

Abstract
Molecular genetic analyses in Schizosaccharomyces pombe rely on selectable markers that are used in cloning vectors or to mark targeted gene deletions and other integrated constructs. In this study, we used genetic mapping data and genomic sequence information to predict the identity of the S. pombe lys2+ gene, which is homologous to Saccharomyces cerevisiae LYS4+. We confirmed this prediction, showing that the cloned SPAC343.16 gene can complement a lys2-97 mutant allele, and constructed the lys2+-based cloning vector pRH3. In addition, we deleted the S. pombe his7+ gene with a lys2+-marked polymerase chain reaction (PCR) product and the S. pombe lys2+ gene with a his7+-marked PCR product. Strains carrying these deletions of lys2+ or his7+ serve as relatively efficient hosts for the deletion of the ade6+ gene by lys2+- or his7+-marked PCR products when compared with hosts carrying lys2 or his7 point mutations. Therefore, these studies provide plasmids and strains allowing the use of lys2+ as a selectable marker, along with improved strains for the use of his7+ to mark gene deletions.
Keywords: Schizosaccharomyces pombe, Fission yeast, his7+, lys2+, Selectable marker, Deletion
Introduction
Selectable markers are essential to molecular genetic studies in the fission yeast Schizosaccharomyces pombe. They are incorporated into autonomously replicating plasmids and used to mark DNA fragments inserted into chromosomes by homologous recombination in order to delete or alter the sequence of a gene at its endogenous locus. As more molecular genetic tools are developed and strains carrying gene deletions or epitope tags at multiple loci are constructed, the number of usable selectable markers can become limiting.
The first plasmid markers used in S. pombe were Saccharomyces cerevisiae URA3+, which complements an S. pombe ura4- mutation, and Saccharomyces cerevisiae LEU2+, which complements an S. pombe leu1- mutation (Beach and Nurse 1981; Losson and Lacroute 1983). These markers function well on multicopy plasmids; however, single copy URA3+ fails to complement a ura4- mutant allele, while single copy LEU2+ (expressed from its own promoter) only weakly complements a leu1- mutant allele. Therefore, these markers are generally insufficient for marking a gene deletion. Subsequently, the S. pombe ura4+ (Bach 1987; Grimm et al. 1988) and leu1+ (Kikuchi et al. 1988) genes were cloned, although while ura4+ is commonly used, the Saccharomyces cerevisiae LEU2+ gene remains the preferred marker to S. pombe leu1+. In addition, the S. pombe his7+ (Apolinario et al. 1993), his3+ (Burke and Gould 1994) and arg3+ (Waddell and Jenkins 1995) genes have been cloned and shown to fully restore His+ and Arg+ growth in single copy. Very recently, a series of cloning vectors has been constructed carrying the arg12+, tyr1+, ade7+, and his2+ genes, and a vps34+ gene disruption was constructed using each marker (Fujita et al. 2005).
In addition to biosynthetic pathway genes, drug-resistance markers are used in S. pombe. The kanMX6 marker, which confers G418R growth, is extensively used in gene deletions and epitope-tagging of chromosomal genes (Bähler et al. 1998), while the bacterial hph and nat genes confer resistance to hygromycin B and nourseothricin, respectively (Burland et al. 1991; Sato et al. 2005). The advantage of drug resistance markers is that, except for strains already carrying these markers, all S. pombe strains are suitable hosts for transformation with these markers due to their natural sensitivity to the drugs to which these markers confer resistance. In contrast, biosynthetic pathway genes can only be introduced into a host strain that is defective in that step of the pathway.
To facilitate their use in targeted gene deletions, unmarked chromosomal deletions of ura4+ (Grimm et al. 1988), his3+ (Burke and Gould 1994) and arg3+ (Waddell and Jenkins 1995) were constructed. In addition, using a Cre-loxP system, arg12∷loxP, tyr1∷loxP, ade7∷loxP and his2∷loxP deletion alleles have also been constructed (Fujita et al. 2005). These deletion alleles do not possess any sequences homologous to the selectable marker, thus increasing the frequency with which transformants that regain the relevant prototrophy carry the gene deletion. We have also used the SV40 promoter-driven LEU2+ selectable marker present in the pNMT-TOPO vectors (Invitrogen) for gene deletions, as there is insufficient homology between S. pombe leu1+ and Saccharomyces cerevisiae LEU2+ to promote targeting of LEU2+ to the leu1+ locus, while the SV40 promoter allows sufficient expression of Leu2 to confer Leu+ growth in single copy (Wang et al. 2004).
In this study, we cloned the S. pombe lys2+ gene and incorporated it into a versatile cloning vector to create plasmid pRH3. In addition, we carried out polymerase chain reaction (PCR) based gene deletions of lys2+ and his7+, marking each deletion with the other selectable marker. This work allowed us to identify PCR primers capable of amplifying each marker to produce a product that functions well in single copy. In addition, the resulting lys2Δ∷his7+ deletion allele lacks the entire lys2+ sequence that is amplified to mark the his7Δ∷lys2+ deletion, while the his7Δ∷lys2+ deletion allele lacks the entire his7+ sequence that is amplified to mark the lys2Δ∷his7+ deletion. As expected, these strains are superior to lys2-97 and his7-366 strains for the construction of lys2+- or his7+-marked gene deletions when using PCR products, as demonstrated by comparing the efficiency of deleting the ade6+ gene. Thus, these studies make the his7+ marker easier to use in future gene disruptions, and provide new reagents for the use of lys2+ as a selectable marker in S. pombe.
Materials and methods
Yeast strains and growth media
Yeast strains used in this study are listed in Table 1. Only relevant genotypes are given in the text and figures. Standard S. pombe media YEA, PM and EMM (Watanabe et al. 1988; Alfa et al. 1993) were used for culturing cells. Crosses were carried out on MEA medium (Gutz et al. 1974) and tetrads were dissected on YEA medium.
Table 1. Strain list.
| Strain | Genotype |
|---|---|
| 972 | h− |
| FWP42 | h− leu1-32 lys1-131 |
| FWP45 | h+ leu1-32 |
| FWP58 | h+ his7-366 |
| CHP871 | h− his6-365 lys2-97 |
| CHP880 | h− ura4∷fbp1-lacZ leu1-32 his6-365 lys2-97 |
| CHP1028 | h+ lys2-97 |
| CHP1029 | h+ his7-366 lys2Δ∷his7+ |
| CHP1030 | h+ lys2-97 his7Δ∷lys2+ |
| CHP1031 | h− ura4-D18 leu1-32 his7-366 lys2-97 |
| CHP1050 | h+ ura4-D18 leu1-32 ade6-M210 his7Δ∷lys2+ |
| CHP1051 | h−ura4-D18 leu1-32 ade6-M210 his7Δ∷lys2 + |
| CHP1052 | h+ ura4-D18 leu1-32 ade6-M216 his7Δ∷lys2+ |
| CHP1053 | h−ura4-D18 leu1-32 ade6-M216 his7Δ∷lys2 + |
| CHP1054 | h+ ura4-D18 leu1-32 ade6-M210 lys2Δ∷his7+ |
| CHP1055 | h− ura4-D18 leu1-32 ade6-M210 lys2Δ∷his7+ |
| CHP1056 | h+ ura4-D18 leu1-32 ade6-M216 lys2Δ∷his7+ |
| CHP1057 | h− ura4-D18 leu1-32 ade6-M216 lys2Δ∷his7+ |
Cloning of lys2+ gene
SPAC343.16 (lys2+) was PCR-amplified from strain 972 genomic DNA using oligonucleotides lys2-for (5′-CGTAAAAAAGAAAGAGCTGAGCG-3′) and lys2-rev (5′-GCAGTACGCAAAGATCCTAAGCT-3′). The PCR product was cloned into vector pNMT41 (Invitrogen) and used to transform Escherichia coli according to the manufacturer's instructions. Plasmids from E. coli transformants were purified and screened by a HindIII digest to identify pRH1 and pRH2, which carry the insert PCR product in the two orientations relative to the nmt41 promoter.
Construction of the pRH3 lys2+-marked cloning vector
The lys2+ gene was PCR-amplified using oligonucleotides URA3lys2-for (5′-GATTCGGTAATCTCCGAACAGAAGGAAGAACGAAGGAAGGAGCACAGACTTAGTCTGAATAATCATGCTTGAAACC-3′) and URA3lys2-rev (5′-CCTGATGCGGTATTTTCTCCT TACGCATCTGTGCGGTATTTCACACCGCATATTTAGCGATGTTATTCAGCTTTAC-3′) with plasmid pRH1 as template. The 3′ ends of the oligonucleotides (underlined) are identical to sequences flanking lys2+, while the 5′ ends are identical to sequences flanking the URA3+ selectable marker in plasmid pSP2 (Cottarel et al. 1993). This PCR product was used to replace the URA3+ marker in plasmid pSP2 by co-transformation of strain CHP871 (lys2-97) with plasmid pSP2 (digested with NsiI and StuI) and the PCR product, following the marker-swap protocol of Kelly and Hoffman (Kelly and Hoffman 2002), to create plasmid pRH3.
Construction of lys2Δ∷his7+ and his7Δ∷lys2+ deletion alleles
The lys2Δ∷his7+ and his7Δ∷lys2+ deletion alleles were constructed by a PCR-based strategy. A lys2Δ∷his7+ deletion construct was created by PCR amplification of his7+ from plasmid pMN1 (Apolinario et al. 1993), using oligonucleotides lys2∷his7for (5′-ATTTATCCAGATTTAGAGCGTTAACTTGTAAGCTAATAGTGAAATTTATTATTATATCCAAGCAAGGATTTCGCTATGCC-3′) and lys2∷his7rev (5′-AAAACTAAAGTTGAAAAAAATGAAAATCAACGTTCGAAAAGAATGTCAACATATTTAATGACCGACTGCTATCATCTGCG-3′). Sequences complementary to his7+ are underlined, while the oligonucleotide 5′ ends contain sequences flanking lys2+. A his7D∷lys2+ deletion construct was created by PCR amplification of lys2 + from plasmid pRH1, using oligonucleotides his7∷lys2for (5′-TGATTTTATTTCAACTAAATTAAGAGTATTATAAATGCCACCAAAAGACTGTTTCCTAACGCATGCTACGAACTAGTCTG-3′) and his7∷lys2rev (5′-AATAATTACACAAAGTAAAAATTTCCGCATTAGCAAGGTTTTAGTAGACTACACCTTAGTGGGAAGATAGATTGATGGAC-3′). Sequences complementary to lys2+ are underlined, while the oligonucleotide 5′ ends contain sequences flanking his7+. Cells were transformed to His+ or Lys+ (Bähler et al. 1998) and homologous recombination events were confirmed by PCR analysis using his7testfor (5′-GACTCGCATGATTCCATCCATC-3′) and his7testrev (5′-CAGAAGGTATACACAGCGACA-3′) or lys2testfor (5′-CGTAAAAAAGAAAGAGCTGAGCG-3′) and lys2testrev (5′-GCAGTACGCAAAGATCCTAAGCT-3′).
Construction of ade6Δ∷his7+ and ade6Δ∷lys2+ deletion alleles
The ade6+ gene was deleted by a one-step PCR-based method. Oligonucleotides ade6-his7for (5′-CTATGCAGAATAATTTTTCCAACCAACTTTTTCTAATTTATCAGCCTTTTCTAAAACCAAGCAAGGATTTCGCTATGCC-3′) and ade6-his7rev (5′-ATGAGCGAAAAACAGGTTGTAGGGATCCTTGGAGGTGGTCAATTGGGCCGAATGATGGACCGACTGCTATCATCTGCG-3′) were used to PCR-amplify his7+ from plasmid pMN1, while oligonucleotides ade6-lys2for (5′-CTATGCAGAATAATTTTTCCAACCAACTTTTTCTAATTTATCAGCCTTTTCTAAAAACGCATGCTACGAACTAGTCTG-3′) and ade6-lys2rev (5′-ATGAGCGAAAAACAGGTTGTAGGGATCCTTGGAGGTGGTCAATTGGGCCGAATGATGGTGGGAAGATAGATTGATGGAC-3′) were used to PCR-amplify lys2 + from plasmid pRH1. Strains CHP1029 (lys2Δ∷his7+), CHP1030 (his7Δ∷lys2 +), and CHP1031 (lys2-97 his7-366) were transformed to Lys+ and His+, as appropriate (Bähler et al. 1998). The percent of Ade− transformants was scored by replica plating to EMM-ade and to YEA-ade.
Results and discussion
lys2+ Gene prediction and cloning
The initial goal of this study was to clone and develop a new selectable marker for S. pombe, preferably in a biosynthetic pathway for which other markers are not available. While the S. pombe lys1+ gene had been cloned (Beach et al. 1982; Ford et al. 1993), it is too large to be used as a selectable marker. We chose to pursue lys2+, due to mapping data on the website http://www.izb.unibe.ch/res/kohli/Chromo1.html placing it 4.3 cM from rad9+ (164 PD:1 NPD:9 TT). As rad9+ has been cloned (Murray et al. 1991), its physical location on the chromosome is known. Other chromosome 1 cross data (Table 2) indicate that 1 cM is equivalent to approximately 7–12 kb in physical map distance. Examination of the S. pombe GeneDB (http://www.genedb.org/genedb/pombe/index.jsp) for a Saccharomyces cerevisiae LYS gene homolog within 30.5–52.5 kb of rad9+ reveals that SPAC343.16 is approximately 38 kb from rad9+, and is predicted to encode a homoaconitate hydratase that is 62% identical to Saccharomyces cerevisiae Lys4. Based on these data, we set out to test whether SPAC343.16 is lys2+.
Table 2. Genetic and physical map distances for Chromosome 1 gene pairs.
| Gene pair | Map distance (cM) | Physical distance | kb/cM |
|---|---|---|---|
| cdc3-cdc16 | 23.7 | 168 kb | 7.1 |
| cdc12-cdc25 | 26.7 | 195 kb | 7.3 |
| arg3-spo3 | 18.0 | 220 kb | 12.2 |
| rad9-lys2 | 4.3 | 30.5–52.5a |
Values obtained from http://www.izb.unibe.ch/res/kohli/Chromo1.html
Distance range is an estimate based on the ratio of physical to genetic map distance from the other three gene pairs
To determine if SPAC343.16 is lys2+, we PCR-amplified this gene from strain 972 (lys2+) and cloned it into a vector that can transform S. pombe leu1-32 strains to Leu+. The PCR product includes the SPAC343.16 promoter, so that the orientation of insertion into this nmt-driven expression vector will not affect function. Plasmids pRH1 and pRH2, carrying the insert in either orientation, complement lys2-97 in strain CHP880, while failing to complement lys1-131 in strain FWP42 (Fig. 1). Conversely, a clone of the lys1+ gene, plys1+, which we previously obtained from a pWH5-based genomic library (Feilotter et al. 1991), transforms FWP42, but not CHP880, to Lys+. Thus, SPAC343.16 is lys2+.
Fig. 1.
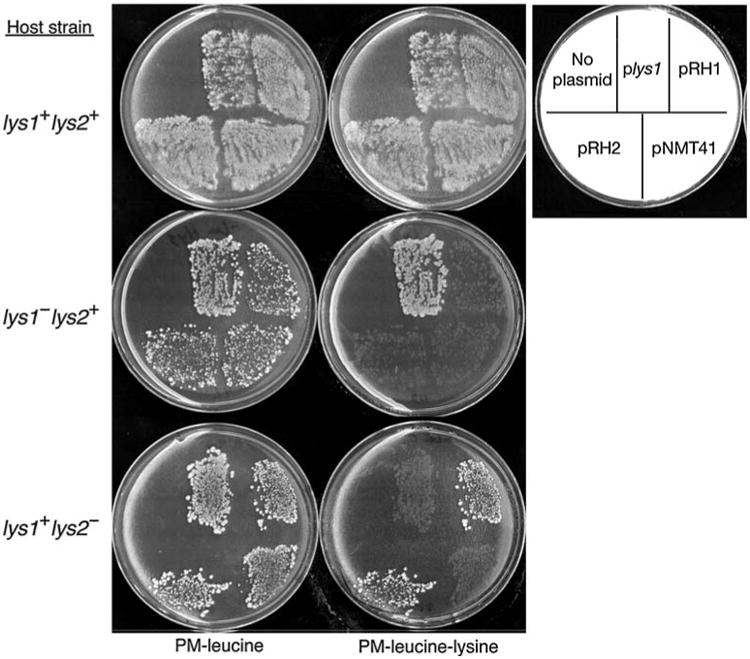
Plasmid complementation of lys1-131 and lys2-97 mutant alleles. Strains FWP45 (lys1+ lys2+), FWP42 (lys1-131), and CHP880 (lys2-97) were transformed to Leu+ with plasmids plys1+ (pWH5 vector carrying lys1+ genomic DNA), pRH1 (pNMT41 carrying lys2+ PCR product), pRH2 (pNMT41 carrying lys2+ PCR product inserted in the opposite orientation from that of pRH1), or pNMT41 (empty vector). A mock transformation (no plasmid) was carried out in the absence of plasmid DNA. Leu+ transformants were replica plated to PM medium lacking leucine and lysine. Plasmid plys1+ is shown to complement the lys1-131 mutant allele, while plasmids pRH1 and pRH2 complement the lys2-97 allele
Construction of a lys2+-based cloning vector
Using a marker-swap protocol (Kelly and Hoffman 2002), we replaced Saccharomyces cerevisiae URA3+ in plasmid pSP2 (Cottarel et al. 1993) with lys2+ to create plasmid pRH3 (Fig. 2). This plasmid possesses the S. pombe ars1 to facilitate high-efficiency transformation, along with a polylinker with unique restriction sites for XhoI, EcoRI, SmaI, XmaI, BamHI, SpeI, NotI, EagI, SacII, and BxtXI. In addition, pRH3 can be used in a blue/white screen in E. coli to identify plasmids containing insert DNA. Plasmid pRH3 can transform CHP871 (lys2-97) cells to Lys+ and produces blue colonies when introduced into E. coli strain TOP10 (data not shown). Thus, pRH3 should be a useful cloning vector for transforming S. pombe strains that carry markers in the more commonly used leucine, uracil and histidine biosynthetic pathways due to the presence of other plasmids or marked chromosomal insertions. Using the lys2+-specific sequences employed in this construction (see Materials and methods), it would be a simple matter to replace the selectable marker in other S. pombe vectors with lys2+.
Fig. 2.
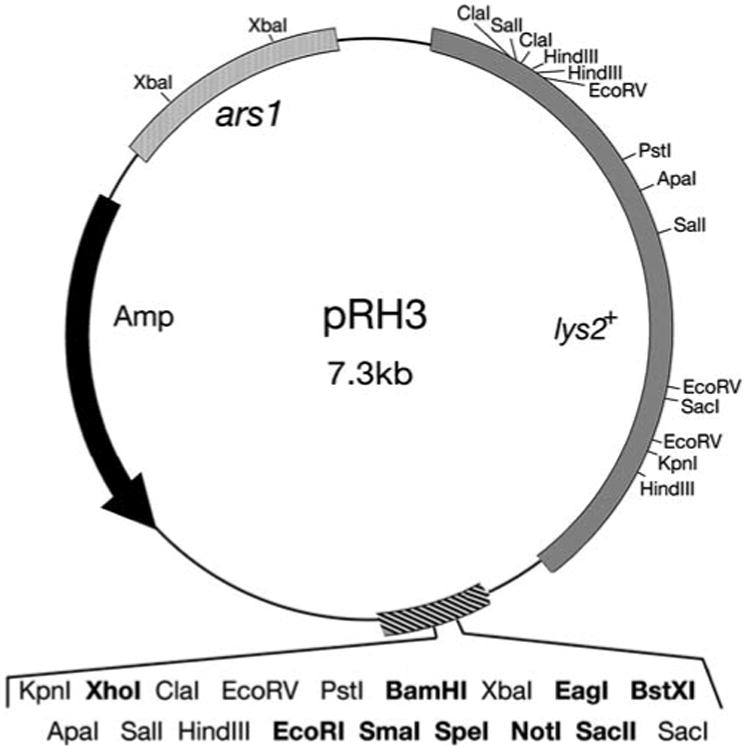
Schematic of the pRH3 cloning vector. Plasmid pRH3 was constructed by marker swap transformation, replacing the URA3+ marker of plasmid pSP2 (Cottarel et al. 1993) with the lys2+ gene. Unique restriction sites in the polylinker are shown in bold. The relative locations of polylinker sites that are present elsewhere is the plasmid are also shown
Construction of lys2Δ∷his7+ and his7Δ∷lys2+ deletion alleles using PCR products
Polymerase chain reaction-based gene deletions generally utilize long oligonucleotides whose 3′ ends prime amplification of a selectable marker and whose 5′ ends provide targeting sequences to target insertion into the genome by homologous recombination. We designed two sets of oligonucleotides; one to target his7+ to the lys2+ locus (lys2Δ∷his7+) and a second to target lys2+ to the his7+ locus (his7Δ∷lys2+). PCR amplification from plasmids pMN1 (his7+, Apolinario et al. 1993) and pRH1 (lys2+) resulted in the expected 1.7 and 2.8 kb products (data not shown), which were used to transform strain FWP58 (his7-366) to His+ and strain CHP871 (lys2-97) to Lys+. Less than one percent of transformants in each transformation acquired the corresponding auxotrophy due to homologous recombination. This low frequency is presumably due to the ability of the transforming DNA to repair his7-366 and lys2-97 at their respective loci. PCR analysis confirmed the homologous recombination events to create the lys2Δ∷his7+ and his7Δ∷lys2+ alleles (Fig. 3a) in strains CHP1029 and CHP1030, respectively. As expected, the 3.2 kb product for the lys2+ locus is reduced to 2.1 kb due to its replacement with the smaller his7+ gene to create the lys2Δ∷his7+ allele. Conversely, the 2.2 kb product for the his7+ locus is increased to 3.4 kb due to its replacement with the larger lys2+ to create the his7Δ∷lys2+ allele (an additional 1.9 kb nonspecific band is also visible in the his7 diagnostic lanes).
Fig. 3.
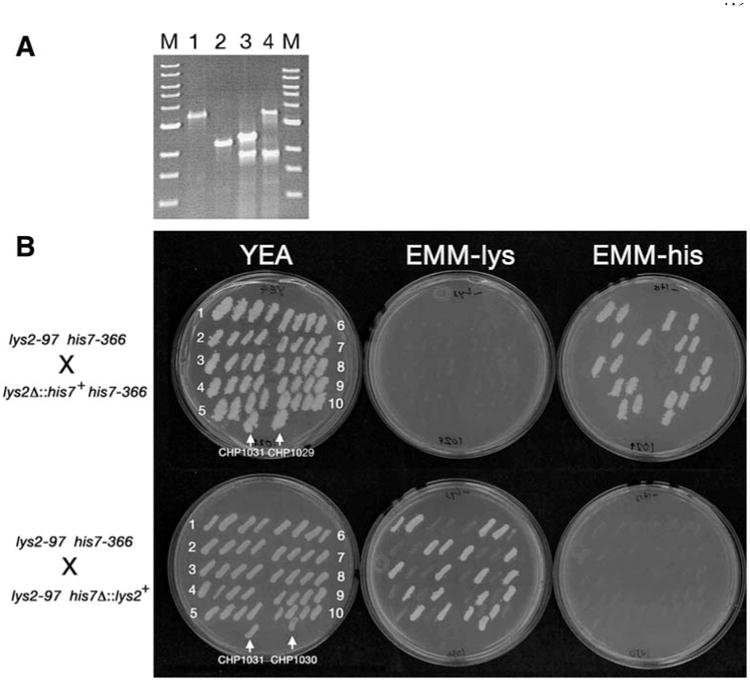
PCR-based gene disruptions using lys2+ and his7+ markers. a PCR analysis of the lys2+ (Lanes 1 and 2) and his7+ (Lanes 3 and 4) loci from strains FWP58 (lys2+; Lane 1), CHP1029 (lys2Δ∷his7+; Lane 2), CHP1028 (his7+; Lane 3), and CHP1030 (his7Δ∷lys2+; Lane 4). Lanes marked M contain a 1 kb ladder size marker. b Tetrad analysis of strains CHP1029 (lys2Δ∷his7+) and CHP1030 (his7Δ∷lys2+) crossed with CHP1031. Ten tetrads from each cross were patched onto a YEA plate and then replica plated to EMM-lys, EMM-his, and YEA. The 2:2 segregation patterns for the growth phenotype conferred by the respective selectable markers demonstrates that CHP1029 and CHP1030 each carry integration at a single genetic locus. The PCR product size in a demonstrates that a single copy of the integration cassette is present at each locus
This was our first attempt to use either lys2+ or his7+ in a PCR-based deletion protocol. We therefore needed to verify that these amplified products confer histidine and lysine prototrophy in single copy, so that future deletion protocols can use the same priming sequences. The PCR analysis in Fig. 3a uses oligonucleotides that flank the targeting sites for each deletion, thus the size of the PCR product demonstrates that each deletion contains a single insertion of the selectable marker, as opposed to a tandem insertion. To determine whether the deletion strains CHP1029 (his7-366 lys2Δ∷his7+) and CHP1030 (his7Δ∷lys2+ lys2-97) carry insertions of the PCR products at additional genetic loci, we crossed each strain with CHP1031 (his7-366 lys2-97) and examined the segregation pattern for the His+ and Lys+ phenotypes in the progeny. A single integration should produce a 2:2 pattern, while integrations at multiple loci would result in other patterns depending on whether a single copy of the marker in question is sufficient to confer a His+ or Lys+ phenotype. As shown in Fig. 3b, all ten tetrads from each cross display a 2:2 pattern indicating the presence of a single copy of the respective markers. More than 30 additional tetrads from each cross also showed a 2:2 segregation pattern (data not shown). Therefore, single copy integrations of these his7+- and lys2+-marked PCR products confer His+ and Lys+ prototrophy, respectively.
Increased efficiency of homologous gene deletion in lys2Δ∷his7+ and his7Δ∷lys2+ strains
As mentioned above, we assumed that the low homologous recombination frequency observed while constructing the lys2Δ∷his7+ and his7Δ∷lys2+ strains was due to relatively high frequency repair of the his7-366 and lys2-97 alleles. To determine whether strains bearing the lys2Δ∷his7+ and his7Δ∷lys2+ deletions could serve as improved hosts for PCR-based gene deletions, we compared the efficiency with which we could delete ade6+ from CHP1029 (his7-366 lys2Δ∷his7+), CHP1030 (his7Δ∷lys2+ lys2-97), and CHP1031 (his7-366 lys2-97). When using CHP1031 cells, >0.3% of the Lys+ transformants (0/350) were Ade-, and 0.2% of the His+ transformants (1/550) were Ade-. In contrast, 14.3% of the Lys+ CHP1029 transformants (1/7) were Ade-, and 13.7% of the His+ CHP1030 transformants (19/139) were Ade-. Therefore, strains bearing these deletion alleles are significantly better for use in PCR-based gene deletion protocols. In addition, we have carried out subsequent crosses to produce sets of strains appropriate for the construction of diploid strains that are homozygous for lys2Δ∷his7+ or his7Δ∷lys2+ to be used in gene knockout experiments (see Table 1, strains CHP1050-1057).
Conclusions
In this study, we used genetic and physical mapping information to identify the S. pombe lys2+ gene, demonstrating that three different plasmid constructs carrying this gene complement lys2-97, but not lys1-131 (Fig. 1; data not shown). One of these plasmids, pRH3 (Fig. 2), should be a useful cloning vector as it contains an S. pombe ars, conferring high efficiency transformation, and a large polylinker to accommodate a variety of cloning strategies. In addition, one can apply a blue/white screen to identify pRH3 derivatives that carry insert DNA.
We have also demonstrated the ability to use both lys2+ and his7+ in PCR-based gene disruption constructs. In doing this, we have identified sequences (see Materials and methods) capable of PCR-amplifying these two markers such that they are functional in single copy. By deleting the his7+ gene with lys2+ and deleting the lys2+ gene with his7+, we have created strains that are useful hosts for subsequent gene deletions. Previous efforts to use his7+ as a selectable marker for gene deletions were hampered by the fact that his7-366 can be directly repair by the transforming DNA (Apolinario et al. 1993; Jin et al. 1995; Landry et al. 2000). As shown here, this problem is greatly enhanced when using a PCR-based strategy, since the deletion cassette possesses little sequence homology to the gene to be deleted relative to the homology between the selectable marker and the spontaneous mutant allele in the host. Since the PCR-amplified markers used in this study do not share homology with the deletion alleles, repair at the site of the deletion allele is not possible. As expected, the PCR products used to delete ade6+ produced more than tenfold fewer total transformants in strains CHP1029 (his7-366 lys2Δ∷his7+) and CHP1030 (his7Δ∷lys2+ lys2-97), than in strain CHP1031 (his7-366 lys2-97) using comparable amounts of transforming DNA. The total number of Lys+ CHP1029 transformants was especially low, possibly indicating suboptimal expression of Lys2 in this particular context, however we were readily able to identify an Ade− transformant among the seven Lys + colonies.
lys2+ is one of many genes whose identities could be predicted using genetic mapping data and genomic sequence information. In fact, a list of gene predictions can be found at http://www.sanger.ac.uk/Projects/S_pombe/genetic_map.shtml. While most genes of the methionine biosynthetic pathway have not been cloned, the fact that methionine added to the growth medium derepresses conjugation and other glucose-regulated behaviors suggests that the use of a marker in this pathway could be problematic (Schweingruber et al. 1998). The list of predicted, but not cloned, genes includes all four tryptophan biosynthesis genes. Thus, while many biosynthetic pathway genes remain to be cloned, either trp3+ or trp4+ may be particularly useful due to their small size and the fact that no other marker from this pathway is currently in use.
Acknowledgments
We thank Val Wood for pointing out other efforts to predict the identities of S. pombe genes based on genetic and map distance information. We thank Doug Ivey for critical reading of the manuscript. This work was supported by National Institutes of Health grant GM46226 to C.S.H. and a Boston College Undergraduate Research Fellowship to R.L.H.
References
- Alfa C, Fantes P, Hyams J, McLeod M, Warbrick E. Experiments with fission yeast. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1993. [Google Scholar]
- Apolinario E, Nocero M, Jin M, Hoffman CS. Cloning and manipulation of the Schizosaccharomyces pombe his7 + gene as a new selectable marker for molecular genetic studies. Curr Genet. 1993;24:491–495. doi: 10.1007/BF00351711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ML. Cloning and expression of the OMP decarboxylase gene URA4 from Schizosaccharomyces pombe. Curr Genet. 1987;12:527–534. doi: 10.1007/BF00419562. [DOI] [PubMed] [Google Scholar]
- Bähler J, et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Beach D, Nurse P. High-frequency transformation of the fission yeast Schizosaccharomyces pombe. Nature. 1981;290:140–142. doi: 10.1038/290140a0. [DOI] [PubMed] [Google Scholar]
- Beach D, Piper M, Nurse P. Construction of a Schizosaccharomyces pombe gene bank in a yeast bacterial shuttle vector and its use to isolate genes by complementation. Mol Gen Genet. 1982;187:326–329. doi: 10.1007/BF00331138. [DOI] [PubMed] [Google Scholar]
- Burke JD, Gould KL. Molecular cloning and characterization of the Schizosaccharomyces pombe his3 gene for use as a selectable marker. Mol Gen Genet. 1994;242:169–176. doi: 10.1007/BF00391010. [DOI] [PubMed] [Google Scholar]
- Burland TG, Pallotta D, Tardif MC, Lemieux G, Dove WF. Fission yeast promoter-probe vectors based on hygromycin resistance. Gene. 1991;100:241–245. doi: 10.1016/0378-1119(91)90374-k. [DOI] [PubMed] [Google Scholar]
- Cottarel G, Beach D, Deuschle U. Two new multi-purpose multicopy Schizosaccharomyces pombe shuttle vectors, pSP1 and pSP2. Curr Genet. 1993;23:547–548. doi: 10.1007/BF00312650. [DOI] [PubMed] [Google Scholar]
- Feilotter H, Nurse P, Young PG. Genetic and molecular analysis of cdr1/nim1 in Schizosaccharomyces pombe. Genetics. 1991;127:309–318. doi: 10.1093/genetics/127.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford RA, Ye ZH, Bhattacharjee JK. Physical and functional characterization of the cloned lys1+ gene of Schizosaccharomyces pombe. J Basic Microbiol. 1993;33:179–186. doi: 10.1002/jobm.3620330308. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Giga-Hama Y, Takegawa K. Development of a genetic transformation system using new selectable markers for fission yeast Schizosaccharomyces pombe. Yeast. 2005;22:193–202. doi: 10.1002/yea.1201. [DOI] [PubMed] [Google Scholar]
- Grimm C, Kohli J, Murray J, Maundrell K. Genetic engineering of Schizosaccharomyces pombe: a system for gene disruption and replacement using the ura4 gene as a selectable marker. Mol Gen Genet. 1988;215:81–86. doi: 10.1007/BF00331307. [DOI] [PubMed] [Google Scholar]
- Gutz H, Heslot H, Leupold U, Loprieno N. Schizosaccharomyces pombe. In: King RC, editor. Handbook of genetics. Plenum; New York: 1974. [Google Scholar]
- Jin M, et al. sck1, a high copy number suppressor of defects in the cAMP-dependent protein kinase pathway in fission yeast, encodes a protein homologous to the Saccharomyces cerevisiae SCH9 kinase. Genetics. 1995;140:457–467. doi: 10.1093/genetics/140.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DA, Hoffman CS. Gap repair transformation in fission yeast to exchange plasmid-selectable markers. Biotechniques. 2002;33:978–982. doi: 10.2144/02335bm02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi Y, Kitazawa Y, Shimatake H, Yamamoto M. The primary structure of the leu1+ gene of Schizosaccharomyces pombe. Curr Genet. 1988;14:375–379. doi: 10.1007/BF00419995. [DOI] [PubMed] [Google Scholar]
- Landry S, Pettit MT, Apolinario E, Hoffman CS. The fission yeast git5 gene encodes a Gβ subunit required for glucose-triggered adenylate cyclase activation. Genetics. 2000;154:1463–1471. doi: 10.1093/genetics/154.4.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losson R, Lacroute F. Plasmids carrying the yeast OMP decarboxylase structural and regulatory genes: transcription regulation in a foreign environment. Cell. 1983;32:371–377. doi: 10.1016/0092-8674(83)90456-7. [DOI] [PubMed] [Google Scholar]
- Murray JM, Carr AM, Lehmann AR, Watts FZ. Cloning and characterisation of the rad9 DNA repair gene from Schizosaccharomyces pombe. Nucleic Acids Res. 1991;19:3525–3531. doi: 10.1093/nar/19.13.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Dhut S, Toda T. New drug-resistant cassettes for gene disruption and epitope tagging in Schizosaccharomyces pombe. Yeast. 2005;22:583–591. doi: 10.1002/yea.1233. [DOI] [PubMed] [Google Scholar]
- Schweingruber AM, Hilti N, Edenharter E, Schweingruber ME. Methionine induces sexual development in the fission yeast Schizosaccharomyces pombe via an ste11-dependent signalling pathway. J Bacteriol. 1998;180:6338–6341. doi: 10.1128/jb.180.23.6338-6341.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell S, Jenkins JR. arg3+, a new selection marker system for Schizosaccharomyces pombe: application of ura4+ as a removable integration marker. Nucleic Acids Res. 1995;23:1836–1837. doi: 10.1093/nar/23.10.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kao R, Ivey FD, Hoffman CS. Strategies for gene disruptions and plasmid constructions in fission yeast. Methods. 2004;33:199–205. doi: 10.1016/j.ymeth.2003.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Lino Y, Furuhata K, Shimoda C, Yamamoto M. The S. pombe mei2 gene encoding a crucial molecule for commitment to meiosis is under the regulation of cAMP. EMBO J. 1988;7:761–767. doi: 10.1002/j.1460-2075.1988.tb02873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]