

Abstract
Signal transducer and activator of transcription 3 (STAT3) is constitutively activated in the majority of lung cancer. This study aims at defining connections between STAT3 function and the malignant properties of non–small cell lung carcinoma (NSCLC) cells. To address possible mechanisms by which STAT3 influences invasiveness, the expression of matrix metalloproteinase-1 (MMP-1) was analyzed and correlated with the STAT3 activity status. Studies on both surgical biopsies and on lung cancer cell lines revealed a coincidence of STAT3 activation and strong expression of MMP-1. MMP-1 and tyrosine-phosphorylated activated STAT3 were found co-localized in cancer tissues, most pronounced in tumor fronts, and in particular in adenocarcinomas. STAT3 activity was constitutive, although to different degrees, in the lung cancer cell lines investigated. Three cell lines (BEN, KNS62, and A549) were identified in which STAT3 activitation was inducible by Interleukin-6 (IL-6). In A549 cells, STAT3 activity enhanced the level of MMP-1 mRNA and stimulated transcription from the MMP-1 promoter in IL-6–stimulated A549 cells. STAT3 specificity of this effect was confirmed by STAT3 knockdown through RNA interference. Our results link aberrant activity of STAT3 in lung cancer cells to malignant tumor progression through up-regulation of expression of invasiveness-associated MMPs.
Introduction
Lung cancer is the leading cause of cancer death in the world [1]. Various molecular dysregulations have been associated with lung cancer, including overexpression or mutation of epidermal growth factor receptors (EGFRs) and their ligands [2], activation of Src kinase [3], elevated expression and autocrine proliferative function of Interleukin-6 (IL-6) [4], and aberrant nuclear expression of survivin [5].
Interestingly, a common denominator of all these situations and processes seen in lung cancer is the involvement of signal transducer and activator of transcription 3 (STAT3). STAT3 can be activated by tyrosine kinase growth factor receptors such as the EGFR as well as nonreceptor tyrosine kinases such as Src and gp130 family member cytokines such as IL-6 [6]. It is known to upregulate genes promoting cell cycle progression and/or preventing apoptosis [7].
We and others have demonstrated aberrant activity of STAT3 in various malignant tumors of both hematopoietic and solid types [8,9]. An important role of STAT3 activity in lung cancer was suggested by constitutive STAT3 activation in a number of lung cancer cell lines [10] as well as in lung cancer tissue [11].
Functional connections between STAT3 activity and dysregulated upstream signaling processes in lung cancer have been revealed. For instance, IL-6 and its receptor were implicated in the progression of non–small cell lung carcinoma (NSCLC) [12] and were shown to induce enhanced STAT3 activity [13,14]. Moreover, the EGFR activates STAT3 in lung cancer cell lines [15] and can promote tumor survival in vivo in NSCLC by signaling through STAT3 [16]. A specific spectrum of STAT3 target genes is overexpressed in lung carcinomas bearing aberrantly active mutant EGFRs [15].
Malignant properties of lung tumor cells such as hyperproliferation, dedifferentiation, and invasiveness are associated with dysregulated EGF, ErbB2, or IL-6/IL-11 signaling [17,18], raising the question of how STAT3 as a downstream mediator contributes to these effects. Invasiveness was correlated in small cell lung carcinoma (SCLC) cell lines with the expression level of the EGFR [17]. It is tempting to assume that STAT3-mediated gene regulation is a constituent of this connection.
STAT3 elevates the expression of matrix metalloproteinases (MMPs) in different tumor cell types and thereby enhances invasive cell properties [19–21]. Among these is the interstitial collagenase MMP-1, which degrades collagen types I, II, and III and is an established determinant of invasive growth of tumor cells [22]. We have shown before that STAT3 directly drives MMP-1 expression in colorectal carcinoma [21]. A role of MMP-1 in lung cancer is suggested by clinical data: Tissue inhibitor of MMP-1 is an independent predictor of prognosis in patients with NSCLC [23]. Moreover, a polymorphism in the MMP-1 promoter enhances lung cancer susceptibility [24].
Here, we have addressed the role of STAT3 activation in lung carcinoma biopsies and cell lines and its spatial and functional association with MMP-1 expression. We present evidence for the notion that STAT3-dependent regulation of MMP-1 expression is involved in the pathogenesis of NSCLC.
Materials and Methods
Tumor Biopsies
Tumor specimens were obtained from lung cancer patients diagnosed with NSCLC in accordance with the local ethics committee. Histologic diagnosis was established according to the guidelines of the World Health Organization; 108 lung carcinoma biopsies [53 squamous cell carcinomas (SCCs), 43 adenocarcinomas (ACs), 6 large cell lung carcinomas (LCCs), 3 SCLCs, and 3 adenosquamous carcinomas] were obtained in the course of tumor resections. A subset of 24 NSCLC biopsies was employed for biochemical studies. Detailed information on the respective patients and tumor types are given in Table 1. Biopsies were processed for Western blot analysis as described previously [9]. The whole collection of 108 biopsies was subjected to immunohistochemical analysis. Tumor types LCC, SCLC, and adenosquamous carcinoma were not included in statistical evaluation due to small sample sizes.
Table 1.
List of Biopsies Included in This Study With Patient Clinicopathologic Parameters and Immunohistochemical Staining Scores for Tyrosine-Phosphorylated (activated) STAT3, STAT3, and MMP-1
| No. | Histology | TNM | Grade | Sex | pYSTAT3 | STAT3 | MMP-1 |
|---|---|---|---|---|---|---|---|
| 1 | SCC | T2, N0, M0 | G2 | m | − | − | +++ |
| 2 | NSCLC | T1, N3, M0 | G2 | w | ++ | ++ | ++ |
| 3 | AC | T1, N0, M0 | G2 | w | +++ | +++ | +++ |
| 4 | SCC | T2, N1, M0 | G2 | m | ++ | ++ | +++ |
| 5 | LCC | T1, N0, M0 | G2 | m | + | + | + |
| 6 | NSCLC | T2, N2, M0 | G2 | m | + | ++ | ++ |
| 7* | NSCLC | n.d. | n. d. | n. d. | +++ | +++ | + |
| 8 | AC | T4, N0, M0 | G2 | m | − | + | ++ |
| 9* | NSCLC | n.d. | n. d. | n. d. | ++ | ++ | + |
| 10 | SCC | T2, N1, M0 | G3 | m | ++ | +++ | + |
| 11 | AC | T1, N0, M0 | G2 | m | − | − | ++ |
| 12* | NSCLC | n.d. | n. d. | n. d. | + | + | ++ |
| 13 | SCC | T1, N1, M0 | G2 | m | ++ | +++ | ++ |
| 14 | SCC | T4, N3, M0 | G3 | m | +++ | +++ | ++ |
| 15 | AC | T1, N1, M0 | G2 | w | ++ | ++ | +++ |
| 16 | SCC | T2, N1, M0 | G3 | w | ++ | +++ | + |
| 17 | SCC | T2, N2, M0 | G3 | m | ++ | +++ | ++ |
| 18 | SCC | T2, N1, M0 | G3 | m | +++ | +++ | +++ |
| 19 | SCC | T2, N0, M0 | G2 | m | ++ | ++ | ++ |
| 20 | NSCLC | T1, N1, M0 | G1 | m | + | + | ++ |
| 21 | AC | T1, N1, M0 | G2 | m | + | +++ | ++ |
| 22 | AC | T2, N0, M0 | G2 | w | − | − | ++ |
| 23 | AC | T2, N0, M0 | G3 | m | − | + | +++ |
| 24 | SCC | T2, N0, M0 | G2 | m | +++ | +++ | +++ |
A tripartite, graded immunoreactive score considering both staining intensities and percentage of signal-positive cells was employed. n.d., not defined. Asterisks indicate samples originating from open biopsies, i.e., from patients who were not treated surgically and were not included in follow-up surveys.
Cell Lines and Cell Culture
Cell lines NCI-H69 and NCI-H82 were purchased from the American Type Culture Collection (Manassas, VA), and cell lines A549, EPLC-272H, BEN, and LCLC-103H were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). Cell lines Oka C1, KNS-62, and MR56 were gifts from Dr G. Aust, Institute of Anatomy, University of Leipzig. All cell lines except for A549 and BEN were cultured in RPMI 1640 containing 10% fetal calf serum (FCS), 200 mM l-glutamine, 100 mM sodium pyruvate, and 1 mg/ml gentamicin. A549 and BEN were cultured in Dulbecco's modified Eagle's medium. Cells were grown in coated tissue culture plasticware. For cytokine stimulation, cells were incubated with 20 ng/ml recombinant human IL-6 (R&D Systems, Wiesbaden, Germany) for 30 minutes (Western blot analysis), 18 hours (reporter gene assay), or 24 hours (expression analysis) at 37°C.
RNA Interference and Lentiviral Transduction
Lentivirus production and subsequent transduction of cells were carried out as described previously [25]. The following shRNA constructs (Sigma Aldrich Mission TRC Library) were used: shRNA against STAT3 (TRCN0000071456) and control scrambled shRNA (SHC002). The functionality of the shRNAs was validated by quantitative reverse transcription–polymerase chain reaction (PCR) analysis. Transduced cells were selected for puromycin resistance before further analysis.
Western Blot Analysis
Western blot analysis has been described in detail previously [21]. Blots were probed with antibodies to pYSTAT3-Tyr705 (Cell Signaling Technology, Danvers, MA) or MMP-1 (Merck Millipore, Billerica, MA) at 1:1000 dilution.
DNA Constructs
MMP-1 promoter/luciferase constructs were generated starting from plasmid − 4372 hMMP-luci (GenBank: AF023338), obtained from C. E. Brinckerhoff [26]. Construction of pMMP-1-0.6 k and mutations thereof has been described [21,26].
Immunohistology and Fluorescence Immunolabeling
Immunohistologic detection of pYSTAT3 activity and MMP-1 expression in tumor biopsies and fluorescence immunolabeling were performed as described previously [9,27,28]. Negative controls were performed using an irrelevant monoclonal antibody. Staining intensities were classified employing a graded immunoreactive score (−, +, ++, and +++) estimated from the staining intensity and the percentage of positive cells according to a board-certified pathologist opinion [29].
Real-Time PCR
Quantitative determination of MMP-1 mRNA was carried out as described [21]. cDNA was subjected to quantitative real-time PCR analysis using an iCycler Instrument (Bio-Rad, Hercules, CA).
Luciferase Reporter Gene Assay
Exponentially growing cells of A549 (7 × 105) were plated into six-well cluster plates (Greiner) in 2 ml of RPMI 1640/10% FCS and grown to 80% to 90% confluency. The cells were washed, transferred into 1 ml/well fresh medium, and cotransfected with 1 μg of MMP-1 promoter/luciferase constructs and 0.1 μg of the pRL-TK Renilla luciferase plasmid (Promega, Madison, WI) through Polyfect (Qiagen, Hilden, Germany) and optionally stimulated with IL-6 (20 ng/ml) for 8 hours. Luciferase and Renilla activities were determined using a Micro Luminat LB 96 P luminometer and reported as relative light units.
Statistics
Significance of immunohistochemical data was evaluated using the Wilcoxon rank-sum test, and data correlation was examined with the tau-b-test according to Kendall employing the SPSS 11.2 software package for data calculation. Luciferase reporter gene results were evaluated by unpaired t test using GraphPad Prism software.
Results
STAT3 is Persistently Activated and MMP-1 Is Co-Expressed in Lung Cancer Biopsies
Twenty-four NSCLC biopsies were obtained in the course of tumor resections. The patients were staged according to operation and pathologic findings after UICC (Union Internationale Contre le Cancer) criteria. With respect to TNM (Tumor, Node, Metastasis) classification, 8 patients were categorized in stage T1, 11 in stage T2, and 2 in stage T4. Regarding the differentiation grade, one tumor was classified as G1, 14 were classified as G2, and 6 were classified as G3. Details on the tumor specimens are summarized in Table 1.
The surgical specimens were investigated for both pYSTAT3 and STAT3 as well as for MMP-1 expression by Western blot analysis. Figure 1 shows the results for a representative selection of tumor tissue samples. Table 1 gives staining scores for the whole panel of biopsies covered by this study. In total, 19 of 24 examined biopsies (79%) displayed constitutive pYSTAT3, although to different degrees. All samples were positive for the active form of MMP-1.
Figure 1.
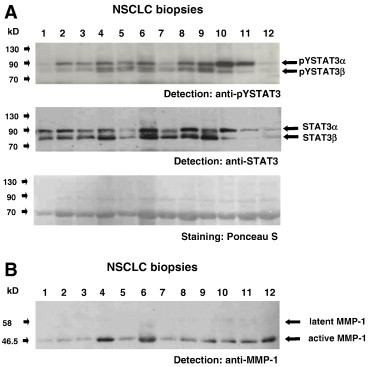
Analysis of STAT activity and MMP-1 expression in extracts of NSCLC biopsies. (A) Examination of tumor sample lysates for activated STAT3 by Western blot analysis. Extracts from 12 arbitrarily chosen tumor biopsies were separated by polyacrylamide gel electrophoresis (PAGE), subjected to Western blot, and then probed with an antibody to tyrosine-phosphorylated STAT3 (top) and reprobed with an antibody to STAT3 (center). Arrows indicate the positions of STAT3 isoforms. Comparable loading of lanes was confirmed by staining with Ponceau S (bottom). (B) Analysis of tumor samples for MMP-1 expression. Lysates from the same NSCLC biopsies as in A were subjected to Western blot and probed with an antibody to MMP-1. Positions of latent and active MMP-1 are indicated by arrows.
STAT3 Activation Coincides With Expression of MMP-1 in Lung Cancer Tissue
We next performed an immunohistologic examination of tumor biopsies with antibodies to pYSTAT3 and MMP-1. Two representative examples for specific staining of consecutively sectioned biopsies are presented in Figure 2A. In the vast majority of cases, pYSTAT3 activity was present in tumor regions that were also positive for MMP-1.
Figure 2.
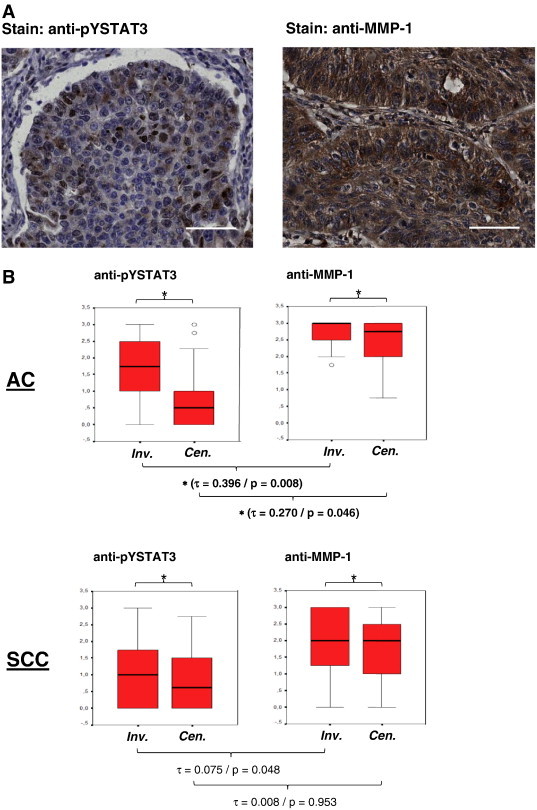
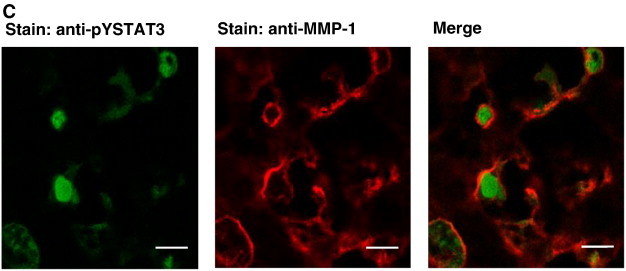
Correlation of STAT3 activation and MMP-1 expression in NSCLC biopsies. (A) Immunohistochemical analysis of tyrosine-phosphorylated STAT3 (left) and MMP-1 expression (right) in tumor tissue of an arbitrarily selected SCC from the patient cohort studied in this manuscript. Paraffin-embedded sections were stained with an antibody to tyrosine-phosphorylated STAT3 or MMP-1, respectively, as indicated, followed by microscopic visualization employing goat anti-goat or anti-mouse biotin, respectively, and streptavidin-HRP. Scale bars represent 50 μm. (B) pYSTAT3 (left) and MMP-1 (right) abundance in NSLCC biopsies. Immunohistochemical staining intensities in AC (N = 43, top part) and SCC (N = 53, bottom part) samples were determined for tumor center (Cen.) and invasive front (Inv.) areas as indicated and expressed by box plots using the scores 0, 1, 2, and 3 as described in the Materials and Methods section. Significance of mean differences in staining intensities between invasive front and tumor center areas (P < .001) is indicated by asterisks. Τ and P values determined by Kendall's tau correlation for the mutual association of pYSTAT3 and MMP-1 in both tumor types are indicated. (C) Immunofluorescence photomicrographs of a representative SCC section immunoreactive for anti-pTyr705 STAT3 (left, green fluorescent due to staining with Cy2-labeled goat anti-rabbit secondary antibody) and anti-MMP-1 (center, red fluorescent due to staining with Cy3-labeled goat anti-mouse secondary antibody). Right: Overlay of green and red fluorescence indicating co-localization of antigens. Images were taken using a Zeiss LSM 510 Meta confocal microscope device; scale bars represent 10 μm.
To assess this relation more thoroughly, we analyzed the complete available collection of roughly 100 lung cancer biopsies for both pYSTAT3 and MMP-1. Staining intensities were determined for tumor center and invasive front areas and expressed using the scores 0, 1, 2, and 3 as described in the Materials and Methods section. AC samples (N = 43) and SCC samples (N = 53) were evaluated separately. As shown in Figure 2B, activity levels of STAT3 (appearance of pYSTAT3) and expression levels of MMP-1 were significantly higher in the invasive fronts of both ACs and SCC compared to the tumor centers (P < .001 in all cases). Interestingly, using Kendall's tau correlation, a significant association was found for pYSTAT3 and MMP-1 in both invasive front and central areas of ACs (Kendall-tau-b 0.346/P = .008 and 0.270/P = .046, respectively). In SCC, such a tendency was also seen, although statistical significance was not reached.
This observation prompted us to study spatial coincidence of pYSTAT3 and MMP-1 by double immunofluorescence microscopy. Figure 2C shows a typical example of pYSTAT3 and MMP-1 signals co-localizing in tumor tissue with phosphorylated, activated STAT3 accumulated in nuclei and MMP-1 expression appearing mostly perinuclearly and extracellularly, in line with the role of MMPs in modifying the extracellular matrix.
STAT3 is Constitutively Active in Lung Cancer Cell Lines
Next, we analyzed the status of pYSTAT3 in nine established lung cancer cell lines. As shown in Figure 3, all cell lines tested showed constitutive STAT3 activation with a wide range of activity spectra, indicative of different autocrine stimulatory situations or transforming tyrosine kinase mutational context. The expression levels of STAT3 were comparable, but the intensity of phosphorylation ranged from barely detectable (NCI-H69) to very strong (NCI-H82). In all cell lines, at least two different STAT3 splice variants appeared in tyrosine-phosphorylated form, most probably STAT3 α and β. These results show that permanent cell lines derived from lung cancer tissue preserve autonomous STAT3 activation independently from a specific in vivo environment, which is quite different to colorectal cancer cell lines as we have published [9,21].
Figure 3.
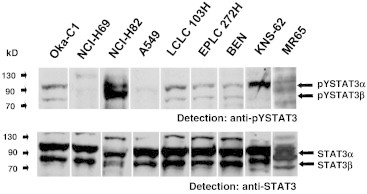
Analysis of STAT3 expression and activity in lung cancer cell lines. Samples of the indicated cell lines were lysed, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and subjected to Western blot analysis onto nitrocellulose mebranes. Membranes were subsequently probed with an antibody specifically recognizing STAT3 phosphorylated at tyrosine 705 (top). Comparable loading of the lanes and identity of STAT3 were confirmed by reprobing with an antibody to STAT3 (bottom). Arrows point to STAT3 isoforms STAT3 α and STAT3 β.
IL-6–Induced STAT3 Activation in Lung Cancer Cell Lines Causes Elevated MMP-1 Expression
Previously, we have shown that in colon carcinoma cell lines STAT3 activation can be induced by IL-6 stimulation [9]. We analyzed lung cancer cell lines for their responsiveness to IL-6 by assaying pYSTAT3 (Figure 4A). Three of the cell lines employed in this study (BEN, KNS62, A549) showed a clear and specific increase in pYSTAT3 in dependence of IL-6. Having established a means to experimentally induce STAT3 activation, we addressed a possible causal link between STAT3 activity and expression of MMP-1 in lung carcinoma cells, and mmp-1 gene transcription was assayed by real-time PCR upon activation of STAT3 by IL-6 in the three lung cancer cell lines BEN, KNS62, and A549. Figure 4B shows that incubation of all three cell lines with IL-6 leads to a clear increase in mmp-1 mRNA. Relative induction factors were 2.7 for BEN, 2.8 for KNS62, and 42.8 for A549. These results suggest that STAT3 is directly involved in the control of mmp-1 expression in lung cancer.
Figure 4.
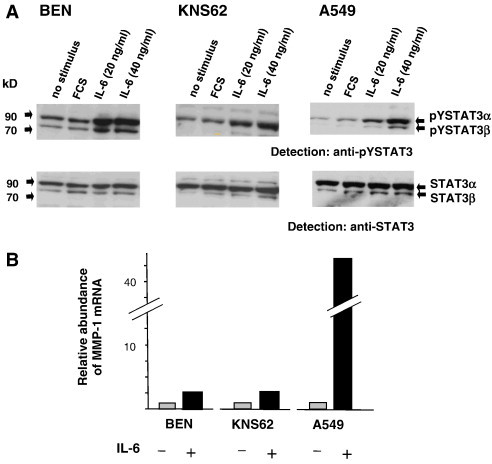
IL-6–inducible tyrosine phosphorylation of STAT3 and concomitant transcriptional activation of the MMP-1 gene in lung cancer cell lines. (A) Cell lines BEN, KNS62, and A549 were grown to 90% confluency in full medium, then starved from FCS for 6 hours, and subsequently treated optionally for 30 minutes with 10% FCS or different concentrations of IL-6 as indicated. Cells were then lysed and extracts were separated, blotted, and analyzed for STAT3 phosphorylation and expression as in Figure 3. (B) IL-6–dependent MMP-1 expression in A549 lung cancer carcinoma cells. Cells were grown for 24 hours in the absence or presence of 20 ng/ml IL-6 as indicated. Total RNA was isolated, reverse transcribed into cDNA, and quantified by reverse transcription–PCR with specific MMP-1 primers and probes. Signal intensities derived from respective non-stimulated cells were set to 1. Results from a typical experiment out of at least three independent experiments are shown.
STAT3 Transcriptionally Activates the MMP-1 Promoter in A549 Cells
To show that STAT3 is directly operative in regulating expression of the MMP-1 gene in lung cancer cells, we generated a pair of A549 cells in which STAT3 expression was either present or abolished by a knockdown approach. STAT3 expression in A549 cells was knocked down by lentivirus-mediated introduction of a STAT3-specific shRNA. For control purposes, a parallel experiment was performed using a scrambled (scr) sh sequence. Specific k.o. of STAT3 expression in the shSTAT3 cell line was confirmed by Western blot (Figure 5A).
Figure 5.
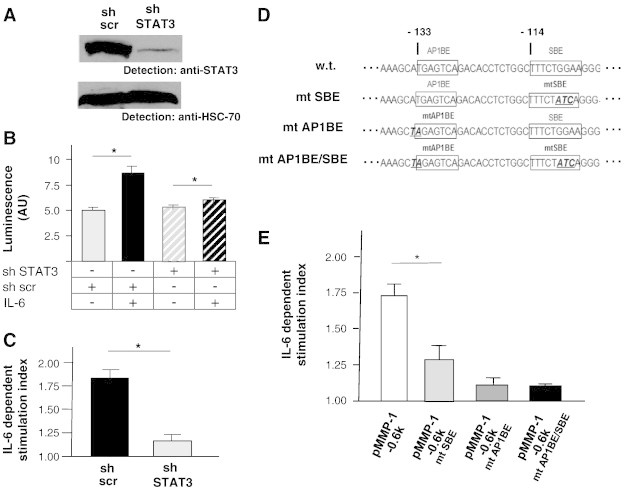
Influence of STAT3 activation in lung cancer cell line A549 cells on wild-type and mutant MMP-1 promoter activities. (A) shRNA-mediated knockdown of STAT3 expression in A549 lung cancer cells. Comparative analysis of STAT3 expression in A549 stably expressing in a STAT3-specific shRNA sequence or a scrambled control shRNA sequence by Western blot and probing with an antibody to STAT3 was performed. The blot was reprobed with an antibody to HSC-70 to verify equal loading. (B) Effect of STAT3 knockdown on IL-6–dependent activation of the MMP-1 promoter. Luciferase reporter gene construct − 4372-hMMP-luci containing a 4.3-kb promoter fragment of the human MMP-1 gene was transfected into STAT3 k.o. A549 cells (shSTAT3) and into control cells (sh scr) along with a Renilla luciferase reporter gene construct for normalization purposes as detailed in the Materials and Methods section. Fourteen hours post-transfection, cell samples were split and incubated for further 8 hours in the absence or presence of 20 ng/ml human IL-6 as indicated. Luciferase activities were determined, normalized against Renilla luciferase activity, and expressed as arbitrary luciferase units (AU). Data represent the means of three independent transfections, each run in triplicate. Error bars show SDs; an asterisk indicates statistical significance (P < .05). (C) Relative representation of data from B. Stimulation indices are expressed as the ratios of specific luciferase activities of IL-6–stimulated and unstimulated cells; basal luciferase measured in unstimulated cells was set to 1. (D) Sequence context of original (“w.t.”) and single or combinatorially mutated (“mt”) SBE and AP1BE in reporter gene constructs based on a 0.6-kb proximal region of the human MMP-1 promoter (pMMP-1-0.6 k). Wild-type and mutant core binding elements are boxed in the respective constructs; numbers indicate nucleotide positions relative to the transcriptional start site of the MMP-1 promoter. Mutated nucleotides appear underlined and in bold italic. (E) IL-6–dependent transcriptional activity of the indicated wild-type and mutant versions of pMMP-1-0.6 k–derived reporter gene constructs. Reporter gene assays were performed as in B; data are presented as in C.
We have previously used a reporter gene construct (− 4372-hMMP-luci) containing a 4.3-kb promoter fragment of the MMP-1 gene to study transcriptional activation [21]. This DNA was transfected into the two A549 cell derivatives described above to study the role of STAT3 in MMP-1 promoter regulation. Figure 5, B and C, shows that IL-6 stimulation evokes reporter gene activity in A549 cells that express STAT3 (“sh scr”), whereas in the STAT3 k.o. A549 line (“sh STAT3”) this responsiveness is heavily impaired. From these findings, we deduce that STAT3 is of prime importance for transcriptional regulation of the MMP-1 cell in lung cancer cells.
Next, we analyzed the contribution of STAT3 to the control of MMP-1 promoter function in A549 cells in some more detail. For this purpose, we employed reporter gene construct pMMP-1-0.6 k, in which a 600-bp fragment of the MMP-1 promoter was placed upstream of a luciferase reporter gene [30].Within a DNA segment between 100 and 150 bp upstream of the transcriptional start site, the human MMP-1 promoter contains both a STAT and an AP-1 recognition element [STAT binding element (SBE) and AP-1 binding element (AP1BE); Figure 5D, “w.t.”].
Transient transfection of pMMP-1-0.6 k into A549 cells and subsequent stimulation with IL-6 led to a significant increase in reporter gene activity compared to non-stimulated cells (Figure 5E, leftmost bar), indicating that STAT3 activation also promotes transcriptional activity of the shortened MMP-1 promoter. A previous report provides evidence for a combined role of STAT3 and AP-1 in transcriptional activity of the MMP-1 promoter in bladder carcinoma cells [31]. To address the respective roles of both factors for MMP-1 expression in lung cancer, we disrupted their binding sites individually and in combination in the context of the 0.6-kb promoter fragment to yield constructs mutated SBE, mutated AP1BE, and mutated AP1BE/SBE (Figure 5D) and tested the activation of the mutated constructs in response to IL-6 in A549 cells. Notably, destruction of the STAT3 binding element clearly reduced transcriptional activation by IL-6. A more pronounced effect, however, resulted from the disruption of the AP1BE, which eliminated activation almost completely, even if the core STAT3 cognate site was still intact (Figure 5E). We deduce from these findings that the STAT3 binding site at the − 133 position relative to the transcriptional start site of the MMP-1 promoter participates in MMP-1 mRNA regulation but can only be engaged if simultaneous AP-1 binding to the neighboring AP1BE occurs.
Discussion
Both STAT3 and MMP-1 have been implicated in lung cancer. The regulation of MMP protease activity in SCLC cells was so far not linked to JAK-STAT or inflammatory cytokine signaling. However, association of STAT3 activity and lung cancer progression is not restricted to NSCLC and has also been described in SCLC [32].
Various findings point to dysregulated STAT3 activity as a determinant of a malignant cell behavior and as an interesting therapeutical target in lung cancer: It has, for instance, been shown that blockade of STAT3 function by a dominant negative mutant in human lung cancer cells leads to suppression of oncogenic growth [11]. The cytokine signaling suppressor SOCS-3 can reduce STAT3-driven cell proliferation in a physiological context and is frequently found silenced by hypermethylation in human lung cancer cells [33]. More recently, it has been demonstrated that oral administration of a bioavailable small molecule STAT3 inhibitor could interfere with the development of xenograft tumors derived from lung cancer cells [34]. STAT3 activity is not only relevant for tumor growth but also influences invasiveness in NSCLC as suggested by a recent kinome profiling approach [35].
Interestingly, STAT3 hyperactivation in lung cancer does not exclusively originate from cytokine receptor signaling, but it was also shown to be connected to EGFR or ErbB2 hyperactivity. The ErbB2 kinase is required for constitutive STAT3 activation in malignant human lung epithelial cells [36]. It has also been observed in other cancer types that IL-6–dependent activation of STAT3 does not necessarily require JAK activity and can be blocked by inhibitors of the Mitogen-Activated Protein Kinase (MAPK) pathway [37]. IL-6 or IL-11 signaling were recently shown to originate from the tumor stroma as a cytokine source [6]. Particularly, cancer-associated fibroblasts are a source for autocrine IL-11 secretion [38], which is triggered by transforming growth factor–β signaling from carcinoma cells. Similarly, IL-6 trans-signaling is triggered by myeloid immune cell infiltration [39]. Altogeher, these findings and our results suggest that in particular targeting and blocking STAT3 activation is relevant for interference of lung cancer invasion downstream of either hyperactive tyrosine kinase or inflammatory cytokine stimuli to ablate MMP-1 expression.
Notably, cooperativity of STAT3- and AP-1–mediated transcriptional regulation of the human MMP-1 promoter has also been shown in colon and bladder cancer cells [30,31]. Moreover, we have analyzed a small collection of colorectal cancer biopsies for STAT3 and AP-1 and observed combined aberrant activities of both factors in the majority of cases [31]. In the light of the present study, it will be important to determine if a significant correlation exists in lung cancer.
Several reports point to an involvement of STAT3 in the development of resistance to chemotherapy and radiotherapy [40–42]. Targeting STAT3, thus, has the potential of improving treatment efficacy in lung cancer [43]. The role of STAT3 activation in lung cancer is probably related to inflammatory processes. Interestingly, chitinase 3-like 1, the product of a STAT3 downstream target gene, is a biomarker of inflammation-induced lung cancer [44].
Elevated MMP-1 expression was associated with unfavorable outcome and progressed lung cancer cases. MMP-1 levels were found increased in tumor samples compared with matched, corresponding normal tissues. Strong MMP-1 expression was correlated significantly with the occurrence of tumor–lymph node metastasis [23]. Moreover, in patients with lymph node metastasis, MMP-1 overexpression was a negative prognostic factor and associated with shorter survival [40].
We showed that transcriptional activity of the MMP-1 promoter is modulated through concerted interaction of STAT3 and AP-1 factors with specific recognition elements [26]. It will, thus, be interesting to address whether aberrant EGFR function may contribute to malignancy in lung cancer through parallel activation of both MAP kinase and JAK/STAT signal transduction pathways.
Taken together, our findings suggest that the combined activities of both STAT3 and MMP-1 should be considered as molecular parameters of NSCLC and, along with EGFR and Src activity, should be further evaluated as potential targets for individualized tumor treatment.
Footnotes
This work was funded by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) Jena, Project B 307-01035, and the Fonds zur Förderung der wissenschaftlichen Forschung Österreich, grant SFB F28, to R.M. We thank Constanze E. Brinckerhoff for the MMP-1 reporter construct and Gabriela Aust for lung cancer cell lines.
References
- 1.Wingo PA, Cardinez CJ, Landis SH, Greenlee RT, Ries LA, Anderson RN, Thun MJ. Long-term trends in cancer mortality in the United States, 1930–1998. Cancer. 2003;97(12 Suppl.):3133–3175. doi: 10.1002/cncr.11380. [DOI] [PubMed] [Google Scholar]
- 2.Rusch V, Baselga J, Cordon-Cardo C, Orazem J, Zaman M, Hoda S, McIntosh J, Kurie J, Dmitrovsky E. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1993;53(10 Suppl.):2379–2385. [PubMed] [Google Scholar]
- 3.Mazurenko NN, Kogan EA, Zborovskaya IB, Kisseljov FL. Expression of pp60c-src in human small cell and non-small cell lung carcinomas. Eur J Cancer. 1992;28:372–377. doi: 10.1016/s0959-8049(05)80056-5. [DOI] [PubMed] [Google Scholar]
- 4.Bihl M, Tamm M, Nauck M, Wieland H, Perruchoud AP, Roth M. Proliferation of human non-small-cell lung cancer cell lines: role of interleukin-6. Am J Respir Cell Mol Biol. 1998;19:606–612. doi: 10.1165/ajrcmb.19.4.3247. [DOI] [PubMed] [Google Scholar]
- 5.Lu B, Gonzalez A, Massion PP, Shyr Y, Shaktour B, Carbone DP, Hallahan DE. Nuclear survivin as a biomarker for non-small-cell lung cancer. Br J Cancer. 2004;91:5735–5740. doi: 10.1038/sj.bjc.6602027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2012;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 8.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal K. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7:545–555. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song L, Turkson J, Karras JG, Jove R, Haura EB. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2003;22:4150–4165. doi: 10.1038/sj.onc.1206479. [DOI] [PubMed] [Google Scholar]
- 11.Seki Y, Suzuki N, Imaizumi M, Iwamoto T, Usami N, Ueda Y, Hamaguchi M. STAT3 and MAPK in human lung cancer tissues and suppression of oncogenic growth by JAB and dominant negative STAT3. Int J Oncol. 2004;24:931–934. [PubMed] [Google Scholar]
- 12.Martín F, Santolaria F, Batista N, Milena A, González-Reimers E, Brito MJ, Oramas J. Cytokine levels (IL-6 and IFN-gamma), acute phase response and nutritional status as prognostic factors in lung cancer. Cytokine. 1999;11:80–86. doi: 10.1006/cyto.1998.0398. [DOI] [PubMed] [Google Scholar]
- 13.Dalwadi H, Krysan K, Heuze-Vourc'h N, Dohadwala M, Elashoff D, Sharma S, Cacalano N, Lichtenstein A, Dubinett S. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clin Cancer Res. 2005;11:7674–7682. doi: 10.1158/1078-0432.CCR-05-1205. [DOI] [PubMed] [Google Scholar]
- 14.Yeh HH, Lai WW, Chen HH. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene. 2006;25:4300–4309. doi: 10.1038/sj.onc.1209464. [DOI] [PubMed] [Google Scholar]
- 15.Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 16.Haura EB, Zheng Z, Song L, Cantor A, Bepler G. Activated epidermal growth factor receptor–Stat-3 signaling promotes tumor survival in vivo in non–small cell lung cancer. Clin Cancer Res. 2005;11:8288–8294. doi: 10.1158/1078-0432.CCR-05-0827. [DOI] [PubMed] [Google Scholar]
- 17.Damstrup L, Rude Voldborg B, Spang-Thomsen M, Brünner N, Skovgaard Poulsen H. In vitro invasion of small-cell lung cancer cell lines correlates with expression of epidermal growth factor receptor. Br J Cancer. 1998;78:631–640. doi: 10.1038/bjc.1998.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang KT, Tsai CM, Chiou YC, Chiu CH, Jeng KS, Huang CY. IL-6 induces neuroendocrine dedifferentiation and cell proliferation in non-small cell lung cancer cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L446–L453. doi: 10.1152/ajplung.00089.2005. [DOI] [PubMed] [Google Scholar]
- 19.Dechow TN, Pedranzini L, Leitch A, Leslie K, Gerald WL, Linkov I, Bromberg JF. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc Natl Acad Sci U S A. 2004;101:10602–10607. doi: 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23:3550–3560. doi: 10.1038/sj.onc.1207383. [DOI] [PubMed] [Google Scholar]
- 21.Tsareva SA, Moriggl R, Corvinus FM, Wiederanders B, Schütz A, Kovacic B, Friedrich K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia. 2007;9:279–291. doi: 10.1593/neo.06820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ossowski L. Invasion of connective tissue by human carcinoma cell lines: requirement for urokinase, urokinase receptor, and interstitial collagenase. Cancer Res. 1992;52:6754–6760. [PubMed] [Google Scholar]
- 23.Gouyer V, Conti M, Devos P, Zerimech F, Copin MC, Créme E, Wurtz A, Porte H, Huet G. Tissue inhibitor of metalloproteinase 1 is an independent predictor of prognosis in patients with nonsmall cell lung carcinoma who undergo resection with curative intent. Cancer. 2005;103:1676–1686. doi: 10.1002/cncr.20965. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Y, Spitz MR, Lei L, Mills GB, Wu X. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter enhances lung cancer susceptibility. Cancer Res. 2001;61:7825–7829. [PubMed] [Google Scholar]
- 25.Kasper M, Regl G, Eichberger T, Frischauf AM, Aberger F. Efficient manipulation of Hedgehog/GLI signaling using retroviral expression systems. Methods Mol Biol. 2007;397:67–78. doi: 10.1007/978-1-59745-516-9_6. [DOI] [PubMed] [Google Scholar]
- 26.Rutter JL, Mitchell TI, Butticè G, Meyers J, Gusella JF, Ozelius LJ, Brinckerhoff CE. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter creates an Ets binding site and augments transcription. Cancer Res. 1998;58:5321–5355. [PubMed] [Google Scholar]
- 27.Schütz A, Schneidenbach D, Aust G, Tannapfel A, Steinert M, Wittekind C. Differential expression and activity status of MMP-1, MMP-2 and MMP-9 in tumor and stromal cells of squamous cell carcinomas of the lung. Tumour Biol. 2002;23:179–184. doi: 10.1159/000064034. [DOI] [PubMed] [Google Scholar]
- 28.Schütz A, Härtig W, Wobus M, Grosche J, Wittekind Ch, Aust G. Expression of ADAM15 in lung carcinomas. Virchows Arch. 2005;446:421–429. doi: 10.1007/s00428-004-1193-z. [DOI] [PubMed] [Google Scholar]
- 29.Remmele W, Hildebrand U, Hienz HA, Klein PJ, Vierbuchen M, Behnken LJ, Heicke B, Scheidt E. Comparative histological, histochemical, immunohistochemical and biochemical studies on oestrogen receptors, lectin receptors, and Barr bodies in human breast cancer. Virchows Arch A Pathol Anat Histopathol. 1986;409:127–147. doi: 10.1007/BF00708323. [DOI] [PubMed] [Google Scholar]
- 30.Zugowski C, Lieder F, Müller A, Gasch J, Corvinus FM, Moriggl R, Friedrich K. STAT3 controls matrix metalloproteinase-1 expression in colon carcinoma cells by both direct and AP-1-mediated interaction with the MMP-1 promoter. Biol Chem. 2011;392:449–459. doi: 10.1515/BC.2011.038. [DOI] [PubMed] [Google Scholar]
- 31.Itoh M, Murata T, Suzuki T, Shindoh M, Nakajima K, Imai K, Yoshida K. Requirement of STAT3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in T24 bladder cancer cells. Oncogene. 2006;25:1195–1204. doi: 10.1038/sj.onc.1209149. [DOI] [PubMed] [Google Scholar]
- 32.Zhao X, Sun X, Li XL. Expression and clinical significance of STAT3, P-STAT3, and VEGF-C in small cell lung cancer. Asian Pac J Cancer Prev. 2012;13:2873–2877. doi: 10.7314/apjcp.2012.13.6.2873. [DOI] [PubMed] [Google Scholar]
- 33.He B, You L, Xu Z, Mazieres J, Lee AY, Jablons DM. Activity of the suppressor of cytokine signaling-3 promoter in human non-small-cell lung cancer. Clin Lung Cancer. 2004;5:366–370. doi: 10.3816/CLC.2004.n.015. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–9628. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azijli K, Yuvaraj S. Kinome profiling of non-canonical TRAIL signaling reveals RIP1-Src-STAT3-dependent invasion in resistant non-small cell lung cancer cells. J Cell Sci. 2012;125:4651–4661. doi: 10.1242/jcs.109587. [DOI] [PubMed] [Google Scholar]
- 36.Fernandes A, Hamburger AW, Gerwin BI. ErbB-2 kinase is required for constitutive stat 3 activation in malignant human lung epithelial cells. Int J Cancer. 1999;83:564–570. doi: 10.1002/(sici)1097-0215(19991112)83:4<564::aid-ijc20>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 37.Kopantzev Y, Heller M, Swaminathan N, Rudikoff S. IL-6 mediated activation of STAT3 bypasses Janus kinases in terminally differentiated B lineage cells. Oncogene. 2002;21:6791–6800. doi: 10.1038/sj.onc.1205815. [DOI] [PubMed] [Google Scholar]
- 38.Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung P, Zhang XH. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–1755. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 40.Ogata Y, Osaki T, Naka T, Iwahori K, Furukawa M, Nagatomo I, Kijima T, Kumagai T, Yoshida M, Tachibana I. Overexpression of PIAS3 suppresses cell growth and restores the drug sensitivity of human lung cancer cells in association with PI3-K/Akt inactivation. Neoplasia. 2006;8:817–825. doi: 10.1593/neo.06409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin YC, Hung MS, Lin CK, Li JM, Lee KD, Li YC, Chen MF, Chen JK, Yang CT. CK2 inhibitors enhance the radiosensitivity of human non-small cell lung cancer cells through inhibition of stat3 activation. Cancer Biother Radiopharm. 2011;26:381–388. doi: 10.1089/cbr.2010.0917. [DOI] [PubMed] [Google Scholar]
- 42.Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, Ha SJ, Soo RA, Christensen JG, Lee JH, Cho BC. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther. 2012;11:2254–2264. doi: 10.1158/1535-7163.MCT-12-0311. [DOI] [PubMed] [Google Scholar]
- 43.Su WP, Cheng FY, Shieh DB, Yeh CS, Su WC. PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int J Nanomedicine. 2012;7:4269–4283. doi: 10.2147/IJN.S33666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan C, Ding X, Wu L, Yu M, Qu P, Du H. Stat3 downstream gene product chitinase 3-like 1 is a potential biomarker of inflammation-induced lung cancer in multiple mouse lung tumor models and humans. PLoS One. 2013;8:e61984. doi: 10.1371/journal.pone.0061984. [DOI] [PMC free article] [PubMed] [Google Scholar]