideation requiring urgent clinical intervention; comorbid substance abuse within the past 3 months; substance dependence within 12 months prior to the screening visit; current or intended pregnancy or breastfeeding; use of antidepressants within 7 days of the screening visit (5 weeks for fluoxetine); current psychotherapy at the time of screening; or receipt of electroconvulsive therapy within 6 months of the screening visit. Patients were also excluded if they had a lifetime history of failure to respond to 6 or more weeks of treatment with escitalopram oxalate (≥10 mg/d) or 4 or more sessions of CBT for depression.
Treatment Protocol
Treatment consisted of 2 phases: a short-term treatment phase (phase 1) and a combination treatment phase (phase 2). Phase 1 provided the data for this report. In phase 1, patients were randomly assigned (1:1 ratio) to receive a 12-week treatment course of either escitalopram oxalate (flexibly dosed from 10–20 mg/d) or manual-based, depression-focused CBT (16 one-hour sessions over 12 weeks) (Figure 1). Prior to study start, the study statistician prepared a permuted-block randomization schedule, with the assignments placed in order and sealed in opaque envelopes. Following acquisition of the pretreatment PET and magnetic resonance imaging scans, patients who continued to meet eligibility criteria were randomized to escitalopram or CBT. Escitalopram oxalate was started at 10 mg/d and could be increased to 20 mg/d at or after week 3 if the patient had an HDRS score more than 7 and was tolerating the medication. Down-titration to 10 mg/d was permitted if adverse effects were intolerable at the 20–mg/d dose. The CBT sessions were scheduled twice weekly for the first 4 weeks, followed by weekly sessions for the subsequent 8 weeks. Changes in symptom severity were assessed using the HDRS conducted by raters blinded to treatment group. Ratings were performed weekly for the first 6 weeks and then biweekly until week 12. Patients who did not remit on completion of their phase 1 treatment were offered enrollment in phase 2 comprising an additional 12 weeks of treatment with combination escitalopram and CBT.
Figure 1. Study Design and Outcomes.
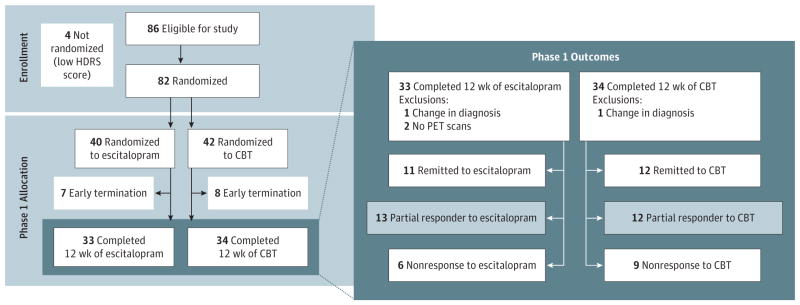
Outcome groups defined by Hamilton Depression Rating Scale (HDRS) scores. Remission was defined as an HDRS score of 7 or less; partial response, as an HDRS score decrease of more than 30% but not achieving remission; and nonresponse, as an HDRS score decrease of 30% or less. Escitalopram was given as escitalopram oxalate. CBT indicates cognitive behavior therapy and PET, positron emission tomography.
Outcome Metrics
Clinical outcomes were defined using the HDRS, with the target end point being remission, defined as an HDRS score of 7 or less at both weeks 10 and 12 of phase 1 treatment,47 to ensure stability of remission beyond a single “good week.” Nonresponse was defined as a 30% or less HDRS score change from baseline to the phase 1 end point.48 Partial responders (change in HDRS score >30% but not achieving remission) and dropouts were not included in the analyses for this report to avoid potential dilution of either the remission or the nonresponse groups.
Imaging Acquisition
Prior to treatment randomization, brain glucose metabolism was measured using PET (High-Resolution Research Tomograph scanner; Siemens), using standard methods without arterial blood sampling.49 For each scan, a 370-MBq dose of fludeoxyglucose F18 (FDG) was administered intravenously, with a 20-minute 3-dimensional image acquisition beginning 40 minutes after tracer injection. During uptake, patients remained supine, awake, and resting with eyes closed and ears uncovered. Patients were given no explicit cognitive instructions but were asked to avoid ruminating on any 1 topic during the 40-minute FDG uptake period.21 Raw emission images were corrected for injected dose and attenuation (using cesium 137, 6-minute transmission scan), reconstructed, and smoothed to an in-plane resolution of 4.0 mm Full-Width Half-Maximum.34 A high-resolution T1-weighted structural magnetic resonance imaging scan was separately acquired for spatial normalization procedures and anatomical reference (TIM Trio 3-T whole-body scanner; Siemens) (3-dimensional magnetization-prepared rapid acquisition with gradient echo optimized at echo time = 5 milliseconds, repetition time = 35 milliseconds, matrix = 256 × 208 × 196, and 1-mm isotropic resolution).
Image Preprocessing
Attenuation-corrected PET images were coregistered to corresponding T1-weighted magnetic resonance imaging anatomical images using a 6-df linear transform and subsequently written into standard space using a nonlinear transform calculated from the T1-weighted image (DARTEL50 and SPM8; Wellcome Trust Centre for Neuroimaging, http://www.fil.ion.ucl.ac.uk/spm/). Four patients had no anatomical scan and were normalized using a study-specific FDG template. Spatially normalized images were smoothed with an 8-mm Full-Width Half-Maximum Gaussian kernel and corrected for differences in whole-brain global mean activity.23 Relative glucose metabolic rates were used for all analyses.
Image Analysis
A 2-way analysis of variance (ANOVA) with treatment (escitalopram or CBT) and outcome (remission or nonresponse) was performed to identify a putative escitalopram/CBT remission TSB using the baseline pretreatment FDG-PET scans (analyses performed with AFNI [National Institute of Mental Health, National Institutes of Health] and SPSS [IBM SPSS], statistical threshold P < .001 uncorrected, and a minimum cluster volume of 100 voxels, 0.34 mL). With this approach, a main effect of remission would identify brain regions associated with remission to treatment independent of randomization group, ie, a nonspecific biomarker. The treatment × outcome interaction would identify brain regions where the CBT treatment effect (remission or nonresponse) was distinguished from the escitalopram treatment effect (remission or nonresponse). Average normalized glucose metabolism values were extracted from clusters identified by the ANOVA (mean cluster activity) for further analysis.
Post hoc analyses of the extracted regions from the ANOVA interaction were used to refine selection of a potential TSB pattern by examining the effect sizes of the group differences for each region. We defined a region as a true TSB if it differentiated both the remission and nonresponse differences (by treatment) and the escitalopram and CBT differences (by outcome); thus, there were 4 comparisons of interest to consider when evaluating each region of interest as a stratification tool for treatment recommendation. Given the limited sample size, we report these comparisons using effect size, rather than statistical significance, to quantify their actual potential use as an eventual TSB. The 2-group effect size can be interpreted as the difference in metabolic activity between specified groups in units of standard deviation.51 Because each region had a different magnitude of glucose metabolic activity and variation, each individual value was standardized using a z score, with regional z score means plotted to illustrate the nature of the regional interaction effects. Because these data are already standardized to the level of variation, there are no “error bars” in the related graphs.
To further assess the generalizability of findings identified in this restricted analysis to the full sample of study completers, metabolic activity was correlated with percentage of change in HDRS score within each treatment group to determine if the putative biomarkers identified in the ANOVA showed the predicted general pattern in the full cohort of phase 1 treatment completers.
Results
Clinical Effects
Eighty-two patients were randomized to treatment; however, 2 patients received a change in their psychiatric diagnosis during the trial, and they were not used in the analyses. This resulted in 41 randomized to CBT and 39 to escitalopram. Sixty-five patients completed phase 1; 63 of the completers (79% of the total sample) had baseline FDG-PET scans available for analysis. Phase 1 remission rates were similar for both treatments: CBT = 12 of 33 (36.3%) and escitalopram = 12 of 30 (40.0%) (Figure 1 and Table 1). Nonresponse rates were also similar for both treatments: CBT = 9 of 33 (27.3%) and escitalopram = 6 of 30 (20.0%). Thirty-eight patients with clear outcomes and usable PET scans were included in the primary analysis: 12 patients with CBT remission, 11 patients with escitalopram remission, 9 patients with CBT nonresponse, and 6 patients with escitalopram nonresponse. There were no statistical differences in age, sex, or demographic or illness characteristics between randomization groups (escitalopram vs CBT). There were also no baseline demographic differences among the treatment-specific phase 1 outcome groups (Table 1). However, CBT nonresponders had higher baseline anxiety ratings (Hamilton Anxiety Rating Scale total score).
Table 1.
Group Comparisons of Clinical Characteristics
| Variable | CBT
|
Escitalopram Oxalate
|
Significance of Group × Treatment Interaction | ||
|---|---|---|---|---|---|
| Nonresponders (n = 9) | Remitters (n = 12) | Nonresponders (n = 6) | Remitters (n = 12)a | ||
| Age, y, mean (SD) | 45.4 (8.8) | 42.5 (10.8) | 40.3 (5.2) | 39.8 (6.3) | .67b |
|
| |||||
| Male, No. (%) | 3 (33.3) | 7 (58.3) | 2 (33.3) | 5 (41.7) | 0.628c |
|
| |||||
| White, No. (%) | 7 (77.8) | 8 (66.7) | 5 (100) | 8 (72.7) | 0.345c |
|
| |||||
| Education, y, mean (SD) | 14.7 (1.7) | 15.6 (1.6) | 15.4 (1.7) | 16.2 (2.1) | .94b |
|
| |||||
| Age at onset of MDD, y, mean (SD) | 28.4 (12.3) | 28.7 (11.2) | 24.0 (11.6) | 25.5 (10.7) | .88b |
|
| |||||
| ≥3 Lifetime episodes, No. (%) | 4 (50.0) | 4 (33.3) | 2 (33.3) | 4 (33.3) | 0.623c |
|
| |||||
| Duration of current episode, wk, mean (SD) | 124.5 (118.5) | 257.3 (308.8) | 299.8 (620.0) | 135.3 (266.2) | .20b |
|
| |||||
| No. of previous AD trials in current episode, mean (SD) | 1.2 (1.5) | 1.2 (1.0) | 1.5 (1.4) | 1.0 (1.1) | .59b |
|
| |||||
| Melancholic subtype, No. (%) | 4 (44.4) | 5 (45.5) | 3 (50.0) | 6 (50.0) | 0.976c |
|
| |||||
| Current anxiety disorder, No. (%) | 3 (33.3) | 2 (16.7) | 1 (16.7) | 5 (41.7) | |
|
| |||||
| Baseline HDRS score, mean (SD) | 19.9 (3.8) | 17.9 (2.7) | 18.0 (2.1) | 19.3 (3.7) | .13b |
|
| |||||
| Baseline HAMA total score, mean (SD) | 18.9 (7.6) | 12.8 (2.8) | 13.3 (2.3) | 15.3 (3.0) | .009b |
|
| |||||
| BDI total score, mean (SD) | 19.2 (4.6) | 18.7 (7.2) | 20.0 (3.8) | 21.5 (7.7) | .64b |
Abbreviations: AD, antidepressant; BDI, Beck Depression Inventory; CBT, cognitive behavior therapy; HAMA, Hamilton Anxiety Rating Scale; HDRS, 17-item Hamilton Depression Rating Scale; MDD, major depressive disorder.
Only 11 of 12 escitalopram remitters had usable positron emission tomography scans.
P values from 2-way analysis of variance.
P values from test of the homogeneity of the odds ratio (Breslow-Day).
Neuroimaging Results
Treatment×Outcome ANOVA
There was no significant main effect of remission, ie, no treatment-nonspecific biomarker was identified. Significant treatment × outcome interaction effects were demonstrated for 6 regions: right anterior insula, right inferior temporal cortex (Brodmann area [BA] 20), left amygdala, left premotor cortex (BA 6), right motor cortex (BA 4), and precuneus (BA 7) (Table 2 and Figure 2).
Table 2.
Treatment by Outcome Interaction Results and Post hoc Analyses of Extracted Regions of Interest
| Region | MNI Coordinates, Peak
|
Side | Cluster Size,a Voxels | Effect Sizesb
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| X | Y | Z | REM-NR to CBT | REM-NR to sCIT | CBT-sCIT in NR | CBT-sCIT in REM | Average Marginal ES | |||
| Anterior insula | +30.0 | +24.0 | −13.5 | R | 529 | 1.69 | 1.17 | 1.52 | 1.33 | 1.43 |
| Inferior temporal (BA 20) | +42.0 | −33.0 | −25.5 | R | 469 | 1.23 | 1.45 | 2.09 | 0.59 | 1.34 |
| Amygdala | −27.0 | −7.5 | −27.0 | L | 233 | 0.98 | 1.61 | 1.89 | 0.69 | 1.29 |
| Premotor (BA 6) | −27.0 | +1.5 | +58.5 | L | 233 | 1.40 | 1.03 | 1.75 | 0.68 | 1.22 |
| Motor (BA 4) | +25.5 | −27.0 | +60.0 | R | 147 | 0.61 | 1.78 | 1.80 | 0.58 | 1.19 |
| Precuneus (BA 7) | −18.0 | −67.5 | +43.5 | L | 101 | 1.18 | 1.27 | 1.37 | 1.08 | 1.23 |
Abbreviations: BA, Brodmann area; CBT, cognitive behavior therapy; CBT-sCIT, mean difference between CBT and escitalopram oxalate (treatment effect); MNI, Montreal Neurological Institute; NR, nonresponders; REM, remitters; REM-NR, mean difference between remitters and nonresponders (remission effect); sCIT: escitalopram oxalate.
Whole-brain 2-way analysis of variance with P < .001 uncorrected; voxel size, 1.5 mm × 1.5 mm × 1.5 mm.
Effect size = mean difference divided by pooled standard deviation.
Figure 2. Potential Treatment-Specific Biomarker Candidates.

Mean regional activity values for remitters and nonresponders segregated by treatment arm are plotted for the 6 regions showing a significant treatment × outcome analysis of variance interaction effect. Regional metabolic activity values are displayed as region/whole-brain metabolism converted to z scores. Regions match those shown in Table 2. Escitalopram was given as escitalopram oxalate. CBT indicates cognitive behavior therapy.
Post hoc Analyses of Extracted Regions of Interest
The average effect sizes of each region for the various contrasts are shown in Table 2 in order of cluster size from the ANOVA. This was used to rank the regions of interest in the order of their potential utility as a discriminator. Only the insula and precuneus showed differences larger than 1 SD in all 4 contrasts, with the insula showing the largest average difference across all 4 comparisons. These findings indicate that metabolic activity of the right anterior insula is the most viable TSB candidate (Table 2 and Figure 3). Further, the anterior insula was the only region that showed relative hypometabolism in 1 group (region/whole-brain mean <1.0) and hypermetabolism in the other (region/whole-brain mean >1.0), adding support for potential use as a treatment stratification tool.
Figure 3. Right Anterior Insula as the Optimal Treatment-Specific Biomarker Candidate.

Expanded view of findings. A, Scatterplot of insular activity from individual subjects in the remitter (REM) and nonresponder (NR) groups. Note: the anterior insula is the only region where the interaction subdivides patients into hypermetabolic (region/whole-brain mean >1.0) and hypometabolic (region/whole-brain mean <1.0) subgroups. B, Correlations of insula activity with percentage of change in Hamilton Depression Rating Scale (HDRS) score in the full cohort of subjects treated with cognitive behavior therapy (CBT) and escitalopram oxalate.
Assessment of the Insula TSB Across the Full Sample
There was a significant correlation between baseline insula activity and percentage of change in HDRS scores in both the CBT and escitalopram groups. A positive correlation was shown for the CBT group (r = 0.55; df = 31; P = .001) (Figure 3). In contrast, the escitalopram-treated patients showed an opposite but less significant correlation (r = −0.31; df = 28; P = .09). Both correlations are consistent with the more restricted findings in the binarized remitter-nonresponder analyses.
Although not a primary planned analysis, the presence of multiple regions identified in the ANOVA suggests that a combination rather than a single TSB might be more accurate in discriminating the groups. Although underpowered, a principal component analysis was performed using the 6 identified regions of interest. All regions loaded on 1 factor, which did not provide superior internal consistency to the insula alone (data not shown).
Discussion
This 12-week randomized study of 2 first-line treatments for MDD identified 2 FDG-PET–defined brain pattern subtypes that differentially predicted remission to either CBT or escitalopram. Among the 6 identified cortical and limbic regions, the anterior insula metabolism best discriminated treatment outcome: insula hypometabolism was associated with remission to CBT and poor response to escitalopram, while insula hypermetabolism was associated with remission to escitalopram and poor response to CBT. These data suggest that insula metabolism alone (relative to each person’s whole-brain mean metabolism) may serve as a pretreatment biomarker to guide initial treatment selection (medication vs CBT) for a patient presenting with a major depressive episode. To validate the insula TSB, a prospective replication study in which patients are treated according to brain type will be required. That said, this forced-choice analytic strategy establishes a potential stratification algorithm for managing patients with MDD based on brain state rather than patient or professional preference, anticipating the real-world decision-making process faced by clinicians, namely, choosing a first treatment that will most likely lead to remission while also avoiding a treatment that is likely to fail.
A role for the anterior insula in major depression is well established. The insula is crucial in mediating the translation of visceral experiences to subjective feeling states.52 Additionally, anterior insula activity is linked to behaviors relevant to depression including interoception, emotional self-awareness, decision making, and cognitive control.53–55 The anterior insula is extensively connected to various frontal, limbic, and brainstem regions, including the anterior cingulate cortex, amygdala, and hypothalamus.56 Volume reductions of the anterior but not posterior insula have been described in currently ill patients with MDD as well as patients with remitted MDD compared with healthy controls.57 Changes in insula activity occur with a variety of treatments for MDD, including medication,58 vagus nerve stimulation,59 deep brain stimulation,60 and mindfulness training,61 suggesting a role for this region in mediating antidepressant response and remission more generally.62 Notably, past studies have reported both increases33 and decreases39 in baseline resting-state activity relative to never-depressed control subjects. This is consistent with the presence of at least 2 baseline patterns within the broader population of depressed patients. Most recently, baseline insula activity has been correlated with response to vagus nerve stimulation.26 These previous studies taken together with the current findings support the anterior insula as a potential candidate for an imaging TSB.
Contrary to past published studies,63 the rostral anterior cingulate did not discriminate the outcome subgroups in either the main effect or interaction analyses. A post hoc examination of responder and nonresponder differences within each treatment arm did reveal a nonsignificant rostral cingulate activity difference, with metabolism in responders greater than nonresponders, but solely in the escitalopram group. While consistent with past reports, this finding did not meet the TSB criteria defined for the current study, ie, a region whose activity can differentiate both good and poor outcomes for both treatments.
Critical to the stated aims, remission (rather than response) was the targeted end point in this study because the presence of residual symptoms is a known predictor of clinical relapse, even in patients with significant improvement.64,65 Because the primary aim of this study was to identify distinct brain patterns that optimally predict remission to each of 2 specific treatments, patients with unclear treatment outcomes were excluded from the primary analysis (ie, responders without remission and partial responders). This enriched sample allowed for detection of clear remission and nonresponse signals; as such, these analyses did not attempt to characterize the neurobiological variability of patients with more ambiguous clinical outcomes. This is a commonly used approach when the goal is to develop or test a biological signal for stratifying subjects.66,67 Nevertheless, baseline insula activity did correlate significantly with change in depression severity across all subjects, supporting the interpretation that insula activity is a plausible TSB suitable for further testing. Based on the correlational analysis across all subjects, the data further suggest that the anterior insula TSB may most optimally identify those patients who require CBT.
If confirmed with prospective testing, this putative TSB has both clinical and pathophysiological implications. At present, treatment failure with antidepressant medication often leads to the addition of a second drug and not a categorical switch to an evidence-based psychotherapy.68 Results herein suggest that patients who require CBT have a distinct neurophysiology that differs categorically from patients who require escitalopram and knowledge of such may help to improve current clinical practice patterns. Further, using this or any other imaging-based TSB to define patient subgroups provides a brain-based platform to investigate genetic, immune, neuroendocrine, and behavioral variations from a new perspective.
While these first results are encouraging, there are several limitations. Clearly, there are patients who are not successfully treated with either of these 2 options, either alone or in combination.69 Therefore, our strategy can be best seen as a first-line stratification approach to treatment selection. Future studies, in addition to testing this insula biomarker prospectively, should include a design that works to identify patients resistant to both of these first-line treatments.60,70
The lack of a placebo arm could be considered a limitation, but given the randomized design of the study, there is no reason to believe that placebo responders would be unevenly distributed between the 2 groups. Thus, even if present, placebo effects on remission rates would be expected to be similar in both treatment groups. Although inclusion of a placebo arm might have provided further insights into mediators of improvement during treatment, the absence of a placebo arm does not diminish the potential clinical utility of the identified TSB.
It is also possible that these results are specific to the cohort recruited for this trial. As such, a stratification strategy based on insula metabolism will require prospective testing in a new group of comparably depressed patients. Similarly, additional studies will be required to determine if remitters to other medications have a similar or different TSB from that seen with escitalopram or if remitters to other evidence-based psychotherapies have a similar TSB to that seen with CBT.71,72 Such studies are critical next steps toward the development of biology-based algorithms to guide treatment selection for MDD at all stages of illness. Still, if replicated, the insula TSB defined in this study would provide the first objective marker to guide initial treatment selection for major depression, an important advance in potentially reducing the costs and disability associated with this highly prevalent disorder.
Acknowledgments
Funding/Support: This work was supported by National Institutes of Health grants R01 MH073719 (Dr Mayberg), T32 GM08695 (Ms McGrath), K23 MH086690 (Dr Dunlop), and K23 MH077869 (Dr Holtzheimer).
Footnotes
Author Contributions: Dr Mayberg had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Additional Contributions: Treating psychotherapists: Sheethal Reddy, PhD, Patrick Sylvers, PhD, Lorie Ritschel, PhD, Meredith Jones, PhD, Mary Heekin, LCSW, Maryrose Gerardi, PhD, and Jill Rosenberg, LCSW. Treating physicians: Ebrahim Haroon, MD, Jeffrey Rakofsky, MD, Dylan Wint, MD, and Corey Beck, MD. Clinical coordinators: Ronald Chismar, RN, Melanie Galanti, BA, Rachelle Gibson, RN, Lauren Marx, BS, Melissa McKenzie, BA, and Tanja Mletzko, MA. Imaging team: Rebecca DeMayo, MA, Eundria Hill, MSW, Kiseung Choi, MS, and Justin Rajendra, BA. Raters: Margo Aaron, BA, Yara Betancourt, BA, Cristina Velasquez Delgado, BA, Novall Khan, BS, Ximena Marincic, BS, and Christopher Vaughan, BA.
Conflict of Interest Disclosures: Dr Dunlop has received honoraria for consulting work performed for Bristol-Myers Squibb, MedAvante, Pfizer, and Roche. He has received research support from AstraZeneca, Bristol-Myers Squibb, Evotec, Forest, GlaxoSmithKline, Novartis, Ono Pharmaceuticals, Pfizer, Shire, Takeda, and Transcept. Dr Holtzheimer has received consulting fees from St Jude Medical Neuromodulation and Cervel Neurotech and an honorarium from Johnson & Johnson. Dr Craighead is a board member of Hugarheill, an Icelandic company dedicated to the prevention of depression, and he receives book royalties from John Wiley & Sons. He is a consultant to the George West Mental Health Foundation that oversees Skyland Trail, a residential treatment facility in Atlanta, Georgia. Dr Mayberg has received consulting and intellectual property licensing fees from St Jude Medical Inc.
References
- 1.Kessler RC, Berglund P, Demler O, et al. National Comorbidity Survey Replication. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289(23):3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Birnbaum HG, Kessler RC, Kelley D, Ben-Hamadi R, Joish VN, Greenberg PE. Employer burden of mild, moderate, and severe major depressive disorder: mental health services utilization and costs, and work performance. Depress Anxiety. 2010;27(1):78–89. doi: 10.1002/da.20580. [DOI] [PubMed] [Google Scholar]
- 3.Judd LL, Akiskal HS, Zeller PJ, et al. Psychosocial disability during the long-term course of unipolar major depressive disorder. Arch Gen Psychiatry. 2000;57(4):375–380. doi: 10.1001/archpsyc.57.4.375. [DOI] [PubMed] [Google Scholar]
- 4.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychiatric Association; 2000. text revision. [Google Scholar]
- 5.Kennedy SH, Lam RW, Parikh SV, Patten SB, Ravindran AV. Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults: introduction. J Affect Disord. 2009;117(suppl 1):S1–S2. doi: 10.1016/j.jad.2009.06.043. [DOI] [PubMed] [Google Scholar]
- 6.American Psychiatric Association. Treating Major Depressive Disorder: A Quick Reference Guide. Washington, DC: American Psychiatric Association; 2010. pp. 1–28. [Google Scholar]
- 7.National Institute for Health and Clinical Excellence. Depression: The Treatment and Management of Depression in Adults. London, England: National Institute for Health and Clinical Excellence; 2009. NICE clinical guideline 90. [Google Scholar]
- 8.Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. 2009;60(11):1439–1445. doi: 10.1176/ps.2009.60.11.1439. [DOI] [PubMed] [Google Scholar]
- 9.Holtzheimer PE, Mayberg HS. Stuck in a rut: rethinking depression and its treatment. Trends Neurosci. 2011;34(1):1–9. doi: 10.1016/j.tins.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessler RC, Akiskal HS, Ames M, et al. Prevalence and effects of mood disorders on work performance in a nationally representative sample of U.S. workers. Am J Psychiatry. 2006;163(9):1561–1568. doi: 10.1176/appi.ajp.163.9.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunlop BW, Reddy S, Yang L, Lubaczewski S, Focht K, Guico-Pabia CJ. Symptomatic and functional improvement in employed depressed patients: a double-blind clinical trial of desvenlafaxine versus placebo. J Clin Psychopharmacol. 2011;31(5):569–576. doi: 10.1097/JCP.0b013e31822c0a68. [DOI] [PubMed] [Google Scholar]
- 12.Kapur S, Phillips AG, Insel TR. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol Psychiatry. 2012;17(12):1174–1179. doi: 10.1038/mp.2012.105. [DOI] [PubMed] [Google Scholar]
- 13.Saijo N. Critical comments for roles of biomarkers in the diagnosis and treatment of cancer. Cancer Treat Rev. 2012;38(1):63–67. doi: 10.1016/j.ctrv.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 14.Welch TD, Yang EH, Reeder GS, Gersh BJ. Modern management of acute myocardial infarction. Curr Probl Cardiol. 2012;37(7):237–310. doi: 10.1016/j.cpcardiol.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Quitkin FM, Stewart JW, McGrath PJ, et al. Columbia atypical depression: a subgroup of depressives with better response to MAOI than to tricyclic antidepressants or placebo. Br J Psychiatry Suppl. 1993;(21):30–34. [PubMed] [Google Scholar]
- 16.Irwin MR, Miller AH. Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun. 2007;21(4):374–383. doi: 10.1016/j.bbi.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Müller N, Myint A-M, Schwarz MJ. Inflammatory biomarkers and depression. Neurotox Res. 2011;19(2):308–318. doi: 10.1007/s12640-010-9210-2. [DOI] [PubMed] [Google Scholar]
- 18.Arana GW, Baldessarini RJ, Ornsteen M. The dexamethasone suppression test for diagnosis and prognosis in psychiatry: commentary and review. Arch Gen Psychiatry. 1985;42(12):1193–1204. doi: 10.1001/archpsyc.1985.01790350067012. [DOI] [PubMed] [Google Scholar]
- 19.Huezo-Diaz P, Uher R, Smith R, et al. Moderation of antidepressant response by the serotonin transporter gene. Br J Psychiatry. 2009;195(1):30–38. doi: 10.1192/bjp.bp.108.062521. [DOI] [PubMed] [Google Scholar]
- 20.Ising M, Lucae S, Binder EB, et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch Gen Psychiatry. 2009;66(9):966–975. doi: 10.1001/archgenpsychiatry.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Empaire I, Guico-Pabia CJ, Preskorn SH. Antidepressant treatment and altered CYP2D6 activity: are pharmacokinetic variations clinically relevant? J Psychiatr Pract. 2011;17(5):330–339. doi: 10.1097/01.pra.0000405363.95881.01. [DOI] [PubMed] [Google Scholar]
- 22.Konarski JZ, Kennedy SH, Segal ZV, et al. Predictors of nonresponse to cognitive behavioural therapy or venlafaxine using glucose metabolism in major depressive disorder. J Psychiatry Neurosci. 2009;34(3):175–180. [PMC free article] [PubMed] [Google Scholar]
- 23.Kennedy SH, Konarski JZ, Segal ZV, et al. Differences in brain glucose metabolism between responders to CBT and venlafaxine in a 16-week randomized controlled trial. Am J Psychiatry. 2007;164(5):778–788. doi: 10.1176/ajp.2007.164.5.778. [DOI] [PubMed] [Google Scholar]
- 24.DeBattista C, Kinrys G, Hoffman D, et al. The use of referenced-EEG (rEEG) in assisting medication selection for the treatment of depression. J Psychiatr Res. 2011;45(1):64–75. doi: 10.1016/j.jpsychires.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Leuchter AF, Cook IA, Marangell LB, et al. Comparative effectiveness of biomarkers and clinical indicators for predicting outcomes of SSRI treatment in Major Depressive Disorder: results of the BRITE-MD study. Psychiatry Res. 2009;169(2):124–131. doi: 10.1016/j.psychres.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Conway CR, Chibnall JT, Gangwani S, et al. Pretreatment cerebral metabolic activity correlates with antidepressant efficacy of vagus nerve stimulation in treatment-resistant major depression: a potential marker for response? J Affect Disord. 2012;139(3):283–290. doi: 10.1016/j.jad.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siegle GJ, Thompson WK, Collier A, et al. Toward clinically useful neuroimaging in depression treatment: prognostic utility of subgenual cingulate activity for determining depression outcome in cognitive therapy across studies, scanners, and patient characteristics. Arch Gen Psychiatry. 2012;69(9):913–924. doi: 10.1001/archgenpsychiatry.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayberg HS, Brannan SK, Mahurin RK, et al. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. 1997;8(4):1057–1061. doi: 10.1097/00001756-199703030-00048. [DOI] [PubMed] [Google Scholar]
- 29.Pizzagalli D, Pascual-Marqui RD, Nitschke JB, et al. Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am J Psychiatry. 2001;158(3):405–415. doi: 10.1176/appi.ajp.158.3.405. [DOI] [PubMed] [Google Scholar]
- 30.Wu J, Buchsbaum MS, Gillin JC, et al. Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. Am J Psychiatry. 1999;156(8):1149–1158. doi: 10.1176/ajp.156.8.1149. [DOI] [PubMed] [Google Scholar]
- 31.Dougherty DD, Weiss AP, Cosgrove GR, et al. Cerebral metabolic correlates as potential predictors of response to anterior cingulotomy for treatment of major depression. J Neurosurg. 2003;99(6):1010–1017. doi: 10.3171/jns.2003.99.6.1010. [DOI] [PubMed] [Google Scholar]
- 32.Brody AL, Saxena S, Stoessel P, et al. Regional brain metabolic changes in patients with major depression treated with either paroxetine or interpersonal therapy: preliminary findings. Arch Gen Psychiatry. 2001;58(7):631–640. doi: 10.1001/archpsyc.58.7.631. [DOI] [PubMed] [Google Scholar]
- 33.Ketter TA, Kimbrell TA, George MS, et al. Baseline cerebral hypermetabolism associated with carbamazepine response, and hypometabolism with nimodipine response in mood disorders. Biol Psychiatry. 1999;46(10):1364–1374. doi: 10.1016/s0006-3223(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 34.Goldapple K, Segal Z, Garson C, et al. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61(1):34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- 35.Beck A, Rush A, Shaw B, Emery G. Cognitive Therapy of Depression. New York, NY: Guilford; 1979. [Google Scholar]
- 36.Beck AT. The current state of cognitive therapy: a 40-year retrospective. Arch Gen Psychiatry. 2005;62(9):953–959. doi: 10.1001/archpsyc.62.9.953. [DOI] [PubMed] [Google Scholar]
- 37.Bartlett EJ, Barouche F, Brodie JD, et al. Stability of resting deoxyglucose metabolic values in PET studies of schizophrenia. Psychiatry Res. 1991;40(1):11–20. doi: 10.1016/0925-4927(91)90025-l. [DOI] [PubMed] [Google Scholar]
- 38.Brody AL, Saxena S, Silverman DH, et al. Brain metabolic changes in major depressive disorder from pre- to post-treatment with paroxetine. Psychiatry Res. 1999;91(3):127–139. doi: 10.1016/s0925-4927(99)00034-7. [DOI] [PubMed] [Google Scholar]
- 39.Kimbrell TA, Ketter TA, George MS, et al. Regional cerebral glucose utilization in patients with a range of severities of unipolar depression. Biol Psychiatry. 2002;51(3):237–252. doi: 10.1016/s0006-3223(01)01216-1. [DOI] [PubMed] [Google Scholar]
- 40.Drevets WC, Price JL, Bardgett ME, Reich T, Todd RD, Raichle ME. Glucose metabolism in the amygdala in depression: relationship to diagnostic subtype and plasma cortisol levels. Pharmacol Biochem Behav. 2002;71(3):431–447. doi: 10.1016/s0091-3057(01)00687-6. [DOI] [PubMed] [Google Scholar]
- 41.Saxena S, Brody AL, Ho ML, Zohrabi N, Maidment KM, Baxter LR., Jr Differential brain metabolic predictors of response to paroxetine in obsessive-compulsive disorder versus major depression. Am J Psychiatry. 2003;160(3):522–532. doi: 10.1176/appi.ajp.160.3.522. [DOI] [PubMed] [Google Scholar]
- 42.Little JT, Ketter TA, Kimbrell TA, et al. Bupropion and venlafaxine responders differ in pretreatment regional cerebral metabolism in unipolar depression. Biol Psychiatry. 2005;57(3):220–228. doi: 10.1016/j.biopsych.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 43.Milak MS, Parsey RV, Lee L, et al. Pretreatment regional brain glucose uptake in the midbrain on PET may predict remission from a major depressive episode after three months of treatment. Psychiatry Res. 2009;173(1):63–70. doi: 10.1016/j.pscychresns.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition With Psychotic Screen (SCID-I/P W/PSY SCREEN) New York: Biometrics Research, New York State Psychiatric Institute; 2002. [Google Scholar]
- 45.Dunlop BW, Kelley ME, Mletzko TC, Velasquez CM, Craighead WE, Mayberg HS. Depression beliefs, treatment preference, and outcomes in a randomized trial for major depressive disorder. J Psychiatr Res. 2012;46(3):375–381. doi: 10.1016/j.jpsychires.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rush AJ, Trivedi MH, Stewart JW, et al. Combining Medications to Enhance Depression Outcomes (CO-MED): acute and long-term outcomes of a single-blind randomized study. Am J Psychiatry. 2011;168(7):689–701. doi: 10.1176/appi.ajp.2011.10111645. [DOI] [PubMed] [Google Scholar]
- 48.Nierenberg AA, Farabaugh AH, Alpert JE, et al. Timing of onset of antidepressant response with fluoxetine treatment. Am J Psychiatry. 2000;157(9):1423–1428. doi: 10.1176/appi.ajp.157.9.1423. [DOI] [PubMed] [Google Scholar]
- 49.Phelps ME, Huang SC, Hoffman EJ, Selin C, Sokoloff L, Kuhl DE. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: validation of method. Ann Neurol. 1979;6(5):371–388. doi: 10.1002/ana.410060502. [DOI] [PubMed] [Google Scholar]
- 50.Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38(1):95–113. doi: 10.1016/j.neuroimage.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 51.Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2. Hillsdale, NJ: Lawrence Erlbaum Associated; 1988. [Google Scholar]
- 52.Critchley HD, Wiens S, Rotshtein P, Ohman A, Dolan RJ. Neural systems supporting interoceptive awareness. Nat Neurosci. 2004;7(2):189–195. doi: 10.1038/nn1176. [DOI] [PubMed] [Google Scholar]
- 53.Farb NA, Segal ZV, Anderson AK. Attentional modulation of primary interoceptive and exteroceptive cortices. Cereb Cortex. 2013;23(1):114–126. doi: 10.1093/cercor/bhr385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Craig AD. How do you feel—now? The anterior insula and human awareness. Nat Rev Neurosci. 2009;10(1):59–70. doi: 10.1038/nrn2555. [DOI] [PubMed] [Google Scholar]
- 55.Critchley HD. Neural mechanisms of autonomic, affective, and cognitive integration. J Comp Neurol. 2005;493(1):154–166. doi: 10.1002/cne.20749. [DOI] [PubMed] [Google Scholar]
- 56.Augustine JR. Circuitry and functional aspects of the insular lobe in primates including humans. Brain Res Brain Res Rev. 1996;22(3):229–244. doi: 10.1016/s0165-0173(96)00011-2. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi T, Yücel M, Lorenzetti V, et al. Volumetric MRI study of the insular cortex in individuals with current and past major depression. J Affect Disord. 2010;121(3):231–238. doi: 10.1016/j.jad.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 58.Kennedy SH, Evans KR, Krüger S, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158(6):899–905. doi: 10.1176/appi.ajp.158.6.899. [DOI] [PubMed] [Google Scholar]
- 59.Conway CR, Sheline YI, Chibnall JT, George MS, Fletcher JW, Mintun MA. Cerebral blood flow changes during vagus nerve stimulation for depression. Psychiatry Res. 2006;146(2):179–184. doi: 10.1016/j.pscychresns.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 60.Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 61.Farb NAS, Segal ZV, Mayberg H, et al. Attending to the present: mindfulness meditation reveals distinct neural modes of self-reference. Soc Cogn Affect Neurosci. 2007;2(4):313–322. doi: 10.1093/scan/nsm030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu CH, Steiner H, Costafreda SG. Predictive neural biomarkers of clinical response in depression: a meta-analysis of functional and structural neuroimaging studies of pharmacological and psychological therapies. Neurobiol Dis. 2013;52:75–83. doi: 10.1016/j.nbd.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 63.Pizzagalli DA. Frontocingulate dysfunction in depression: toward biomarkers of treatment response. Neuropsychopharmacology. 2011;36(1):183–206. doi: 10.1038/npp.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paykel ES, Ramana R, Cooper Z, Hayhurst H, Kerr J, Barocka A. Residual symptoms after partial remission: an important outcome in depression. Psychol Med. 1995;25(6):1171–1180. doi: 10.1017/s0033291700033146. [DOI] [PubMed] [Google Scholar]
- 65.Judd LL, Paulus MJ, Schettler PJ, et al. Does incomplete recovery from first lifetime major depressive episode herald a chronic course of illness? Am J Psychiatry. 2000;157(9):1501–1504. doi: 10.1176/appi.ajp.157.9.1501. [DOI] [PubMed] [Google Scholar]
- 66.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342(12):836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 67.Ridker PM. Cardiology Patient Page. C-reactive protein: a simple test to help predict risk of heart attack and stroke. Circulation. 2003;108(12):e81–e85. doi: 10.1161/01.CIR.0000093381.57779.67. [DOI] [PubMed] [Google Scholar]
- 68.Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114–119. doi: 10.1097/JCP.0b013e31823f705d. [DOI] [PubMed] [Google Scholar]
- 69.Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry. 2007;164(5):739–752. doi: 10.1176/ajp.2007.164.5.739. [DOI] [PubMed] [Google Scholar]
- 70.Rush AJ, Warden D, Wisniewski SR, et al. STAR*D: revising conventional wisdom. CNS Drugs. 2009;23(8):627–647. doi: 10.2165/00023210-200923080-00001. [DOI] [PubMed] [Google Scholar]
- 71.Dunlop BW, Binder EB, Cubells JF, et al. Predictors of remission in depression to individual and combined treatments (PReDICT): study protocol for a randomized controlled trial. Trials. 2012;13(1):106. doi: 10.1186/1745-6215-13-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kennedy SH, Downar J, Evans KR, et al. The Canadian Biomarker Integration Network in Depression (CAN-BIND): advances in response prediction. Curr Pharm Des. 2012;18(36):5976–5989. doi: 10.2174/138161212803523635. [DOI] [PubMed] [Google Scholar]