
Figure.
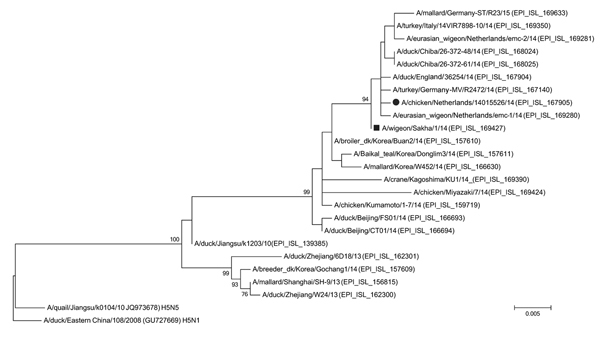
Phylogenetic tree of hemagglutinin gene of highly pathogenic avian influenza A(H5N8) viruses. The evolutionary history was inferred by using the maximum-likelihood method based on the Tamura-Nei model in MEGA6 (14). The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-joining and BIONJ (15) algorithms to a matrix of pairwise distances estimated by using the maximum composite likelihood approach and then selecting the topology with superior log likelihood value. The Tamura-Nei model was used by assuming a gamma distributed rate among nucleotide sites. The tree is drawn to scale; scale bar indicates the number of nucleotide substitutions per site. The analysis involved 25 nt sequences. All positions containing gaps and missing data were eliminated. There were a total of 761 nt positions in the final dataset. Black dot indicates A/chicken/Netherlands/14015526/2014; black square indicates A/wigeon/Sakha/1/2014.