

Abstract
The goal of molecular cytogenetic testing for children presenting with developmental delay is to identify or exclude genetic abnormalities that are associated with cognitive, behavioral, and/or motor symptoms. Until 2010, chromosome analysis was the standard first-line genetic screening test for evaluation of patients with developmental delay when a specific syndrome was not suspected. In 2010, The American College of Medical Genetics and several other groups recommended chromosomal microarray (CMA) as the first-line test in children with developmental delays, multiple congenital anomalies, and/or autism. This test is able to detect regions of genomic imbalances at a much finer resolution than G-banded karyotyping. Until recently, no CMA testing had been approved by the United States Food and Drug Administration (FDA). This review will focus on the use of the Affymetrix CytoScan® Dx Assay, the first CMA to receive FDA approval for the genetic evaluation of individuals with developmental delay.
Keywords: Affymetrix CytoScan® Dx Assay, global developmental delay, mental retardation, chromosomal microarray, genetic testing
Introduction
The goal of molecular cytogenetic testing for children presenting with developmental delay (DD) is to identify or exclude genetic abnormalities in the child that are associated with cognitive, behavioral, and/or motor symptoms. Either the recognition of a clinically relevant genetic abnormality or the exclusion of a suspected genetic syndrome can allow for improved accuracy of developmental prognosis, neurodevelopmental treatment plan, and management of potential coexisting somatic concerns. Additionally, identification of a specific genetic cause of a child's developmental delay can provide important information to the family regarding recurrence risk for future pregnancies. Until 2010, chromosome analysis was the standard first-line genetic screening test for evaluation of patients with developmental delay when a specific syndrome was not suspected. With chromosome analysis, a cytogeneticist is able to visualize the chromosomes allowing for reliable detection of chromosomal aneuploidies, large structural rearrangements, and large deletions or duplications that are greater than 10 Mb in size. However, in 2010, The American College of Medical Genetics (ACMG), now known as The American College of Medical Genetics and Genomics, and several other groups recommended chromosomal microarray (CMA) as the first-line test in patients with developmental delay, intellectual disability, multiple congenital anomalies, and/or autism.[1-3] This test is able to detect regions of genomic imbalances termed copy number variation (CNV) at a much finer resolution than G-banded karyotyping. There are currently a few different platforms for CMA testing with some differences in methodology, resolution, and detection across platforms. Additionally, there may be differences in the clinical interpretation of CNVs among laboratories.[4] Until recently, no CMA testing had been approved by the United States Food and Drug Administration (FDA). This review will focus on the use of the Affymetrix CytoScan® Dx Assay, the first CMA to receive FDA approval,[5] for the genetic evaluation of individuals with developmental delay.
Developmental Delay
Developmental delay is a common reason for referral for genetics evaluation in childhood. Global developmental delay (GDD) is the diagnostic term used to describe children under six years of age who perform more than two standard deviations below age-matched peers in two or more aspects of early childhood development.[6] Typically, children with GDD demonstrate intellectual disability in addition to problems with expressive and/or receptive language, motor function, social interaction, or behavior. As formal intelligence testing is not always accurate in children five years old and younger,[7] and as intellectual disability may be paralleled by disability in other domains that are particularly apparent early in life, GDD is a more accurate term than intellectual disability in this population. Missed developmental milestones and/or developmentally abnormal behaviors are typically noted by a child's family, pediatric care provider, or preschool/daycare teachers. Preliminary assessment usually begins with a comparison of the individual child's abilities in multiple domains to age-based developmental milestones. Skills-based screening tools such as the Denver Developmental Screening Test[8] and the Cognitive Adaptive Test/Clinical Linguistic and Auditory Milestone Scale[9] are well-validated, easy to administer non-invasive examinations that can be completed rapidly in the outpatient setting prior to referral to a developmental pediatrician, pediatric neurologist, or geneticist. GDD affects up to 3% of the general population, and is a common reason for neurologic and genetic testing early in life.[10]
Developmental delay may have genetic, environmental, complex, or multifactorial etiologies. If history and physical examination of a child with DD does not lead the provider to suspect a specific genetic condition or environmental etiology and if family history is negative for multiple miscarriages or other history suggestive of chromosomal rearrangement, the first-line genetic test to be performed should be a CMA. In fact, CMA has replaced G-banded karyotyping or chromosome analysis as the first-line test not only for DD, but also in the setting of autism spectrum disorders and/or multiple congenital anomalies.[1, 2] The CMA has improved resolution over G-banded karyotyping, which is unable to discriminate anomalies less than 3-5 Mb in size. A review of 33 studies comprising 21,698 patients with DD, ASD, or multiple congenital anomalies revealed that CMA had a much higher diagnostic yield of 15-20% compared to the ∼3% yield of G-banded karyotyping when patients with Down syndrome and other recognizable chromosomal disorders were excluded.[2] CMA is also considered to be cost-effective in this setting as the price is less than the combination of G-banded karyotyping and a customized fluorescence in situ hybridization (FISH) test.[2] In rare cases when a geneticist suspects a particular genetic disorder when evaluating a patient with DD, the specific molecular test available for the suspected condition should be completed first, prior to general genetic screening tests.[10] However, CMA is preferable to FISH when a specific microdeletion or microduplication is suspected because CMA offers information regarding the size of the aberration and also surveys the whole genome for additional abnormalities. On occasion a child's medical history may be suggestive of an environmental etiology for the DD such as when there has been prenatal exposure to specific drugs or infections, preterm birth, delivery complications resulting in hypoxic-ischemic encephalopathy, and/or meningitis. In these situations, genetic screening tests may not be necessary, although the possibility of a genetic underpinning of DD should always be considered.
Molecular testing for DD can be extensive as well as expensive. The recommended sequence of events for evaluation of a child presenting with DD is presented in Figure 1. Additional molecular tests beyond CMA may be pursued in an effort to obtain a specific diagnosis. These molecular tests include targeted next generation sequencing panels and/or whole exome/genome sequencing. In most cases, finding a genetic cause of DD does not lead to specific therapy or different management practices than available for DD without a specific diagnosis. Nevertheless, making a genetic diagnosis of the etiology of DD can have numerous benefits to the individual child and the child's family. Discovery of an underlying genetic cause of DD may enable accurate prognosis of developmental potential, awareness of associated medical or behavioral abnormalities prompting specific therapies or intervention, and possibly even knowledge of predicted life expectancy. Additionally, identification of the genetic etiology enables accurate genetic counseling and determination of recurrence risk. There are also psychosocial benefits to providing a specific molecular diagnosis that may also be helpful for parents of the affected child as it may alleviate significant guilt or worry that specific historical events or parenting practices caused the neurologic impairment. Additionally, making a specific genetic diagnosis may provide future cost savings as it alleviates further diagnostic testing and can reduce future treatments unlikely to be necessary and/or successful.
Figure 1.
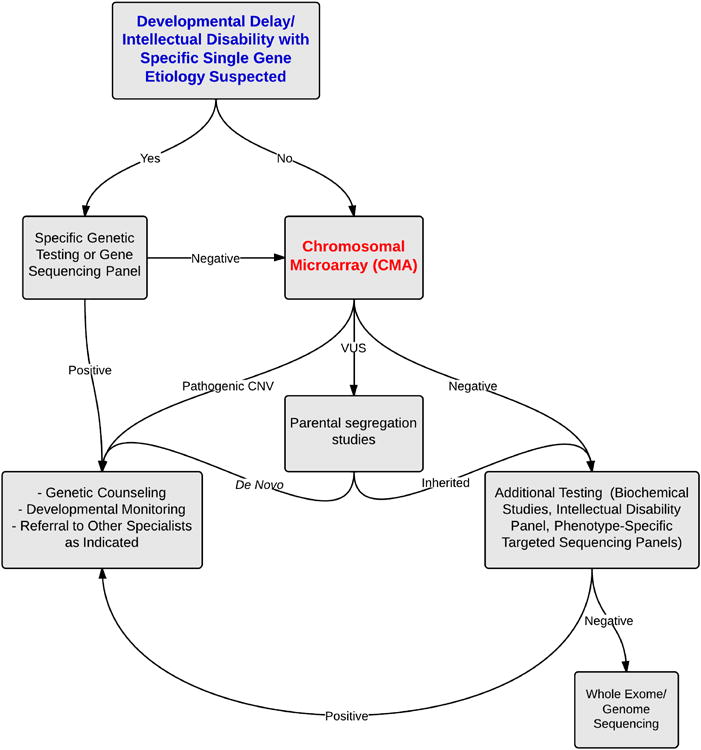
Recommended initial molecular testing strategy for a child presenting with developmental delay.
Chromosomal Microarray Platform
The CMA, also termed cytogenetic microarray, molecular karyotyping, or genomic copy number array, is a deoxyribonucleic acid (DNA)-based testing method used to identify CNVs, which are either gains (i.e. microduplications) or losses (i.e. microdeletions) of genomic material. CMA platforms were historically designed to use either array CGH or single nucleotide polymorphism (SNP) genotyping to assess CNVs; however, platforms that provide both CNVs as well as copy neutral absence of heterozygosity (AOH) have become standard practice in clinical laboratories. For both methodologies, DNA segments or probes are immobilized to a solid support, the array. Array CGH requires the use of non-polymorphic DNA fragments, which are designed to detect only CNV, whereas SNP arrays utilize allele-specific oligonucleotide (ASO) probes, which enable determination of genotype information in addition to detection of CNV. In array CGH, the DNA fragments may be generated from clones, as when using bacterial artificial chromosomes (BACs), or may be synthesized oligonucleotides. Traditionally, in array CGH, two DNAs, the test sample and the reference sample, are labeled with different fluorophores (typically Cyanine 3 and Cyanine 5), and after denaturation, the DNAs are competitively co-hybridized to the array. Fluorescence is measured and any difference in fluorescent intensity for the two samples at a given location highlights a region of CNV. For SNP arrays, historically only the test sample is hybridized to the array and the fluorescent intensity is compared with a database of controls. Because SNP arrays also enable genotype determination, in addition to identifying regions of CNV, they can also be utilized to reveal AOH. Array CGH platforms may also have SNP information supplemented to the array; however, the density is typically lower than what is present on SNP arrays. Multiple stretches of AOH throughout the genome may indicate consanguinity and raises the suspicion for autosomal recessive disease. A large stretch of AOH in a single chromosomal location may indicate uniparental disomy (UPD). For both methodologies, probe density and probe spacing affect the functional resolution of CMA. Additionally, the specific statistical algorithms utilized to interpret the data and for setting calling criteria may also affect resolution.[1]
CMA may result in detection of thousands of CNVs in a single individual. In one study, using a NimbleGen array with ∼2.1 million oligonucleotide probes and stringent calling criteria of a minimum of 10 consecutive probes involved, the average number of CNVs identified in an individual Utah resident with ancestry from Northern and Western Europe was 1,117.[11] Notably, not all CNVs identified are pathogenic or disease-causing. Analysis of CMA CNV results includes classification of CNVs into pathogenic, benign, and variants of uncertain significance (VUS) categories. Classification of CNVs is complex, and is best completed by a clinical laboratory with molecular and cytogenetic expertise.[12] Factors influencing CNV classification include CNV frequency, size, location, copy number state, and gene content. Knowledge of CNV inheritance is also helpful, if available.[13] Classification is often aided by the use of several publically available databases, laboratory internal databases, and literature review. The Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) is a catalog of known structural variation in healthy control samples. Two databases with information on clinically relevant variants are ECARUCA (http://www.ecaruca.net) and CAGdb (http://www.cagdb.org), and databases with information on a combination of common and clinically relevant variants are dbVAR (http://www.ncbi.nlm.nih.gov/dbvar), DECIPHER (http://decipher.sanger.ac.uk), and ISCA CNV (https://www.iscaconsortium.org).[13]
Pathogenic or disease-causing CNVs are likely to include gene-rich regions and/or known disease-causing genes. Additionally, deletions are generally more poorly tolerated, and thus more likely to be pathogenic, than duplications. A number of recurrent, pathogenic CNVs associated with GDD and/or ID have been identified (Table 1). The majority of identified CNVs are benign CNVs, or variants that do not result in clinical disease. These CNVs should be considered as normal human variation, and well-characterized benign CNVs may be excluded from CMA reports.[14] A CNV is classified as a VUS when a definitive assessment of pathogenic or benign cannot be made. In these instances, parental samples are often requested so that segregation studies may be performed. As an example, a variant that arose de novo in an affected patient is likely to be considered more suspicious than a variant that was inherited from an unaffected parent, although all such generalizations should be made with caution, as certain CNVs may demonstrate incomplete penetrance and/or variable expressivity.
Table 1. Example common recurrent pathogenic CNVs associated with GDD/ID [15].
| 1q21.1 deletion syndrome | 16p11.3 duplication syndrome |
| 3q29 deletion syndrome | 22q11.2 microduplication syndrome |
| 15q13.3 deletion syndrome | Xq28 duplication syndrome |
| 16p11.2 deletion syndrome | Angelman syndrome |
| 1q21.1 duplication syndrome | DiGeorge/Velocardiofacial syndrome |
| 3q29 duplication syndrome | Potocki-Lupski syndrome |
| 7q11.23 duplication syndrome | Prader-Willi syndrome |
| 15q11-q13 duplication syndrome | Smith-Magenis syndrome |
| 15q13.13 duplication syndrome | Williams-Beuren syndrome |
One limitation of CMA testing is that there can be significant variability in the categorization of CNVs by different clinical laboratories. In 2009, Tsuchiya et al queried 11 different clinical laboratories to assess 13 CNVs, and for none of the CNVs assessed was their complete agreement in classification. The authors proposed the classification differences may have arisen due to differences in content of internal databases, differing opinions as to clinical consequences of specific gene deletions or duplications, and differences in comfort level of the reporting clinical laboratory directors.[4] Additional limitations of CMA testing include inability to detect balanced chromosomal rearrangements or differentiate certain trisomies from unbalanced Robertsonian translocations.[1] Additionally, the presence of marker chromosomes and low-level mosaicism may be missed.[16]
Affymetrix CytoScan® Dx Assay Methods
The Affymetrix CytoScan® Dx Assay utilizes a high density combined CGH and SNP array platform, which assesses approximately 2,696,550 markers, including approximately 750,000 SNP markers. Each oligonucleotide is approximately 25 base pairs long. Intragenic probe spacing is approximately 1 probe every 880 base pairs and intergenic probe spacing is approximately 1 probe every 1700 base pairs. This assay is FDA approved for CNV assessment of genomic DNA (gDNA) isolated from peripheral whole blood samples. To perform the assay, gDNA is digested with the Nsp1 restriction enzyme and digested DNA is then ligated to Nsp1 adapters. The ligation product is then amplified via polymerase chain reaction (PCR) to produce amplicons in the 200 to 1100 bp range. The amplicons are then purified and digested with DNAse I to produce 25 to 125 bp fragments. The fragments are end-labeled with a modified biotinylated base and the sample is then hybridized to the array. The array is washed and then stained with a streptavidin-coupled dye and a biotinylated anti-streptavidin antibody. The array is then scanned with the GeneChip Scanner and the signal intensity for each marker is assessed. Using the Chromosome Analysis Suite (ChAs Dx) software, the signal for the sample is then compared to a reference set, which is based on the average of over 400 samples. Differences in signal between the sample and reference are expressed as a log2 ratio and represents relative intensity for each marker. A discrete copy number value is determined from the relative intensity data and is displayed. Genotype information for the SNP markers is visualized with the Allele Track.
Guidelines are included in this out-of-box assay regarding specific criteria for CNV reporting. The specific Affymetrix CytoScan® Dx Assay CNV evaluation criteria for inclusion of a CNV in the clinical report was not publically available for our review at the time of this publication.
Affymetrix CytoScan® Dx Assay Specifications
The ACMG has published standards regarding CMA design and performance.[12] As part of these standards, the ACMG recommends that any CMA platform should have premarket analytical validation to confirm that there is a >99% sensitivity for detection of CNVs ≥400 kb and that there is sufficient analytical specificity so that the false positive rate (FPR) for CNVs called of any size is <1%.[12, 14] At the time of this publication it is difficult to rate the Affymetrix CytoScan® Dx Assay in performance for these suggested parameters because limited information regarding the assay is currently publically available. We have, however, utilized the FDA submission material for the Affymetrix CytoScan® Dx Assay to provide a limited assessment.[17]
Regarding sensitivity, the Affymetrix CytoScan® Dx Assay FDA submission describes a study of 960 subjects, in whom 680 genetic aberrations were identified by predicate methods. Of these aberrations, the Affymetrix CytoScan® Dx Assay could have identified 646 (the remainder were non-identifiable; for example, balanced translocations, aberrations of the Y TER, or aberrations outside the analytical claims of the test). The CytoScan® Dx identified 639/646 of those aberrations; therefore, the sensitivity was 99%. The identified genetic abnormalities were of different sizes, types and genomic locations.[17]
Regarding analytical specificity, one can determine the FPR per CNV call. Any CNV call, regardless of pathogenicity, should be confirmed by an independent methodology with a FPR <1%.[12] FPR is traditionally defined as 1-specificity, where specificity = TN/(TN+FP), and therefore, FPR = 1-(TN/(TN+FP)) {TN=true negative, FP=false positive}.[2] For purposes of regulatory approval, the FDA defines the false positive rate as FPRFDA = 1-agreement with an orthogonal technology. Affymetrix has compared performance of the CytoScan® Dx Assay to sequencing and composite methods. 132 samples were used for the comparison analysis and the CNVs in these samples covered 63.5% of the genome. For the sequencing method, the criterion for accuracy was ≥50% overlap in size of the detected CNV and the same copy number state (either gain or loss) between the two methods. The composite method was defined as “another analytically validated molecular method” and included routine patient care studies (excluding Affymetrix microarray), qPCR, and sequencing.[18] Results of the sequencing and composite method comparisons with Affymetrix-defined hypervariable regions excluded are presented in Table 2. While the FPR for CNV losses ≥ 400kb may meet the suggested ACMG criteria of <1% FPR, the FPR for gains may be much higher. The ACMG guidelines also suggest that CNVs less than 400 kb in size should be called only if the FPR does not rise.[12] It should also be noted that no information regarding the false positives detected during the FDA submission, such as log2 ratio of individual probes in the aberration, was available at the time of this review. The overall performance for gains was lower than that for losses. In practice, it is possible that review of the raw data by a trained laboratory geneticist would have led to suspicion of a false positive in many of the calls. For example, a number of the false-positives for the gains were a result of marker placement (e.g. across the centromere) that would have been easily visualized by a trained laboratory geneticist.[18] These results suggest that confirmation of CytoScan® Dx Assay results by a second method may be necessary, as per the intended use statement.
Table 2. Sequencing and Composite Method Comparisons [17]*.
| % agreement (95% CI) | False Positive Rate (95% CI) | ||
|---|---|---|---|
| Sequencing Method | CNV loss 400kb to 1 Mb | 94.3% (81.4%, 98.4%) | 5.7% (1.6%, 18.6%) |
| CNV loss > 1 Mb | 97.3% (92.3%, 99.1%) | 2.7% (0.9%, 7.7%) | |
| CNV gain 400 kb to 1 Mb | 86.5% (72.0%, 94.1%) | 13.5% (5.9%, 28.0%) | |
| CNV gain >1 Mb | 98.8% (93.5%, 99.8%) | 1.2% (0.2%, 6.5%) | |
| Composite Method | CNV loss 400kb to 1 Mb | 100.0% (65.3%, 100.0%) | 0.0% (0.0%, 34.7%) |
| CNV loss > 1 Mb | 100.0% (76.1%, 100.0%) | 0.0% (0.0%, 23.9%) | |
| CNV gain 400 kb to 1 Mb | 94.6% (71.5%, 95.9%) | 5.4% (4.1%, 28.5%) | |
| CNV gain >1 Mb | 88.6% (62.5%, 96.7%) | 11.4% (3.3%, 37.5%) | |
Affymetrix-defined hypervariable regions excluded
Additionally, the Affymetrix CytoScan® Dx Assay FDA submission describes 3 of 108 phenotypically normal individuals with a “pathogenic CNV” identified by CytoScan® Dx Assay. The 3 pathogenic CNVs were distinguished from 47 VUS, which were also identified.[17] By conventional FPR estimation methods, this would indicate a false positive rate of 2.8%. No further details regarding the identified CNVs in these individuals are provided. It is unlikely that 3 phenotypically normal individuals would have a pathogenic CNV; however, the pathogenic CNVs identified may represent CNVs with reduced penetrance or delayed presentation. More details are needed to assess Affymetrix CytoScan® Dx Assay CNV calling criteria, and it is likely that review of the raw data would have indicated a likely false positive for these calls. Additionally, the pathogenic nature of a call may be influenced by the availability of clinical and parental information, which was not available to the interpreter in this assessment.
The limited data provided in the FDA submission and the unavailability of additional marketing materials at the time of this publication makes it difficult to fully assess the sensitivity and specificity of the Affymetrix CytoScan® Dx Assay platform. The Affymetrix CytoScan® Dx Assay documentation does claim a resolution limit of 3Mb for AOH and a detection of mosaicism if the mosaicism represents 20% or greater. The fact that the interpretation of CNV results described in the FDA submission was done in isolation emphasizes why it is important to:
use the assay only for the intended use population
avoid use as a screening test where the majority of those tested are healthy individuals
use the Affymetrix CytoScan® Dx Assay in conjunction with other analytical and clinical findings by a certified healthcare professional.
The full intended use statement is provided as Appendix A.
Comparison of CytoScan® Dx to Other Platforms
When choosing an array platform, it is important to note array design, including whether the array is targeted or whole genome, whether the array is BAC versus oligonucleotide, and whether SNP detection is included. Because synthesized oligonucleotide probes are much smaller than BAC probes (<60 base pairs versus ∼75,000 to 150,000 base pairs, respectively), oligonucleotide probes generally have improved breakpoint resolution.[1] Oligonucleotide array design also generally allows for placement of multiple adjacent probes, which enables improved accuracy and reproducibility. SNP arrays allow for the detection of the copy number neutral abnormality of AOH, and therefore, should be preferentially utilized in certain settings, such as when consanguinity has been noted.
Design details for several widely-used CMA platforms are shown in Table 3. Of note, the Affymetrix CytoScan® HD Array has a probe content that is very similar if not identical to the Affymetrix CytoScan® Dx Assay. The Affymetrix CytoScan® HD Array is already used by several clinical laboratories. Specific pricing and turn-around-time information for the Affymetrix CytoScan® Dx Assay is likely laboratory-dependent, and was not available at the time of this publication.
Table 3. Design Specifications of Representative Widely-Used CMA Platforms.
| CMA Name | Total # Copy Number Markers | # Oligonucleotide Markers | # SNP Markers | Probe Length | Probes per SNP Marker |
|---|---|---|---|---|---|
| Affymetrix CytoScan® HD Array | >2.6 million | ∼1.9 million | ∼750,000 | 25 base pairs | 6 probes |
| Agilent SurePrint G3 ISCA CGH+SNP 4X180 K array | 170,359 | 110,712 | 59,647 | 60 base pairs | 1 probe |
| Illumina Infinium CytoSNP-850K BeadChip | ∼850,000 | 0 | ∼850,000 | 50 base pairs | ≥15 probes |
List Prices: Affymetrix CytoScan® HD Array, Part #: 901835= $9,480 (includes reagents and arrays for 24 reactions); Agilent SurePrint G3 ISCA CGH+SNP 4X180 K array, Catalog #: G4890A= $3,468 (1 kit, for 4 samples); and Illumina Infinium CytoSNP-850K BeadChip, Catalog #: WG-322-1002= $4,000 (BeadChip for 16 samples). The price of the Affymetrix CytoScan® HD Array is not available at the time of this publication.
FDA Approval
The central non-technical difference between the Affymetrix CytoScan® Dx Assay and other similar microarray platforms is the recent FDA approval for use of the Affymetrix CytoScan® Dx Assay in the diagnosis of children with developmental delay, intellectual disability, congenital anomalies, and/or dysmorphic features. The FDA is an agency within the United States Department of Health and Human Services which acts to protect public health by ensuring safety of drugs, vaccines, biopharmaceuticals, medical devices, and other biological products. In contrast to laboratory-developed tests (LDTs) that are developed for use by a single company or laboratory, commercial laboratory tests are considered medical devices due to the aim of their widespread use, and thus must be evaluated and approved by the FDA. The FDA reviewed the Affymetrix CytoScan® Dx Assay through its de novo classification process, a regulatory pathway for novel low-to-moderate-risk medical devices. The full CytoScan® Dx Assay, including the CytoScan® Dx array, the testing reagents, analysis software, and hardware platform, was approved for the specified indications. The CytoScan® Dx Assay has a similar probe content to the CytoScan® HD Array, which has been used previously by independent clinical laboratories and for research purposes. The FDA's review of the test provides clinical laboratories with information about the expected performance of the device and quality of the results, but it does not provide the level of detailed test performance information needed to assure compliance with the ACMG microarray platform performance recommendations.[12, 14] Additionally, the package insert for the Affymetrix CytoScan® Dx Assay is not yet publically available for review. The FDA approval also comes with the disclaimer that CytoScan® Dx results
should not be used for stand-alone diagnostic purposes, pre-implantation or prenatal testing or screening, population screening, or for the detection of, or screening for acquired or genetic aberrations occurring after birth, such as cancer. The test results should only be used in conjunction with other clinical and diagnostic findings, consistent with professional standards of practice, including confirmation by alternative methods, evaluation of parental samples, clinical genetic evaluation, and counseling as appropriate. Interpretation of test results is intended to be performed only by health care professionals who are board certified in clinical cytogenetics or molecular genetics.[5]
Since the early 1990s, microarrays have been widely used in biomedical research. Ten years later, CMAs began to be widely used for clinical diagnostic purposes. Although independent laboratories offering clinical microarray testing are regulated and certified by the Centers for Medicare and Medicaid Services through the Clinical Laboratory Improvement Amendments (CLIA) program, and therefore, must provide detailed information regarding test accuracy, precision, sensitivity, and specificity for regulatory review, no microarray testing platform itself has previously been FDA approved for widespread commercial use. FDA approval for use of the Affymetrix CytoScan® Dx Assay for genetic evaluation of children diagnosed with DD enables individual laboratories to more easily validate the diagnostic assay given that specific instructions and guidelines for use of the device and information regarding expected device performance are available. In addition, all required device components are manufactured in a certified Good Manufacturing Practices (GMP) and International Organization for Standardization (ISO) facility, which may improve quality control measures. FDA approval is particularly helpful for laboratories that may be inexperienced in the use of a certain test or assay; however, laboratories that choose to use a commercial test are unable to make any modifications to the approved device protocol and thus may have reduced flexibility.
FDA approval of the Affymetrix CytoScan® Dx Assay may also help improve third-party reimbursement for this testing by improving awareness of the clinical indications of CMA testing. At present, not all insurance companies are providing reimbursement for this testing despite clear clinical guidelines indicating CMA as a first-line test.
Expert Commentary
Developmental delay is a prevalent problem with profound medical, economic, and social consequences. Achieving a genetic diagnosis as the underlying cause of idiopathic DD in both syndromic and non-syndromic cases has great benefit for optimizing management strategies for developmental progress and associated somatic disorders. Additionally, making an accurate genetic diagnosis for children with DD can provide families with prognostic information for both the child with DD and future pregnancies, and can provide a measure of peace to families who may have significant guilt or anxiety over the idiopathic nature of their child's neurologic dysfunction.
CMA is the first-line testing strategy for the genetic evaluation of children with DD in the absence of suspicion of a particular genetic diagnosis or when there has been a history of multiple miscarriages, and thus, this test has widespread use and utility. Until now, each U.S. clinical laboratory that offered this test chose their own CMA platform and developed their own data analysis strategy, which was independently submitted to regulatory agencies, including CLIA, for test approval and review. With the availability of an FDA-approved commercial CMA test, independent laboratories are more easily able to adopt a pre-approved platform, making test validation less cumbersome. For this reason, it may be easier for a smaller laboratory or a laboratory with limited experience with this testing methodology to offer this particular test. It is worth noting, however, that use of the approved product requires all instructions and analysis procedures to be followed and no changes in methodology by an individual lab may be made.
At the time of this publication, there is limited information regarding CNV reporting criteria for the FDA-approved CytoScan® Dx Assay and resolution limitations. The CytoScan® Dx Assay is unlikely to provide improved resolution over other readily available clinical CMA platforms. In fact, the Affymetrix CytoScan® HD Array (an array similar to the chip used in the CytoScan® Dx Assay) has already been validated and approved by independent laboratories for use as a clinical LDT. FDA approval of Affymetrix's CytoScan® Dx Assay does give this platform a market advantage with clinical laboratories that currently do not have an established CMA methodology over other similar microarray-based genetic diagnostic tests. This may have the effect of standardizing results and reporting among laboratories with limited CMA experience.
Five-Year View
In the next five years, CMA will continue to be a first-line molecular diagnostic test for children with uncharacterized DD. With continued widespread use of CMA and deposition of identified variants into public databases, CNV classification will improve and allow for more definitive calling. Additionally, improvement in microarray technology will continue to allow for improved sensitivity, specificity, and resolution. However, as the cost of next generation sequencing decreases and next generation sequencing CNV calling algorithms improve, it is likely that a single test that assesses both copy number and gene sequence will replace CMA. Whether this occurs in the next five years or later will depend on the rate of technological advances and future reimbursement practices by insurance providers.
Key Issues.
Chromosomal microarray is the first-line test of choice for genetic evaluation of developmental delay when a specific single gene disorder is not suspected.
The Affymetrix CytoScan® Dx Assay platform is the first FDA-approved microarray technology for use in the diagnosis of postnatal developmental delay.
The Affymetrix CytoScan® Dx Assay techonology is similar to CMAs already in use by clinical laboratories. FDA approval may offer a market advantage, particularly for laboratories with limited prior experience with microarray technology.
The Affymetrix CytoScan® Dx Assay design and performance are difficult to assess using ACMG-recommended criteria at this time due to limited publically-available data.
Acknowledgments
Financial and Competing Interests Statement: Dr. Webb is supported by NIH training grant T32 GM082773-06. Dr. Stroustrup is supported by grant K23ES022268-01 from the National Institutes of Environmental Health Sciences. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was utilized in the production of this manuscript.
Funding Source: Dr. Webb is supported by NIH training grant T32 GM082773-06. Dr. Stroustrup is supported by grant K23ES022268 from the National Institutes of Environmental Health Sciences.
Abbreviations
- ACMG
The American College of Medical Genetics and Genomics
- AOH
absence of heterozygosity
- ASO
allele-specific oligonucleotide
- BAC
bacterial artificial chromosome
- CGH
comparative genomic hybridization
- CLIA
Clinical Laboratory Improvement Amendments
- CMA
chromosomal microarray
- CNV
copy number variant
- DD
developmental delay
- DNA
deoxyribonucleic acid
- FDA
Federal Drug Administration
- GDD
global developmental delay
- gDNA
genomic deoxyribonucleic acid
- FDA
United States Food and Drug Administration
- GMP
good manufacturing practices
- FISH
fluorescence in situ hybridization
- FPR
false positive rate
- ISO
International Organization for Standardization
- LDT
laboratory-developed test
- SNP
single nucleotide polymorphism
- PCR
polymerase chain reaction
- UPD
uniparental disomy
- VUS
variant of uncertain significance
Appendix A
CytoScan® Dx Assay is a qualitative assay intended for the postnatal detection of chromosomal copy number variants (CNV) in genomic DNA (gDNA) obtained from peripheral whole blood in patients referred for chromosomal testing based on clinical presentation. CytoScan® Dx Assay is indicated for the detection of CNVs associated with developmental delay and/or intellectual disability (DD/ID), congenital anomalies, and/or dysmorphic features. Assay results are intended to be used in conjunction with other clinical and diagnostic findings, consistent with professional standards of practice including confirmation by alternative methods, parental evaluation, clinical genetic evaluation, and counseling as appropriate. Interpretation of assay results is intended to be performed only by healthcare professionals board certified in clinical cytogenetics or molecular genetics. The assay is intended to be used on the GeneChip® System 3000Dx and analyzed by Chromosome Analysis Suite Dx Software (ChAS Dx Software). This device is not intended to be used for standalone diagnostic purposes, pre-implantation or prenatal testing or screening, population screening, or for the detection of, or screening for, acquired or somatic genetic aberrations.
Footnotes
Financial Disclosure: The authors have no financial relationships relevant to this article to disclose.
Conflict of interest: The authors have no conflicts of interest to disclose.
References
* of interest
** of considerable interest
- 1.Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12(11):742–5. doi: 10.1097/GIM.0b013e3181f8baad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2**.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64. doi: 10.1016/j.ajhg.2010.04.006. This consensus statement of The International Standard Cytogenomic Array (ISCA) Consortium provides an evidence-based summary of clinical cytogenetic testing comparing clinical microarry to G-banded karyotyping with respect to technical advantages and limitations, diagnostic yield for various types of chromosomal aberrations, and issues that affect test interpretation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–35. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4*.Tsuchiya KD, Shaffer LG, Aradhya S, Gastier-Foster JM, Patel A, Rudd MK, et al. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet Med. 2009;11:866–73. doi: 10.1097/GIM.0b013e3181c0c3b0. This study assess the variability in interpretation and reporting of copy number changes that are detected by array-based technology in the clinical laboratory. [DOI] [PubMed] [Google Scholar]
- 5.United States Food and Drug Administration, US Department of Health and Human Services. FDA allows marketing for first of-its-kind post-natal test to help diagnose developmental delays and intellectual disabilities in children. FDA News Release. 2014 Jan 17; Available at: http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm382179.htm.
- 6.Petersen MC, Kube DA, Palmer FB. Classification of developmental delays. Semin Pediatr Neurol. 1998;5:2–14. doi: 10.1016/s1071-9091(98)80012-0. [DOI] [PubMed] [Google Scholar]
- 7.Topcu M, Yalnizoglu D. Developmental abnormalities and mental retardation: diagnostic strategy. In: Dulac O, Lassonde M, Sarnat HB, editors. Handbook of Clinical Neurology. New York: Elsevier; 2013. pp. 211–7. [DOI] [PubMed] [Google Scholar]
- 8.Frankenburg WK, Dodds JB. The Denver developmental screening test. J Pediatr. 1967;71:181–91. doi: 10.1016/s0022-3476(67)80070-2. [DOI] [PubMed] [Google Scholar]
- 9.R C, Shapiro BK, Palmer FB, Allen MC, Capute AJ. CAT/CLAMS. A tool for the pediatric evaluation of infants and young children with developmental delay. Clinical Adaptive Test/Clinical Linguistic and Auditory Milestone Scale. Clin Pediatr. 1994;33:410–5. doi: 10.1177/000992289403300706. [DOI] [PubMed] [Google Scholar]
- 10*.Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol. 2012;19:173–80. doi: 10.1016/j.spen.2012.09.004. The review provides an updated diagnostic approach to a child with unexplained global developmental delay or intellectual disability. [DOI] [PubMed] [Google Scholar]
- 11.Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–12. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12**.Kearney HM, South ST, Wolff DJ, Lamb A, Hamosh A, Rao KW. American College of Medical Genetics recommendations for the design and performance expectations for clinical genomic copy number microarrays intended for use in the postnatal setting for detection of constitutional abnormalities. Genet Med. 2011;13(7):676–9. doi: 10.1097/GIM.0b013e31822272ac. In this paper, the American College of Medical Genetics, the professional organization of board-certified clinical laboratory geneticists, outlines recommendations for the design and performance expectations for clinical genomic copy number microarrays and associated software intended for use in the postnatal setting for detection of constitutional abnormalities. [DOI] [PubMed] [Google Scholar]
- 13.Hehir-Kwa JY, Pfundt R, Veltman JA, de Leeuw N. Pathogenic or not? Assessing the clinical relevance of copy number variants. Clin Genet. 2013;84:415–21. doi: 10.1111/cge.12242. [DOI] [PubMed] [Google Scholar]
- 14**.South ST, Lee C, Lamb AN, Higgins AW, Kearney HM. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet Med. 2013;15:901–9. doi: 10.1038/gim.2013.129. This paper outlines revised professional standards and guidelines for clinical genomic copy number microarrays and associated software intended for use in the postnatal setting for detection of constitutional abnormalities. [DOI] [PubMed] [Google Scholar]
- 15.Morrow EM. Genomic copy number variation in disorders of cognitive development. J Am Acad Child Adol Psych. 2010;49:1091–104. doi: 10.1016/j.jaac.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Scott SA, Cohen N, Brandt T, Toruner G, Desnick RJ, Edelmann L. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet Med. 2010;12:85–92. doi: 10.1097/GIM.0b013e3181cc75d0. This study evaluates the sensitivity of whole-genome oligonucleotide array comparative genomic hybridization for the detection of mosaic cytogenetic abnormalities. [DOI] [PubMed] [Google Scholar]
- 17**.United States Food and Drug Administration, US Department of Health and Human Services. Evaluation of Automatic Class III Designation for Affymetrix® CytoScan® Dx Assay Decision Summary. Silver Spring, MD: 2014. http://www.accessdata.fda.gov/cdrh_docs/reviews/k130313.pdf. Publically available FDA submission information for the Affymetrix® CytoScan® Dx Assay. [Google Scholar]
- 18.Cruz Cuevas J. Clinical Cytogenetic Applications and the Affymetrix Medical Affairs Department. Santa Clara, CA: 2014. Personal Communication. [Google Scholar]