

Abstract
Purpose.
Methylenetetrahydrofolate reductase (Mthfr) is a key enzyme in homocysteine-methionine metabolism. We investigated Mthfr expression in retina and asked whether mild hyperhomocysteinemia, due to Mthfr deficiency, alters retinal neurovascular structure and function.
Methods.
Expression of Mthfr was investigated at the gene and protein level using quantitative (q) RT-PCR, in situ hybridization, immunoblotting, and immunohistochemistry (IHC). The Mthfr+/+ and Mthfr+/− mice were subjected to comprehensive evaluation using ERG, funduscopy, fluorescein angiography (FA), spectral-domain optical coherence tomography (SD-OCT), HPLC, and morphometric and IHC analysis of glial fibrillary acidic protein (GFAP) at 8 to 24 weeks.
Results.
Gene and protein analyses disclosed widespread retinal expression of Mthfr. Electroretinography (ERG) revealed a significant decrease in positive scotopic threshold response in retinas of Mthfr+/− mice at 24 weeks. Fundus examination in mice from both groups was normal; FA revealed areas of focal vascular leakage in 20% of Mthfr+/− mice at 12 to 16 weeks and 60% by 24 weeks. The SD-OCT revealed a significant decrease in nerve fiber layer (NFL) thickness at 24 weeks in Mthfr+/− compared to Mthfr+/+ mice. There was a 2-fold elevation in retinal hcy at 24 weeks in Mthfr+/− mice by HPLC and IHC. Morphometric analysis revealed an approximately 20% reduction in cells in the ganglion cell layer of Mthfr+/− mice at 24 weeks. The IHC indicated significantly increased GFAP labeling suggestive of Müller cell activation.
Conclusions.
Mildly hyperhomocysteinemic Mthfr+/− mice demonstrate reduced ganglion cell function, thinner NFL, and mild vasculopathy by 24 weeks. The retinal phenotype is similar to that of hyperhomocysteinemic mice with deficiency of cystathionine-β-synthase (Cbs) reported earlier. The data support the hypothesis that hyperhomocysteinemia may be causative in certain retinal neurovasculopathies.
Keywords: methylation pathway, homocysteine, retina, retinal degeneration, ERG, mouse
Homocysteine is implicated in several retinal diseases. The authors used a mouse with deficiency of the enzyme Mthfr to study the effects of excess homocysteine on retina and discovered decreased inner retinal function accompanied by mild vasculopathy in Mthfr-deficient mice.
Homocysteine (hcy), an intermediate in methionine metabolism and a sulfur-containing nonproteinogenic amino acid, resides at the intersection of the remethylation and transsulfuration metabolic pathways (Fig. 1). When methionine levels are elevated, hcy is catabolized to cystathionine by cystathionine-β-synthase (Cbs) via the transsulfuration pathway, which requires pyridoxal phosphate (vitamin B6) as a cofactor. Cystathionine subsequently is used in synthesis of downstream products, such as taurine, glutathione (GSH) and hydrogen sulfide (H2S). When the diet is deficient in methionine, hcy is remethylated back to methionine via the remethylation pathway involving the enzymes Mthfr and methionine synthase (MeSe). The remethylation pathway requires folate and cobalamin (vitamin B12) as co-factors.1,2 Perturbance in hcy metabolism can lead to elevated plasma Hcy levels. Homozygous mutations in Cbs and Mthfr enzymes lead to homocystinuria characterized by very high levels of plasma hcy, severe mental and skeletal abnormalities, premature thromboembolism, and lens dislocation.3 Heterozygous mutations in these enzymes or nutritional deficiency of cofactors can lead to moderate increase in plasma hcy known as hyperhomocysteinemia (Hhcy). Hyperhomocysteinemia is an independent risk factor for cardiovascular diseases,4,5 pregnancy complications,6 neural tube defects,7 sickle cell anemia,8,9 osteoporosis,10 diabetes,11,12 schizophrenia,13 Parkinson's and Alzheimer's disease.14,15 Hyperhomocysteinemia is implicated in various ocular pathologies, including diabetic retinopathy,16,17 age-related macular degeneration,18,19 glaucoma (primary and secondary open-angle glaucoma, exfoliation glaucoma, pigmentary glaucoma),20–22 retinal degeneration,23 branch retinal vein occlusion (BRVO), and central retinal vein and artery occlusion (CRVO).24–26 The clinical literature suggests the potential of excess hcy in inducing retinal damage.
Figure 1.
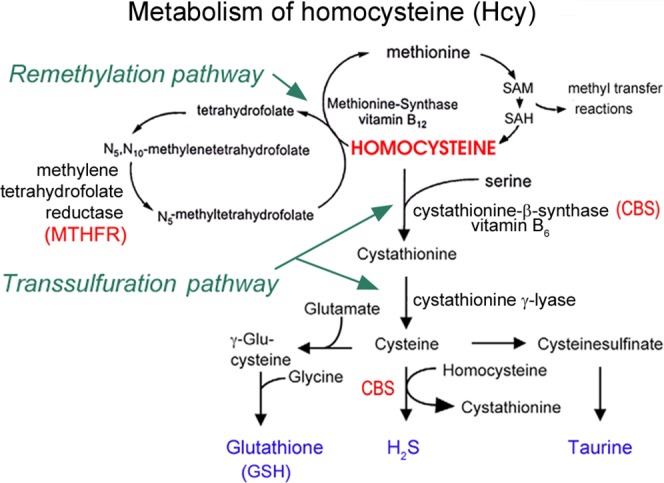
The metabolism of homocysteine. Homocysteine sits at the intersection of the remethylation and the transsulfuration pathways. In the remethylation pathway, Mthfr converts N5, N10-methylenetetrahydrofolate to N5-methyltetrahydrofolate, which then donates a methyl group to homocysteine in the presence of methionine synthase and vitamin B12 for formation of methionine, an amino acid used in many methylation reactions. In the transsulfuration pathway, excess homocysteine can be converted eventually to cysteine in the presence of CBS for the formation of GSH, H2S, and taurine. Deficiencies of enzymes, including Mthfr or CBS, can lead to accumulation of Hcy in a condition known as Hhcy.
Over the past several years, our laboratory has explored various in vitro and in vivo model systems to understand the effects of Hhcy on retina. Our first in vitro report of hcy toxicity using primary mouse ganglion cells incubated with 50 μM D, L-hcy thiolactone resulted in approximately 50% to 60% cell death within 18 hours.27 In vivo studies demonstrated profound ganglion cell loss and inner retinal disruption within 5 days of intravitreal injection of high dosage (200 μM) of D, L-hcy thiolactone.28 Marked photoreceptor cell loss within 15 days and complete ablation of the outer nuclear layer (ONL) within 90 days after hcy-thiolactone intravitreal injection was reported by Chang et al.29
To understand the effects of endogenous elevation of hcy, we characterized the retinal phenotype in Cbs-deficient mice. Homozygous mice have a short life span (approximately 3–5 weeks), approximately 7-fold elevation in retinal hcy, and profound retinal neurovasculopathy.29–31 There is marked inner/outer nuclear retinal layer disruption, ganglion cell loss, and RPE hypertrophy. Vascular alterations include central retinal ischemia, neovascularization, and incompetent blood–retinal barrier.32 Functional studies show severe functional deficits with significant reduction of the ERG.33 A milder phenotype is observed in heterozygous Cbs mice. These mice have a normal life span and approximately 2-fold increase in retinal hcy. The retinal morphology is comparable to that of age-matched wild type animals for many weeks. By 1 year, retinal morphological changes are observed, including modest loss of cells in the ganglion cell layer (GCL), and decreased thickness of inner plexiform and nuclear layers.31 Functional studies reveal a gradual decrease in ERG amplitudes by 10 weeks of age.33
The data from Cbs−/− and Cbs+/− mice raise an important conundrum: is the retinal neurovasculopathy observed in absence/deficiency of Cbs due to elevated hcy levels or is it due to decreased levels of potentially beneficial products (taurine, GSH, and H2S) of the transsulfuration pathway (Fig. 1). This can be addressed by studying the retinal phenotype in a genetic model of Hhcy with intact transsulfuration pathway. In the present study, we used mice with deficiency of Mthfr. The Mthfr mutation is the most common genetic cause of Hhcy,33 more prevalent in the general population than the Cbs mutation. Various polymorphisms have been identified and investigated. One of the most common single nucleotide polymorphisms (SNP) is 677C>T. The normal allele includes C (cytosine) at the 677 position leading to alanine at amino acid 222. This is substituted by T (thymine) leading to valine at amino acid 222 yielding a thermolabile enzyme with reduced activity. Of all Americans 44% are heterozygous for this mutation and 12% are homozygous. The 677C>T mutation is prevalent globally with a frequency of 24% to 40%, 26% to 37%, and 11% in Europeans, Japanese, and African-American population, respectively. The 677C>T mutation predisposes an individual to mild or moderate Hhcy.34–36 The Mthfr mutant mouse provides a powerful model system to study deficiency of Mthfr. Depending upon whether the mice are heterozygous (Mthfr+/−) or homozygous (Mthfr−/−) for the deletion, a mild-moderate to severe hyperhomocysteinemia is present. The Mthfr mutant mice were developed by R. Rozen, PhD. When the mice are bred on the Balb/c background, the homozygous mice have very low survival and reproductive rates, 10-fold increase in plasma hcy, neuropathology, aortic lipid deposits, and decreased methylation capacity. The heterozygous mice have 1.6-fold higher plasma hcy.2 When bred on the C57BL/6 genetic background, there is improved survival and reproduction. Homozygous mice have a 10-fold increase in plasma hcy.37 To our knowledge, there have been no comprehensive studies of retinal function or structure of Mthfr-deficient mice. There was a single study of ERG analysis of Mthfr−/− mice reporting that rod/cone-mediated a- and b- wave amplitudes were significantly decreased compared to Mthfr+/+ at 6 weeks.37 Curiously, when older homozygous mice (13 weeks) were evaluated, these amplitude differences were no longer detected. Apart from these intriguing ERG data, no information is available regarding retinal phenotype in these mice. Given the importance of Hhcy, coupled with high prevalence of Mthfr mutations, the present study systematically investigated the retinal phenotype in Mthfr-deficient mice.
Methods
Animals
We used 98 mice in the study (Table 1). Breeding pairs of Mthfr+/− mice were shipped from the Rozen lab (McGill University Montreal, Canada) to the animal facility of Georgia Regents University. Genotyping was performed routinely to confirm the animal model. Mice were screened for the rd8 mutation in the Crb1 gene and were negative. Maintenance and treatment of animals adhered to the institutional guidelines for humane treatment of animals and to the ARVO Statement for Use of Animals in Ophthalmic and Vision Research.
Table 1.
Animals Used in This Study

Analysis of Mthfr Gene Expression in Mouse Retina
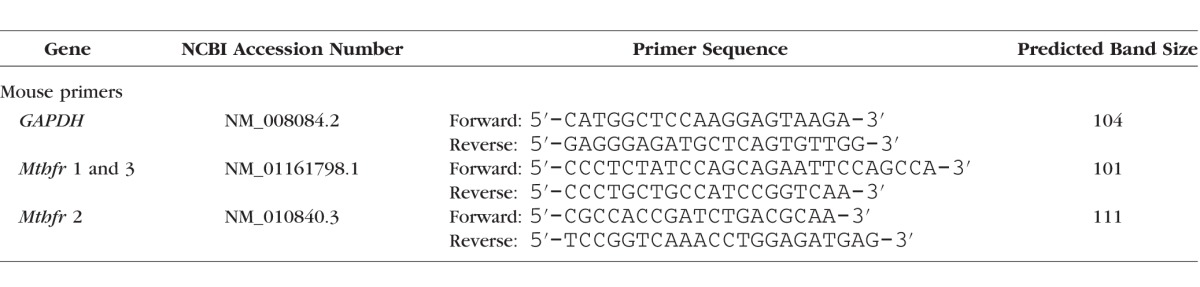
To determine whether Mthfr is expressed in mouse retina, RNA was isolated from neural retina using TRIzol (Invitrogen, Carlsbad, CA, USA) and from testis (positive control). Then, 2 μg RNA was converted to cDNA using SuperScript II Reverse Transcriptase (Invitrogen). The primers were obtained from Integrated DNA Technologies (Coralville, IA, USA; Table 2). Two primers were used to identify the Mthfr variant expressed in mouse retina. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as internal control. Quantitative (q) RT-PCR was performed in triplicate per our method.31 To localize mRNA transcripts encoding Mthfr in mouse retina, riboprobes were prepared and subjected to fluorescent in-situ hybridization analysis according to our method.38
Table 2.
The qRT PCR Primers Used for Mthfr Gene Expression Studies
Analysis of Mthfr Protein in Mouse Retina
To analyze Mthfr protein expression in mouse retina, neural retinal tissue was isolated from C57BL/6 mice for immunoblotting analysis using anti-Mthfr (1:1000; Mr approximately 75 kDa; Abcam, Cambridge, MA, USA) per our method.31 To identify in which retinal cell layers Mthfr was present, 8-week-old C57BL/6 (Mthfr+/+) and Mthfr−/− mice were euthanized by CO2 asphyxiation followed by cervical dislocation. Eyes were harvested and flash-frozen in Tissue-Tek OCT (Miles Laboratories, Elkhart, IN, USA). Cryosections 10 μm thick were incubated with anti-Mthfr antibody and with antibodies specific for several cell types (Neu-N for neurons, vimentin for Müller cells, glial fibrillary acidic protein [GFAP] for astrocytes, and RPE65 for RPE cells, Table 3). Sections were viewed by epifluorescence using the Axioplan-2 microscope equipped with the Axiovision Program and high-resolution camera (Carl Zeiss, Oberkochen, Germany) as described.31 Expression of Mthfr was examined in other ocular tissues, including cornea, lens, ciliary body, and optic nerve.
Table 3.
Antibodies Used for Immunoblotting and Immunohistochemical Analyses

Electroretinogram (ERG)
To assess visual function, Mthfr+/+ and Mthfr+/− mice (12 and 24 weeks) were subjected to ERG analysis. Mice were dark adapted overnight. Each mouse was tested on two separate days, once under isofluorane anesthesia (10 mg/cc) using relatively bright stimuli, and a week later under anesthesia (ketamine 100 mg/cc, xylazine 30 mg/cc, acepromazine) using relatively dim stimuli. The bright stimuli were generated by a white light-emitting diode (LED), and the dim stimuli with a blue LED that was filtered and defocused. Light from the LED was presented to the eye by 1-mm diameter optic fibers that were placed just in front of the pupils. Signals were acquired by DTL fibers placed gently on the corneas, with a drop of hypromellulose to keep eyes moist and enhance electrical contact. These signals were transferred to a Psylab amplifier, with a gain of 10,000, filtered between 0.3 and 400 Hz, with a notch filter at 60 Hz. The amplified signals were digitized by a National Instruments 6323 data acquisition module (National Instruments, Austin, TX, USA), and read into custom software written in Igor Pro. Stimulus intensity was calibrated in scotopic lumens for each of the LED setups.
For the bright stimuli, a series of increasing intensities of 5-ms flashes were presented while dark adapted. Amplitude and timing were measured for a-, b-, and c-waves. Following light adaptation, a variety of stimuli were presented, including 8-second long presentations of pseudorandom luminance values. These stimuli were sampled at 512 Hz and had natural temporal statistics. Responses were analyzed by correlating them with the stimulus to obtain kernels describing the retinal transformation between stimulus and response. For the dim stimuli, an interleaved set of intensities just below and above threshold were presented. Averaged responses were analyzed to obtain positive and negative scotopic threshold responses (pSTRs and nSTRs). These are the amplitudes at 110 and 200 ms following the flash, respectively.39
In Vivo Retinal Imaging (Funduscopy, Fluorescein Angiography, SD-OCT)
Funduscopy and fluorescein angiography were performed in Mthfr+/+ and Mthfr+/− mice using the Micron III camera (Phoenix Research Laboratories, Inc., Pleasanton, CA, USA) according to our methods.32 Retinal morphology was analyzed in vivo using spectral-domain ocular coherence tomography (SD-OCT). Mice were anesthetized as described for ERG analysis. Pupils were dilated with 1% tropicamide (Bausch & Lomb, Tampa, FL, USA) followed by application of GenTeal Lubricant Eye Gel, 10 g (Alcon, Ft. Worth, TX, USA). Systane lubricant eye drops (Alcon) were applied to keep the cornea moist. The SD-OCT images were obtained using the Bioptigen Spectral Domain Ophthalmic Imaging System (SDOIS; Bioptigen Envisu R2200; Bioptigen, Inc., Durham, NC, USA). Imaging included averaged single B scan and volume intensity scans (VIP) with images centered on optic nerve head. Postimaging analysis included auto segmentation report analysis and manual assessment of all retinal layers using InVivoVueTM Diver 2.4 software (Bioptigen, Inc.), which enables auto segmentation measurements of retinal images acquired using SD-OCT. The auto segmentation feature gives an average thickness of retinal layers (calculated from 8 different measurements). Details of the capabilities of this software can be found at in the public domain at www.bioptigen.com. The autosegmentation feature has been reported previously.40,41
Measurement of IOP
The IOP was measured in 24-week-old Mthfr+/+ and Mthfr+/− mice, which were anesthetized using inhaled isofluorane (Butler Animal Health Supply, Dublin, OH, USA). The IOP was measured using a handheld tonometer (Tonolab; Icare Laboratory; Finland Oy, Espoo, Finland) positioned at the center of the cornea. Measurements were repeated twice in each animal.
Evaluation of Retinal Hcy Levels
The HPLC analysis was used to quantify hcy levels in neural retina isolated from 24-week-old Mthfr+/+ and Mthfr+/− mice per our published method.31,42 We evaluated Hcy immunohistochemically as described above in retinal cryosections from 24-week-old Mthfr+/+ (n = 4) and Mthfr+/− (n = 4) mice using an antibody specific for homocysteine (Table 3).
Microscopic Evaluation and Measurement Procedures
Retinal structure was evaluated in hematoxylin and eosin–stained retinal cryosections of 12 and 24-week-old Mthfr+/+ and Mthfr+/− mice for evidence of gross pathology. Morphometric evaluation of retinas involved measurement of the thickness of retinal layers and counting cells in the GCL as described.31
Statistical Analysis
For morphometric and OCT studies, 2-way ANOVA was used to determine whether there were significant differences between mouse groups (factors of mouse group and age). Bonferroni post hoc test was used to compare means. For immunofluorescence and HPLC analysis, Student's t-test was used to determine whether there were significant differences in between mouse groups. Data were analyzed using the GraphPad Prism software (version 6; GraphPad Software, Inc., La Jolla, CA, USA). A P value < 0.05 was considered significant.
Results
Analysis of Mthfr Gene and Protein in Mouse Retina
Representative mouse genotyping data are shown in Figure 2A. To evaluate retinal Mthfr gene expression, neural retina (and testis, which has very high Mthfr enzyme activity) were harvested and subjected to qRT-PCR using mouse-specific Mthfr primers (Table 2) normalized to GAPDH. The analysis revealed Mthfr gene (variants 1 and 3) expression in mouse retina (Fig. 2B), which was approximately 70% that of testis. Similar results were observed with Mthfr gene variant 2 (data not shown). To determine in which retinal types Mthfr gene was expressed, FISH was performed using an Mthfr-specific antisense probe. In situ hybridization demonstrated robust positive staining in the GCL, inner nuclear layer (INL), ONL, and RPE layers (Fig. 2C). Limited fluorescence was observed in sections incubated with sense probe (Fig. 2C). To determine whether Mthfr protein was present in retina, we isolated protein from neural retina (and testis, positive control) of Mthfr+/+ mice and performed immunoblotting using anti-Mthfr antibody. We detected a band of the expected molecular size (approximately 75 kD) in both tissues (Fig. 2D). To confirm the antibody specificity, Western blotting was performed using protein isolated from retina of Mthfr+/+ and Mthfr−/− mice (Fig. 2E). Immunohistochemistry with anti-Mthfr antibody detected the Mthfr protein in several retinal layers of wild type mouse (Fig. 2F), while incubation of Mthfr−/− retina had minimal labeling.
Figure 2.

Assessment of Mthfr gene and protein expression. (A) Representative genotyping data for four offspring; wild type mice expressing only the 145-base pair (bp) product have both copies of the Mthfr gene, heterozygous mice expressing the 216-bp and 145-bp have one copy of the Mthfr gene, and homozygous mice expressing only the 216-bp product do not express the Mthfr gene. (B) Total RNA was isolated from neural retina and testis (positive control) and subjected to qRT-PCR with mouse-specific Mthfr primers. The GAPDH served as the internal control. (C) Retinal Mthfr mRNA localization was investigated by FISH. Retinal cryosections were incubated with Mthfr anti-sense and sense probes. Red fluorescence represents positive staining. (D) Protein was extracted from Mthfr+/+ mouse retina and testis, and subjected to immunoblotting with antibody against Mthfr (Mr = 5 kD). The β-actin (Mr = 42 kD) was the internal loading control. (E) To confirm the specificity of the Mthfr antibody, protein was extracted from Mthfr+/+ and Mthfr−/− retina and subjected to immunoblotting with the Mthfr antibody. The GAPDH (Mr = 37 kD) was the internal loading control. (F) Retinal cryosections from Mthfr+/+ and Mthfr−/− mice were incubated with an antibody against Mthfr followed by incubation with Alexa Fluor 488 (green)–labeled secondary antibody. We used 4′6-dimidino-2-phenylindole (DAPI) to label nuclei (blue fluorescence). Calibration bar: 50 μM. gcl, GCL; inl, INL; onl, ONL; rpe, RPE.
Analysis of Mthfr Protein in Mouse Retina, Cornea, Lens, Ciliary Body, and Optic Nerve
To investigate which retinal cell types are positive for Mthfr, we performed dual immunolabeling experiments using the Mthfr antibody in conjunction with antibody markers for inner retinal neurons (Neu-N), astrocytes (GFAP), Müller cells (vimentin), and RPE cells (RPE65). We observed Mthfr labeling of the GCL (comprised of ganglion and amacrine cells) and cells of the INL (Fig. 3A). We observed Mthfr labeling of astrocytes (Fig. 3B), Müller cells (Fig. 3C), and RPE cells (Fig. 3D). The Mthfr protein is present in other ocular tissues, including the corneal epithelium though it is minimally expressed in corneal stroma (Figs. 3E, 3F). The Mthfr protein was detected in the equatorial lens epithelial cells and in few lens fiber cells (Figs. 3G, 3H). It was detected in superficial and deep epithelial layers of the ciliary body (Figs. 3I, 3J), and in optic nerve head (Figs. 3K, 3L). The data indicate widespread expression of Mthfr in the eye.
Figure 3.

Immunofluorescent co-localization of Mthfr in retina and in ocular tissues. Retinal cryosections prepared from Mthfr+/+ mice were co-immunostained with antibody against Mthfr (green fluorescence) and antibodies against various cell types (red fluorescence). (A) Neu-N to label retinal neurons of gcl and inl. (B) GFAP to label astrocytes. (C) Vimentin to label Müller cells. (D) RPE-65 to label RPE cells. DAPI was used to label nuclei (blue fluorescence). (E) Eye cryosections prepared from Mthfr+/+ mice were immunostained with antibody against Mthfr (green fluorescence). Companion sections were stained with hematoxylin and eosin. Mthfr was detected in cornea (E, F), lens (G, H), ciliary body (I, J), and optic nerve (K, L). Calibration bar: 50 μM (A–J) and 100 μM (K–L). We used DAPI to label nuclei (blue fluorescence). epi, epithelium; st, stroma; c, capsule; sc, sclera.
ERG Results
To assess visual function in Mthfr+/− compared to Mthfr+/+ mice, electrophysiological studies were performed at 12 and 24 weeks. Response to scotopic and photopic stimuli were obtained; there were no significant differences in a- or b-wave amplitudes between Mthfr+/− mice compared to Mthfr+/+ mice at either age examined. Data are shown for the scotopic responses at 12 and 24 weeks (Figs. 4A, 4B). Similar photopic flash responses were obtained (data not shown). We measured kernels from pseudorandom luminance modulations using natural stimuli and detected stronger amplitudes in Mthfr+/+ mice compared to Mthfr+/− mice at 12 and 24 weeks (Fig. 4C). This difference was significant at 12 weeks, but not at 24 weeks. We evaluated STRs and detected a slight, but significant, decrease in the positive STRs of Mthfr+/− mice at 12 weeks, which was more pronounced at 24 weeks (Fig. 4D). These data suggested a modest decrease in ganglion cell function in mild Hhcy mice due to deficiency of Mthfr.
Figure 4.

Electrophysiological assessment of retinal function in 12- and 24-week Mthfr+/− mice. We performed ERG under scotopic conditions over a range of bright flash intensities in Mthfr+/+ (n = 3) and Mthfr+/− (n = 3) at 12 weeks, and Mthfr+/+ (n = 4) and Mthfr+/− (n = 4) at 24 weeks. Graphic depiction of the average amplitudes of a- and b-waves (A, B), kernels (C, D) in Mthfr+/+ compared to Mthfr+/− mice. The pSTR and nSTR amplitudes are shown for Mthfr+/+ compared to Mthfr+/− mice. *Significantly different from Mthfr+/− mice; P < 0.05.
Funduscopy and Fluorescein Angiography (FA)
We examined Mthfr+/+ and Mthfr+/− mice by retinal funduscopy and FA at ages 8 to 24 weeks. The fundus was normal in appearance with no apparent disruption, abnormal spots, or evidence of debris in either mouse group (Figs. 5A, 5C, 5E, 5G, 5I, 5K, 5M, 5O). To visualize retinal vessels, fluorescein dye was injected and images were acquired 30 seconds after injection and every minute thereafter for 5 minutes. Images were arranged according to capture time and data were compared for Mthfr+/+ and Mthfr+/− mice at the same time interval. The FA performed in Mthfr+/+ (Fig. 5B) and Mthfr+/− (Fig. 5D) mice at 8 weeks showed normal vessel filling, no leakage, and uniform capillary network around blood vessels. By 12 weeks, there was evidence of vascular leakage (Fig. 5H, arrows) and vascular tortuosity (Fig. 5H) in Mthfr+/− retinas, which also was evident at 16 weeks in the mutant mice (Fig. 5L). Approximately 30% of mutant mice demonstrated theses alterations through 16 weeks. By 24 weeks of age, significantly more Mthfr+/− mice (approximately 60%) had areas of focal leakage and vascular tortuosity (Fig. 5P). The Mthfr+/+ mice showed no evidence of leakage or vessel tortuosity throughout the 24-week period. The data provided in vivo evidence of increased vascular permeability in mice with deficiency in Mthfr.
Figure 5.
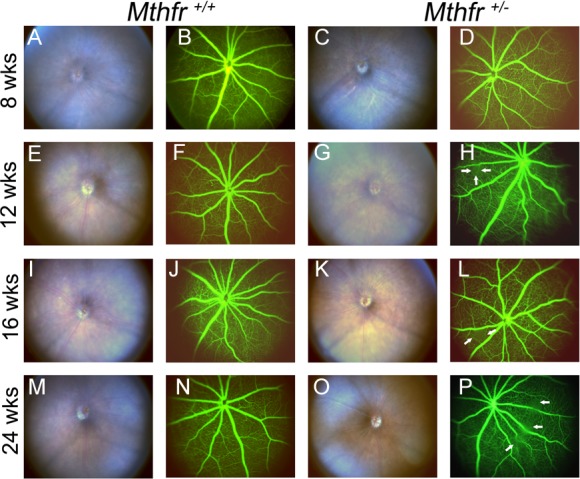
Funduscopy and FA in Mthfr+/− mice. Representative fundus images and FA data are provided for Mthfr+/+ and Mthfr+/− mice at ages 8 weeks (A–D), 12 weeks (E–H), 16 weeks (I–L) and 24 weeks (M–P). At 8 weeks, Mthfr+/+ and Mthfr+/− mice displayed normal fundus and FA (A–D). At 12 weeks, the fundus appeared normal, focal area of fluorescein leakage was observed in Mthfr+/− (indicated by white arrows; [H]). The leakage persisted through 16 weeks (white arrows; [L]) and intensified in appearance and number by 24 weeks of age (P).
SD-OCT Imaging
To obtain information about retinal structure in living animals, SD-OCT was performed. The Mthfr+/+ and Mthfr+/− mice were evaluated at 12, 16, and 24 weeks. Volume image projections and the B-scans of Mthfr+/+ mice showed normal retinal architecture and no evidence of disruption in any retinal layers (data not shown). Postimage analysis of retinal layers used InVivoVue 2.4 DIVER software. A representative segmentation report is provided for Mthfr+/+ mice (Fig. 6A) and Mthfr+/− mice (Fig. 6B). Parameters measured in the automated program include whole retinal thickness (RT), and thickness of the nerve fiber layer (NFL), inner plexiform layer (IPL), INL, outer plexiform layer (OPL), ONL + inner segment (IS), outer segment (OS), and RPE. The RT was approximately 220 μM for Mthfr+/+ and Mthfr+/− mice at 12, 16, and 24 weeks. There was a significant difference between mouse groups at 24 weeks for thickness of the NFL (Mthfr+/+, 14.00 ± 0.72 μM; Mthfr+/−, 10.11 ± 0.87 μM). All other measurements were similar at the three ages studied between Mthfr+/+ and Mthfr+/− mice (data not shown). To validate the automated segmentation analysis, we performed manual measurement of retinal layers with DIVER software using the 5 × 5 grid retinal template. This analysis confirmed the statistically significant decrease in NFL thickness for Mthfr+/− compared to Mthfr+/+ mice (Fig. 6C). The other retinal measurements were similar at all ages studied (data not shown).
Figure 6.

Automated segmentation analysis of retinal nerve fiber layer thickness (RNFL) in retinas of Mthfr+/− mice at 24 weeks. To evaluate thickness of RNFL in retinas of Mthfr+/− mice compared to Mthfr+/+ mice, automated segmentation analysis of RNFL was performed using SD-OCT. Representative SD-OCT heat maps of RNFL in 24-week-old (A) Mthfr+/+ and (B) Mthfr+/− mice are provided. Heat maps are color coded, such that white color reflects thicker RNFL, while dark blue represents thinner RNFL. The EDTRS in Mthfr+/− demonstrated even distribution of thinner RNFL in 300 and 600 μM zones in all quadrants. (C) For validation of automated segmentation analysis, we performed manual OCT measurements with 5 × 5 grid retinal template. Graphical depiction of the RNFL (mean ± SEM) is shown. *Significantly different from Mthfr+/+ mice; P < 0.01. ETDRS, Early Treatment of Diabetic Retinopathy Study; S, superior; I, inferior; N, nasal; T, temporal.
Measurement of IOP
Elevated IOP is a feature of some optic neuropathies, including some types of glaucoma; Hhcy also has been implicated in some forms of glaucoma. To determine whether deficiency of Mthfr is associated with increased pressure, IOP was determined in Mthfr+/+ and Mthfr+/− mice at 24 weeks, an age when there was decreased thickness of the NFL. The average IOP for Mthfr+/+ and Mthfr+/− mice was 9.5 ± 0.65 and 9.6 ± 0.24 mm Hg, respectively, suggesting no significant alterations in IOP in mice with mild Hhcy.
Levels of Homocysteine in Retinas of Mthfr+/− Mice
The Mthfr+/− mice have approximately 1.6-fold higher plasma hcy levels than Mthfr+/+ mice.2 We measured retinal hcy levels in 24-week Mthfr+/+ and Mthfr+/− mice by HPLC. Figure 7A depicts the standard curve used to derive the hcy levels in retinas. Retinas of Mthfr+/+ mice had low levels of hcy (0.2 ± 0.004 pmol/μg protein); retinas of Mthfr+/− mice had a, approximately 2-fold increase in retinal hcy (0.4 ± 0.073 pmol/μg protein), which was statistically significant (P < 0.01, Fig. 7B). We performed immunohistochemistry in retinal cryosections and detected increased hcy in Mthfr+/− retinas (Fig. 7D) compared to Mthfr+/+ (Fig. 7C). Quantification of immunofluorescence levels revealed an approximately 2-fold increase in retinal hcy in Mthfr+/− compared to Mthfr+/+ mice (Fig. 7E).
Figure 7.

Analysis of retinal hcy levels and morphology in Mthfr+/+ and Mthfr+/− mice. Retinal hcy levels were quantified by HPLC and IHC analysis. (A) TheHcy standard curve used for calculation of retinal hcy levels. (B) Graphical representation of retinal hcy levels in Mthfr+/+ and Mthfr+/− mice (24 weeks) expressed as pmol/μg protein. ***Significantly different from wildtype mice, P < 0.001. (C, D) Retinal cryosections from Mthfr+/+ and Mthfr+/− mice (24 weeks) were subjected to immunodetection of hcy; Alexa Fluor 555-conjugated antibody (red) was used to detect positive signals. (E) The immunopositive signals were quantified using Metamorph software. **Significantly different from wild type mice, P < 0.01. (F) Representative hematoxylin and eosin–stained sections of retinas harvested from Mthfr+/+ and (G, H) Mthfr+/− mice (24 weeks). Areas of cellular dropout observed in retinas from Mthfr+/− mice are noted by the black arrow (H). Calibration bar: 50 μM. ipl, IPL; opl, OPL; is, IS; os, outer segment.
Retinal Morphometric Analysis
Eyes from Mthfr+/+ and Mthfr+/− mice were processed for light microscopic evaluation and retinal morphometric analysis at 12, 16, and 24 weeks. The retinal architecture of Mthfr+/+ mice (Fig. 7F) was similar to age-matched Mthfr+/− mice (Figs.7G, 7H). There was no evidence of gross disruption (Fig. 7G). There was, however, evidence of cellular dropout in the GCL regions of Mthfr+/− retinas (Fig. 7H). Morphometric analysis revealed significantly fewer cells in this layer in the 24-week Mthfr+/− retinas (10.33 ± 0.50) compared to Mthfr+/+ retinas (12.04 ± 0.45) per 100 μm retinal length. There were no significant differences in any other retinal measurements between groups. Thus, not only was NFL thickness decreased by 24 weeks (determined by OCT, Fig. 6) in the Mthfr+/− mice, so also was the number of cells in the GCL.
We also evaluated the microscopic appearance of nonretinal ocular tissues (cornea, lens, ciliary body, optic nerve) in the Mthfr+/− mice compared to wild type. There were no notable differences in appearances of lens, ciliary body, or optic nerve (data not shown). Interestingly, in two of five Mthfr+/− mice, corneal disruption was observed by 24 weeks. In one case, the basal epithelial cells were greatly enlarged (Supplementary Fig. S1B) and in another there was evidence of vascularization of the cornea (Supplementary Fig. S1C).
Assessment of Gliosis
The GFAP, an intermediate filament protein expressed in retinal astrocytes under normal conditions, is upregulated in Müller glial cells under stress conditions, such as retinal degeneration, retinal detachment, and ischemia.43 Retinal cryosections from Mthfr+/+ and Mthfr+/− mice were subjected to immunofluorescence analysis of anti-GFAP. In Mthfr+/+ retinas, GFAP staining was positive in astrocytes only (Fig. 8A, left). In contrast, in the Mthfr+/− retinas, there was marked increase in GFAP expression in the radial Müller cells (Fig. 8A, right). Fluorescence intensity increased significantly in the retinas of Mthfr+/− mice at 24 weeks (Fig. 8B) though not at earlier ages.
Figure 8.
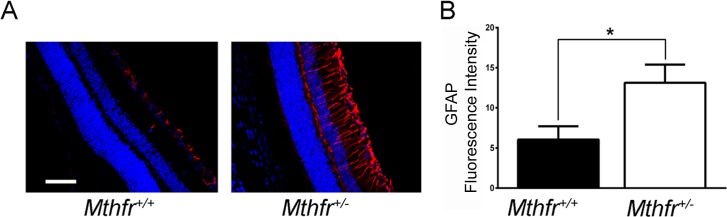
Immunofluorescent detection of GFAP. (A) Retinal cryosections from Mthfr+/+ and Mthfr+/− mice at 24 weeks were subjected to immunofluorescence analysis using anti-GFAP followed by incubation with Alexa Fluor 555 (red-labeled secondary antibody), showing marked increase in GFAP levels in astrocytes and Müller cells (radial labeling) in the Mthfr+/− retina compared to Mthfr+/+, in which GFAP expression was limited to astrocytes. (B) Quantification of GFAP color intensity data obtained from metamorphic analysis. *Significantly higher than Mthfr+/+, P < 0.05, n = 4.
Discussion
This study was done to evaluate the consequence of Hhcy on retinal function and structure using Mthfr+/− mice that manifest moderate elevation of endogenous Hcy.37 Before examining the Mthfr+/− retina we determined that Mthfr gene and protein are expressed in retina. Mthfr converts the N5, N10-methylenetetrahydrofolate to the circulatory form of folate (N5-methyltetrahydrofolate), which is essential for normal retinal health. Deleterious ocular effects of folate deficiency include optic neuropathy and nutritional amblyopia with central vision loss.44,45 It is noteworthy that Cbs is also expressed in several retinal layers.46 thereby demonstrating the underlying importance of folate–hcy–methionine metabolism in the retina.
The most significant findings that emerged from the present study were decreased positive scotopic threshold responses detected by ERG consistent with decreased ganglion cell function, decreased thickness of the NFL layer detected by SD-OCT in Mthfr+/− mice compared to Mthfr+/+, and approximately a 2-fold increase in hcy levels in Mthfr+/− compared to Mthfr+/+ mice. These findings were accompanied by a reduction in the number of ganglion cells detected in systematic morphometric analyses of fixed retinal tissue of Mthfr+/− compared to Mthfr+/+ mice. We also detected focal areas of vascular leakage by FA and increased GFAP levels in Müller glial cells in Mthfr+/− retinas. Our studies of the Mthfr mutant mouse did not detect decreased photoreceptor cell function (or fewer photoreceptor cells in the ONL) nor any significant alterations in the INL. Thus, it appears that the endogenous elevation of Hcy due to deficiency of Mthfr impacts the inner retina most significantly, particularly the ganglion cells. The loss of ganglion cells is not associated with an increase in IOP.
These data contribute to our understanding of the potential effects of Hhcy on retina. Many studies in humans have investigated whether various retinal diseases (diabetic retinopathy, macular degeneration, CRVO, BRVO, glaucoma) are linked to Hhcy.16–25 Despite the considerable evidence incriminating Hhcy in disease, controversy exists as to whether Hhcy is actually pathogenic or merely a biomarker of disease,47,48 including retinal disease.49
Our laboratory has been intrigued with the effects of Hhcy on retinal structure and function. In earlier studies we explored this using Cbs+/− mice, a mutation that renders the mice mildly Hhcy. Those studies, which examined morphology,31 vasculature,32,50 and retinal function by ERG33 suggested that mild Hhcy is associated with moderate reduction (approximately 20%) of ganglion cells, gliosis as evident from upregulation of GFAP, and vascular/functional alterations by 30 weeks of age. The functional abnormalities were observed in Cbs+/− mice at 15 weeks when reductions in amplitude were noted for the ERG a- and b-wave and the light peak component. The later onset functional defects in Cbs+/− mice are consistent with a slow ganglion cell loss. The data obtained from Cbs+/− mice could be interpreted as direct evidence that Hhcy is deleterious to retinal neurons and vasculature. However, such an interpretation ignores the fact that a key metabolic pathway is disrupted in that model system. The transsulfuration pathway mediated by Cbs (Fig. 1) is an endogenous pathway for coupling toxic hcy removal with protective H2S and GSH production. That is, the data from Cbs mice mutation studies may reflect a loss of critical downstream biochemical products rather than direct consequences of elevated hcy.
The current study recognized this possibility and takes advantage of a second model system, one in which levels of hcy are elevated, but the transsulfuration pathway is intact. Using mice deficient/lacking Mthfr, we were able to evaluate effects of Hhcy on retinal structure/function without the loss of potentially beneficial byproducts of the transsulfuration pathway. In Mthfr+/− mice, the transsulfuration pathway is not affected, whereas the remethylation pathway is interrupted. The conclusions of the present study, that ganglion cells are lost, but the remainder of the retina is relatively spared and that there is a mild vasculopathy associated with this model, are similar to our findings with the Cbs mutant mouse.31,32,50 The data provided strong evidence that Hhcy has deleterious effects on ganglion cells and retinal vasculature. It is noteworthy that our immunohistochemical data suggested elevations of hcy throughout the retina, not solely in ganglion cells; Hcy is known to be excitotoxic (much like glutamate51 and so ganglion cells may be particularly vulnerable to its effects when elevated.
The present work sets the stage to investigate comprehensively the mechanism(s) by which Hhcy affects retinal neurovascular integrity. Future studies will determine which retinal cell(s) are directly affected by Hhcy, including ganglion cells, and whether there is protein homocysteinylation in these cells that might compromise survival. In addition, retinal Müller glial cells may be a target for the deleterious consequences of Hhcy, since they provide important trophic support for ganglion cells. Future studies should compare the levels of GSH, H2S, and other antioxidant/protective factors in retinas of Mthfr+/− and Cbs+/− mice. Using models of HHcy also will allow therapeutic intervention strategies to be tested, including dietary augmentation using folate/B6/B12 supplementation and pharmacologic intervention.
Studies of excess hcy on retinal function continue to be reported in the clinical literature. A recent report of 20 subjects with Eales' disease, an idiopathic retinal disorder characterized by inflammation and neovascularization, revealed significant elevation of plasma hcy and hcy-thiolactone compared to control subjects. In the affected patients there was an increase in protein homocysteinylation and a decrease in GSH.52 The steady increase in the number of visual diseases in which Hcy is implicated provides strong impetus to understand mechanisms of hcy-mediated retinal dysfunction and to develop appropriate intervention strategies.
Supplementary Material
Acknowledgments
We thank Srinivas Sonne, PhD, for his assistance with FISH analysis and Ming Zhang, PhD, for the use of the Phoenix Micron III imaging system.
Supported by a Grant R01 EY 012830 from the National Institutes of Health (Betheda, MD, USA).
Disclosure: S. Markand, None; A. Saul, None; P. Roon, None; P. Prasad, None; P. Martin, None; R. Rozen, None; V. Ganapathy, None; S.B. Smith, None
References
- 1.Mudd SH,, Skovby F,, Levy HL,, et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet. 1985; 37: 1–31. [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Z,, Karaplis AC,, Ackerman SL, et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet. 2001; 10: 433–443. [DOI] [PubMed] [Google Scholar]
- 3.Mudd SH,, Levy HL,, Kraus JP.“Disorders of Transsulfuration.” The Metabolic and Molecular Basis of Inherited Disease, 8th ed. New York, NY: McGraw-Hill; 2001: 2016–2040.
- 4.Wierzbicki AS.Homocysteine and cardiovascular disease: a review of the evidence. Diab Vasc Dis Res. 2007; 4: 143–150. [DOI] [PubMed] [Google Scholar]
- 5.Austin RC,, Lentz SR,, Werstuck GH.Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004; 11: 56–64. [DOI] [PubMed] [Google Scholar]
- 6.Vollset SE,, Refsum H,, Irgens LM, et al. Plasma total homocysteine, pregnancy complications, and adverse pregnancy outcomes: the Hordaland Homocysteine study. Am J Clin Nutr. 2000; 71: 962–968. [DOI] [PubMed] [Google Scholar]
- 7.Mills JL,, Lee YJ,, Conley MR,, et al. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet. 1995; 345: 149–151. [DOI] [PubMed] [Google Scholar]
- 8.Houston PE,, Rana S,, Sekhsaria S,, Perlin E,, Kim KS,, Castro OL.Homocysteine in sickle cell disease: relationship to stroke. Am J Med. 1997; 103: 192–196. [DOI] [PubMed] [Google Scholar]
- 9.Van Der Dijs FP,, Schnog JJB,, Brouwer DA.Elevated homocysteine levels indicate suboptimal folate status in pediatric sickle cell patients. Am J Hem. 1998; 59: 192–198. [DOI] [PubMed] [Google Scholar]
- 10.Van Meurs JB,, Dhonukshe-Rutten RA,, Pluijm SM, et al. Homocysteine levels and the risk of osteoporotic fracture. N Engl J Med. 2004; 350: 2033–2041. [DOI] [PubMed] [Google Scholar]
- 11.Ndrepepa G,, Kastrati A,, Braun S,, et al. Circulating homocysteine levels in patients with type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis. 2008; 18: 66–73. [DOI] [PubMed] [Google Scholar]
- 12.Soedamah-Muthu SS,, Chaturvedi N,, Teerlink T,, Idzior-Walus B,, Fuller JH,, Stehouwer CD.Plasma homocysteine and microvascular and macrovascular complications in type 1 diabetes: a cross-sectional nested case-control study. J Intern Med. 2005; 258: 450–459. [DOI] [PubMed] [Google Scholar]
- 13.Agulló-Ortuño MT,, Albaladejo MD,, Parra S, et al. Plasmatic homocysteine concentration and its relationship with complications associated to diabetes mellitus. Clin Chim Acta. 2002; 326: 105–112. [DOI] [PubMed] [Google Scholar]
- 14.Geller V,, Friger M,, Sela BA,, Levine J.Elevated homocysteine level in siblings of patients with schizophrenia. Psych Res. 2013; 210: 769–772. [DOI] [PubMed] [Google Scholar]
- 15.Selhub J,, Bagley LC,, Miller J,, Rosenberg IH.B vitamins, homocysteine, and neurocognitive function in the elderly. Am J Clin Nutr. 2000; 71: 614–620. [DOI] [PubMed] [Google Scholar]
- 16.Mattson MP,, Shea TB.Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003; 26: 137–146. [DOI] [PubMed] [Google Scholar]
- 17.Aydemir O,, Türkçüoğlu P,, Güler M, et al. Plasma and vitreous homocysteine concentrations in patients with proliferative diabetic retinopathy. Retina. 2008; 28: 741–743. [DOI] [PubMed] [Google Scholar]
- 18.Yang G,, Lu J,, Pan C.The impact of plasma homocysteine level on development of retinopathy in type 2 diabetes mellitus. Zhonghua Nei Ke Za Zhi. 2002; 41: 34–38. [PubMed] [Google Scholar]
- 19.Axer-Siegel R,, Bourla D,, Ehrlich R, et al. Association of neovascular age-related macular degeneration and hyperhomocysteinemia. Am J Ophthalmol. 2004; 137: 84–89. [DOI] [PubMed] [Google Scholar]
- 20.Seddon JM,, Gensler G,, Klein ML,, Milton RC.Evaluation of plasma homocysteine and risk of age-related macular degeneration. Am J Ophthalmol. 2006; 141: 201–203. [DOI] [PubMed] [Google Scholar]
- 21.Bleich S,, Jünemann A,, Von Ahsen N, et al. Homocysteine and risk of open-angle glaucoma. J Neural Transm. 2002; 109: 1499–1504. [DOI] [PubMed] [Google Scholar]
- 22.Vessani RM,, Ritch R,, Liebmann JM,, Jofe M.Plasma homocysteine is elevated in patients with exfoliation syndrome. Am J Ophthalmol. 2003; 136: 41–46. [DOI] [PubMed] [Google Scholar]
- 23.Jaksic V,, Markovic V,, Milenkovic S,, Stefanovic I,, Jakovic N,, Knezevic M.Mthfr. 677C>T homozygous mutation in a patient with pigmentary glaucoma and central retinal vein occlusion. Ophthalmic Res. 2010; 43: 193–196. [DOI] [PubMed] [Google Scholar]
- 24.Tsina EK,, Marsden DL,, Hansen RM,, Fulton AB.Maculopathy and retinal degeneration in cobalamin C methylmalonic aciduria and homocystinuria. Arch Ophthalmol. 2005; 123: 1143–1146. [DOI] [PubMed] [Google Scholar]
- 25.Semba RD.Retinal Vascular Disease. Nutrition and Health Handbook of Nutrition and Ophthalmology. Totowa NY: Humana Press;2007: 257–280. [Google Scholar]
- 26.Lahey JM,, Kearney JJ,, Tunc M.Hypercoagulable states and central retinal vein occlusion. Curr Opin Pulm Med. 2003; 9: 385–392. [DOI] [PubMed] [Google Scholar]
- 27.Ganapathy PS,, Dun Y,, Ha Y, et al. Sensitivity of staurosporine-induced differentiated RGC-5 cells to homocysteine. Curr Eye Res. 2010; 35: 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore P,, El-Sherbeny A,, Roon P,, Schoenlein PV,, Ganapathy V,, Smith SB.Apoptotic cell death in the mouse retinal ganglion cell layer is induced in vivo by the excitatory amino acid homocysteine. Exp Eye Res. 2001; 73: 45–57. [DOI] [PubMed] [Google Scholar]
- 29.Chang HH,, Lin DP,, Chen YS, et al. Intravitreal homocysteine-thiolactone injection leads to the degeneration of multiple retinal cells, including photoreceptors. Mol Vis. 2011; 17: 1946–1956. [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe M,, Osada J.ArataniY, Kluckman K, Reddick R, Malinow MR. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocysteinemia. Proc Natl Acad Sci U S A. 1995; 92: 1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganapathy PS,, Moister B,, Roon P.Endogenous elevation of homocysteine induces retinal neuron death in the cystathionine-beta-synthase mutant mouse. Invest Ophthalmol Vis Sci. 2009; 50: 4460–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tawfik A,, Al-Shabrawey M,, Roon P, et al. Alterations of retinal vasculature in cystathionine-beta-synthase mutant mice, a model of hyperhomocysteinemia. Invest Ophthalmol Vis Sci. 2013; 54: 939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu M,, Sturgill-Short G,, Ganapathy P,, Tawfik A,, Peachey NS,, Smith SB.Age-related changes in visual function in cystathionine-beta-synthase mutant mice a model of hyperhomocysteinemia. Exp Eye Res. 2012; 96: 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leclerc D,, Sibani S,, Rozen, R.Molecular Biology of Methylenetetrahydrofolate Reductase (Mthfr) and Overview of Mutations/Polymorphisms. Mthfr Polymorphisms and Disease. Georgetown, TX: Landes Bioscience/Eurekah; Com;2005; 1–20. [Google Scholar]
- 35.Gilbody S,, Lewis S,, Lightfoot T.Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007; 165: 1–13. [DOI] [PubMed] [Google Scholar]
- 36.Franco RF,, Araujo AG,, Guerreiro JF,, Elion J,, Zago MA.Analysis of the 677 C--> T mutation of the methylenetetrahydrofolate reductase gene in different ethnic groups. Thromb Haemost. 1998; 79: 119–121. [PubMed] [Google Scholar]
- 37.Lawrance AK,, Racine J,, Deng L,, Wang X,, Lachapelle P,, Rozen R.Complete deficiency of methylenetetrahydrofolate reductase in mice is associated with impaired retinal function and variable mortality, hematological profiles, and reproductive outcomes. J Inherit Metab Dis. 2011; 34: 147–157. [DOI] [PubMed] [Google Scholar]
- 38.Martin PM,, Ananth S,, Cresci G,, Roon P,, Smith S,, Ganapathy V.Expression and localization of GPR109A (PUMA-G/HM74A) mRNA and protein in mammalian retinal pigment epithelium. Mol Vis. 2009; 15; 362. [PMC free article] [PubMed]
- 39.Saszik SM,, Robson JG,, Frishman LJ.The scotopic threshold response of the dark-adapted electroretinogram of the mouse. J Physiol. 2002; 543: 899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aziz MK,, Aiguo N,, Denise AE,, Sai HC.Evidence of early ultrastructural photoreceptor abnormalities in light-induced retinal degeneration using spectral domain optical coherence tomography. Br J Ophthalmol. 2014; 98: 984–989. [DOI] [PubMed] [Google Scholar]
- 41.Ferguson LR,Dominguez Ii JM, Balaiya S, Grover S, Chalam KV. Retinal thickness normative data in wild-type mice using customized miniature SD-OCT. PLoS One. 2013; 8: 67265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawuła W,, Banecka-Majkutewicz Z,, Kadziński L, et al. Improved HPLC method for total plasma homocysteine detection and quantification. Acta Biochim Pol. 2008; 55: 119–125. [PubMed] [Google Scholar]
- 43.Sarthy V,, Ripps H.The Retinal Müller Cell: Structure and Function. New York NY: Kluwer Academic/Plenum Press; 2001. [Google Scholar]
- 44.Golnik KC,, Schaible ER.Folate-responsive optic neuropathy. J Neurophthalmol. 1994; 14: 163–169. [PubMed] [Google Scholar]
- 45.Sadun A,, Rubin R.Residual psychophysical deficits following recovery from the Cuban epidemic of optic neuropathy. : Lakshminarayan V. Basic and Clinical Applications of Vision Science. Dordrecht: Kluwer;1997: 231–234. [Google Scholar]
- 46.Markand S,, Tawfik A,, Ha Y,, et al. Cystathionine beta synthase expression in mouse retina. Curr Eye Res. 2013; 38: 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smulders YM,, Blom HJ.The homocysteine controversy. J Inherit Metab Dis. 2011; 34: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cacciapuoti F.Hyper-homocysteinemia: a novel risk factor or a powerful marker for cardiovascular diseases? Pathogenetic and therapeutical uncertainties. J Thromb Thrombolysis. 2011; 32: 82–88. [DOI] [PubMed] [Google Scholar]
- 49.Wright AD,, Martin N,, Dodson PM.Homocysteine folates, and the eye. Eye. 2008; 22: 989–993. [DOI] [PubMed] [Google Scholar]
- 50.Tawfik A,, Markand S,, Al-Shabrawey M,, et al. Alterations of retinal vasculature in cystathionine-β-synthase heterozygous mice: a model of mild to moderate hyperhomocysteinemia. Am J Pathol. 2014; 184: 2573–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Do KQ,, Herrling PL,, Streit P,, Cuénod M.Release of neuroactive substances: homocysteic acid as an endogenous agonist of the NMDA receptor. J Neural Transm. 1988; 72: 185–190. [DOI] [PubMed] [Google Scholar]
- 52.Bharathselvi M,, Biswas J,, Selvi R,, et al. Increased homocysteine, homocysteine-thiolactone, protein homocysteinylation and oxidative stress in the circulation of patients with Eales' disease. Ann Clin Biochem. 2013; 50: 330–338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.