

Abstract
For schizophrenia, bipolar disorder, and autism, clinical descriptions are precise and reliable, but there is great overlap among diagnoses in associated genetic polymorphisms and rare variants, treatment response, and other phenomenological findings such as brain imaging. It is widely hoped that new diagnostic categories can be developed which are more precise and predictive of important features of illness, particularly response to pharmacological agents. It is the intent of this paper to describe the diagnostic implications of some current genetic findings, and to describe how the genetic associations with diagnosis may be teased apart into new associations with biologically coherent diagnostic entities and scales, based on the various functional aspects of the associated genes and functional genomic data.
Keywords: genetic, genomic, disease, schizophrenia, bipolar disorder, autism, network, diagnosis
Abstract
Las descripciones clínicas para la esquizofrenia, el trastorno bipolar y el autismo son precisas y confiables, pero hay una gran sobreposición entre estos diagnósticos encuanto a polimorfismos genéticos asociados y variantes raras, respuesta a tratamiento y otros hallazgos como las imágenes cerebrales. Hay grandes expectativas de que se puedan desarrollar nuevas categorías diagnósticas, que sean más precisas y predictoras de las características importantes de la enfermedad, en especial de la respuesta a los fármacos. La intención de este artículo es describir las implicancias diagnósticas de algunos hallazgos genéticos actuales y también describir cómo las asociaciones genéticas con los diagnósticos pueden dar origen a nuevas asociaciones con entidades diagnósticas y escalas biológicamente coherentes, basadas en varios aspectos funcionales de los genes asociados y datos genómicos funcionales.
Abstract
Pour la schizophrénie, les troubles bipolaires et l'autisme, les descriptions cliniques sont précises et fiables, mais il existe un chevauchement important entre ces diagnostics pour les variants rares et les polymorphismes génétiques associés, la réponse au traitement et d'autres symptômes et signes comme l'imagerie cérébrale. De nouvelles catégories diagnostiques plus précises et prédictives des caractéristiques importantes de la maladie, en particulier la réponse aux produits pharmacologiques, sont fortement espérées. Cet article a pour but de décrire les implications diagnostiques de certains résultats génétiques actuels et de décrire comment les associations génétiques observées avec ces catégories diagnostiques peuvent être séparées en de nouvelles associations avec des dimensions et des entités diagnostiques biologiquement cohérentes, à partir des nombreux aspects fonctionnels des gènes associés et des données de génomique fonctionnelle.
Introduction
Diagnosis traditionally is a categorical classification that summarizes a unique set of illness features in a single phrase. In the field of psychiatry, a 1972 publication by Feighner and colleagues1 has long served as a basis for highly reproducible diagnostic criteria, based on the conceptual developments of his coauthors Robins and Guze. This system of diagnosis, with criteria based on clearly defined clinical and behavioral observation, was enshrined in the DSM-III (see the article by Shorter in this issue, p 59) and later DSM versions as the standard for US Psychiatry.
However, three of the five major theoretical bases for validating a diagnosis provided in Feighner et al's paper have not coincided with the clinical diagnostic categories in psychiatry. These are “family study,” “course of illness” (particularly response to treatment agents), and “laboratory tests.” At that time, family study as a basis for diagnosis simply meant coaggregation of a diagnosis in relatives, but would now primarily mean genetic markers. Psychiatric genetic associations with schizophrenia (SZ), bipolar disorder (BD), and autism spectrum disorder (ASD) cross diagnostic boundaries in a very complex manner that is currently not understood biologically.2,3 Before the lack of diagnostic specificity was observed for genetic marker associations, observations of diagnoses in families of patients showed an unexpected degree of overlap. Schizoaffective (SA) patients had, in comparison with control families, excesses of relatives with both BD and SZ. This blurred the two major diagnostic categories established by the German psychiatrist Kraepelin at the end of the 19th and beginning of the 20th centuries. Due to coaggregation of multiple diagnoses in families of patients,4,5 the entire Krapelinian diagnostic system was greatly shaken even before there were any consistent genetic marker associations with these disorders. Alternate bases for establishing diagnosis, with a focus on biological observations, were proposed for decades, starting in the 1970s.6,7
Another of Feighner et al's criteria that is generally no longer diagnosis-specific in psychiatry is course of illness, specifically treatment response to medication. Individual drugs are used to treat multiple disorders. Antipsychotics are a major treatment for BD and for depression, anticonvulsants are used in BD, psychosis, and depression, etc. Results of laboratory tests, a third basis of diagnosis, overlap as well—brain regions with similar abnormalities in multiple disorders have been described.8,9
An idea that has great currency at this time is that we should start over again in psychiatric diagnostic classification, basing it not on clinical description but on biological events, and not on categories but on quantitative metrics.9 The bases currently proposed for such biological events include brain imaging functional associations, neurodevelopmental perspectives, and others, which may prove valid and fruitful,8 although here we will consider them only insofar as they interact with genetic findings. It is the intent of this paper to describe the diagnostic implications of some current genetic findings, and to describe how the genetic associations with diagnosis may be teased apart into new associations with biologically coherent diagnostic entities and scales, based on the various functional aspects of the associated genes and functional genomic data.
Common genetic polymorphisms and rare variants as a basis for a reconceptualized diagnosis system in psychiatry
Genetic associations with common diseases largely consist of common polymorphisms with statistically weak effects on probability of illness, and rare variants with strong effect sizes. In the past decade, two novel types of genetic causation have been discovered with particular relevance to psychiatric disorders: i) polygenic variation directly measurable from common polymorphisms identified by genome-wide assays; and ii) subchromosomal deletions, duplications, and inversions (copy number variants, CNVs). CNVs were described earlier, but it was only in 200410,11 that the ubiquity of CNVs was discovered, and within a few years their role as a major source of human genetic variation was demonstrated.12,13
Prior to these developments, much of the research into the genetic bases of common diseases was focused on the identification of common variants with small effect sizes, particularly via genome-wide association studies (GWASs, Figure 1). Rare alleles with large effects were thought unlikely to contribute much to common disease risk: after all, not only were they were rare, but they would be subject to strong purifying selection. However, due to their large size, CNVs can affect multiple genes at once, so they have the potential for major phenotypic effects. Once their ubiquity was discovered, it was reasonable to predict that they could contribute substantially to overall disease risk (Figure 2), and subsequent studies appear to support that (see below).
Figure 1. McCarthy/Manolio model: single common variants = small effects, single rare variants = large effects. Allele frequency and effect size are generally inversely related, with common variants with large effects being rare and subject to strong purifying selection, and rare variants with small effects being difficult to detect.

From ref 45, adapted from ref 46: McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genetics. 2008;9:356-369. Copyright © Nature Publishing Group 2008.
Figure 2. Additional observations in neuropsychiatric disease. An updated version of Figure 1 shows types of genetic variants now thought to explain some of the genetic risk of neuropsychiatric diseases, including certain rare copy number variants (CNVs) with large effects. Falling outside the predicted inverse linear relationship between allele frequency and effect size, are the presence of any de novo CNV and the contribution of common alleles when incorporated into a polygenic model.

Common single-nucleotide polymorphisms and diagnosis
In the past few years, the number of common single-nucleotide polymorphisms (SNPs) associated with SZ in GWASs has catapulted from less than ten to over 100, with a large number of genes and regulatory regions being located close to many of the associated SNPs, and thus implicated in disease pathology. The most recent report is from the Schizophrenia Working Group of the Psychiatric Genomics Consortium,14 and it includes 128 SNP associations with SZ risk, which come from approximately 108 gene loci, of which 83 are novel.
As has been the case for previous SZ genome-wide studies,15-17 the odds ratios for the individual SNPs are low, ranging from 0.843 to 1.125. This indicates that a very low proportion of disease risk variance can be attributed to any one of these SNPs (Figure 1). The authors used risk profile scores (RPSs) to assess the collective contributions of these SNPs to an individual's SZ risk, and they found that RPSs can explain approximately 7% of variation in risk across their samples.
The RPS is one of the methods of assessing polygenic contributions to disease risk. It was first described in a 2009 International Schizophrenia Consortium paper.18 In a GWAS of SZ patients versus controls, all loci passing an arbitrary threshold of significance (with P-values that are not necessarily statistically significant) are used to calculate a polygenic score for each individual, where the number of score alleles are weighted by the log of the odds ratio (for disease) in a discovery sample. In the initial publication, this score derived from one SZ vs controls sample was elevated in several SZ samples, as well as in BD samples, but not in coronary artery disease and several other medical disorders. Since the percentage of variation in risk across their samples explained by the RPS in this first paper was 3%, the 7% reported by Ripke et al in 2014 represents a substantial increase.14
Another method for estimating polygenic contribution to disease risk is genome -wide complex trait analysis (GCTA). It is a variance-covariance analysis approach which incorporates the effects of every marker in the genome on a phenotype.19 With this more inclusive method, which defines heritability as the proportion of phenotypic variance accounted for by the genetic markers studied, the heritability of schizophrenia accounted for is considerably greater than with the RPS, approximately 23 %.20
While these estimates of polygenic contributions to disease risk explain substantially more variation in disease than the contribution of any one of the common SNPs individually, a considerable amount of variation remains unexplained. This might improve somewhat as GWAS sample sizes are increased, and new loci with smaller effect sizes are discovered. However, the coheritability between disorders (that is, the covariance between disorders on a liability scale3) is substantial for several diagnostic groups (Figure 3), most particularly for SZ, BD, and major depressive disorder (MOD).2-3 Whether this overlap in common variants associated with neuropsychiatric disease means that the current diagnostic categories are less than optimal, and that a future categorization based on the shared genetic elements would be more useful, remains to be explored.
Figure 3. Genome-wide pleiotropy between psychiatric disorders. ADHD, attention deficit-hyperactivity disorder; BPD, bipolar disorder; SCZ, schizophrenia; MDD, major depressive disorder; ASD, autism spectrum disorder.
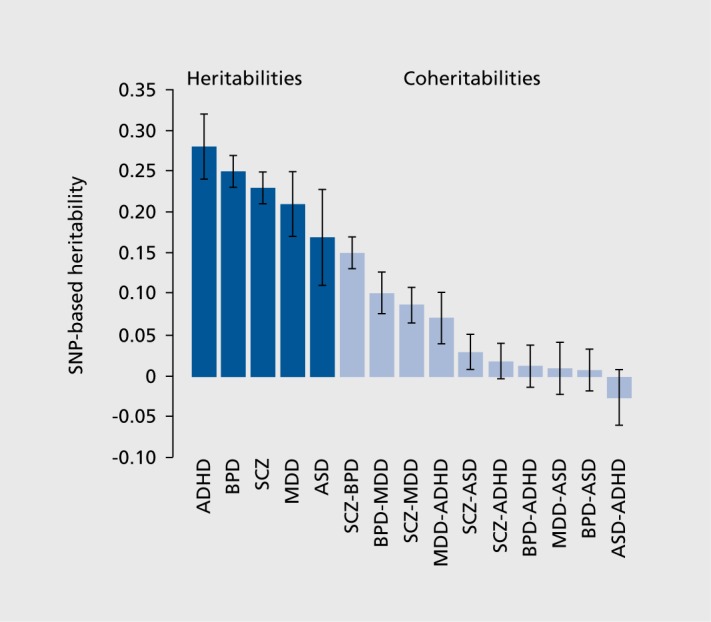
From ref 3: Lee SH, Ripke S, Neale BM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984-994. Copyright © Nature Publishing Group 2013.
Generation of new categorical or quantitative trait diagnoses using genome-wide common SNP data
To create a new diagnosis or quantitative diagnostic trait from genetic data, one might consider the alleles contributing to the coheritability of several diagnoses with consistent two-way correlations, such as SZ, BD, and MDD, and use these alleles as a score, or use the polygenic risk scores that apply to multiple diagnoses.2 This score could then be considered for usefulness as a diagnosis, by testing its applicability to the bases of diagnosis as initially proposed Feighner and colleagues in 1972.1 Similarly, one could define a diagnosis or diagnostic scale, based on location of rare variants or loci consistently impacted by de novo CNVs in several psychiatric disorders, but not in control individuals.21
Members of the Bipolar and Schizophrenia Network for Intermediate Phenotypes Consortium (BSNIP) have recently presented posters with preliminary results,22,23 which illustrate this approach. The consortium is working to identify markers of psychosis across diagnostic boundaries that would generate biologically coherent separation among individuals with psychosis who are diagnosed with BD, SA, or SZ.
Clementz and others in the consortium identified three “biotypes” based on taxometric analyses of electrophysiological, cognitive, and other biological marker data. The measured variables were psychophysiological, and included ERR24,25 eye movements26 including stop-signal tasks, and a cognitive battery.27 SNPs used to calculate polygenic risk scores (PGRS), which appear similar to the RSPs described above, for SZ were derived from very large-scale studies of the Psychiatric Genomics Consortium (PGC) samples, and PGRSs were calculated from whole-genome genotypic arrays for each of the 340 patients studied and for 112 healthy controls. They then compared the three biotype groups on the degree of genetic loading as measured by polygenic risk scores. Essentially, only patients classified as Biotype 1 had significantly greater polygenic risk scores than the healthy controls, but the distribution of diagnoses was the same in each of the three biotypes.
Assuming replication of this initial finding, could this lead to Biotype 1 as a new diagnostic category? We can consider the multiple steps toward that goal as an exercise. A first issue to be resolved is whether the taxometric (categorical) analysis that led to three biotypes is the best way to combine the underlying variables, since the underlying variables are quantitative and the PGRS that was related to biotypes is also a quantitative trait. Apart from that issue, further supporting evidence would be needed to relate the biotypes to the other components of diagnosis. If we consider the “laboratory test” part of diagnosis to mean a biological understanding, then the PGRS, after all, is not a biologically meaningful scale. It needs to be functionally parsed, as in Purcell et al 2014,28 in order to lead to actual biological mechanisms (see discussion below on the relationship of molecular networks to diagnosis). This parsing would only be a first step toward supporting molecular biological hypotheses based on the PGRS. Other types of measurement useful in studying the CNS in psychiatric disorders would also need to be studied for correlation with the biotype groupings, such as the various types of brain imaging measures. In addition to biology, but not unrelated to it, a coherence of other illnessrelated traits is needed to support a diagnostic distinction, as described in Feighner and colleagues' paper.1 This would include whether the biotypes coaggregate in families, whether there are different ages of onset and courses of illness associated with the biotypes, and, perhaps most importantly, whether there were different treatment responses, such as to specific drugs, associated with the different biotypes.
Rare variants as a basis for diagnosis
Several rare copy number CNVs are highly associated with neuropsychiatric diagnoses (Table I).21 CNVs are deletions and duplications that are too small to be observed as classic microscopic bands, although they can be millions of DNA base pairs in size. At certain chromosomal locations, the DNA sequence is predisposed to deletion and/or duplication, and these locations recurrently generate the same CNVs, which individually remain rare in frequency. The most notable of these recurrent deletions occurs on Chromosome 22q11 (long arm [q] of the chromosome, microscopic band 11). This deletion was discovered to be associated with SZ in 1995, and was the first rare variant associated with this disease.29 Since then, it has been shown to be associated not just with SZ, but with ASD and intellectual disability, as well as the cardiovascular, facial, and other malformations originally associated with the deletion.30 Not surprisingly, as it was first to be detected, deletion of 22q11 is the most common rare CNV associated with neuropsychiatric illness, occurring in 1 out of 4000 births. Hie deletion, usually 1 to 3.5 megabases in size, is extremely likely to cause disease, with nearly complete disease penetrance, although the observed diagnoses are quite variable. The resulting phenotype is variably referred to as 22q11 deletion syndrome, DiGeorge syndrome or velocardiofacial syndrome. There are multiple genes with important neurobiological roles in the region, but it has not been possible to assign the pathophysiology to a single gene. Nonetheless, with its definitive laboratory test, and despite the several alternative phenotypes, it arguably meets Feighner's criteria for a valid diagnosis.
Table I. Rare copy number variants (CNVs): risks of illness for autism spectrum disorder (ASD), schizophrenia (SCZ) and bipolar disorder (BD).47 Risks of illness are based on Bayesian probabilities. Data from Malhotra and Sebat's 2012 review.21.
From ref 47: Gershon ES, Alliey-Rodriguez N. New ethical issues for genetic coun seling in common mental disorders. Am J Psychiatry. 2013;170:968-976. Copyright © American Psychiatric Association 2013.
| CNV Locus | Type | ASD | SZ | BD | Risk of any of these disorders |
| 1q21.1 | Deletion | 7.91% | 7.91% | ||
| Duplication | 4.97% | 4.50% | 9.25% | ||
| 3q29 | Deletion | 33.56% | 33.56% | ||
| 7q11.23 | Duplication | 16.05% | 16.05% | ||
| 15q11.2 | Deletion | 2.09% | 2.09% | ||
| 15q11.2-13.1 | Duplication | 20.73% | 20.73% | ||
| 15q13.3 | Deletion | 5.42% | 8.76% | 13.70% | |
| 16p11.2 | Deletion | 5.96% | 5.96% | ||
| Duplication | 7.28% | 9.45% | 4.19% | 19.56% | |
| 17p12 | Deletion | 6.60% | 6.60% | ||
| 22q11.21 | Deletion | 23.06% | 68.25% | 26.37% | 82.01% |
| 22q11.2 | Duplication | 2.07% | 2.07% |
Although the 22q11 deletion syndrome is the most scientifically supported diagnostic entity in psychiatry, it also illustrates how disappointing the practical implications of diagnosis can be. It can be highly useful in risk prediction in rare cases, but it has not led to advances in neurobiology of disease or in treatment approaches. Of course, it is entirely reasonable to expect that these advances will be forthcoming, for this and for each of the rare structural variants associated with neuropsychiatric disease (Table I), but the challenge remains formidable two decades after the initial finding of the psychiatric association.
The presence of any CNV event arising as a de novo mutation (that is, present in a person but not in his/her parents) greatly increases the risk of several psychiatric disorders (Table II, Figure 2). It is possible that there are so many genomic locations whose disruption can cause neuropsychiatric disease that they are ubiquitous throughout the human genome. A more appealing possibility to the scientist is that the polygenic components can be parsed into logical entities (see below in discussion of networks in diagnosis), and that there will be “hot spots” on the genome where psychiatric illness will be caused by a de novo CNV, and other locations where disease will not result, according to the genes affected. This would be consistent with the findings of other types of rare variants in the polygenic components of neuropsychiatric disease, as discussed in the next paragraph.
Table II. De novo copy number variants (DCCNVs): attributable risk and risks of illness for autism spectrum disorder (ASD), schizophrenia (SCZ) and bipolar disorder (BD).47 Data from: Xu 2008,48 Malhotra 2011,49 and Sebat 2007.50 Computation of illness risk of any disorder is 1-(1-P1)(1-P2)(1-P3), where Pi is the risk for each disorder. This calculation indirectly accounts for probability of co-occurrence of more than one disorder in any individual as a product of the probability of each diagnosis. Frequency of DNCNV in normal controls is approximately 1%. Overall burdens of rare and de novo single nucleotide variants (SNVs), and rare small insertions and deletions (indels) are also associated with some neuropsychiatric disorders.28,48 For rare SNVs and indels, the association is not present in the genome as a whole, but in “large set of genes with a higher likelihood of having a role in schizophrenia, on the basis of existing genetic evidence.”28 This offers some hope that parsing of the evidence for genome-wide aggregations of data will lead to neurobio-logically useful diagnostic categories or scales. OR, odds ratio; aBased on Bayesian probability.
From ref 47: Gershon ES, Alliey-Rodriguez N. New ethical issues for genetic coun seling in common mental disorders. Am J Psychiatry. 2013;170:968-976. Copyright © American Psychiatric Association 2013.
| Disease | OR | Rate of DNCNV if Ill | Illness risk if DNCNVa |
| Schizophrenia | 6.27 | 6.10% | 5.67% |
| Bipolar disorder | 4.77 | 4.32% | 4.45% |
| ASD | 7.50 | 7.18% | 4.07% |
| Risk of any one of these disorders | 13.53% |
Molecular networks as a basis for diagnosis
An alternative approach to the single locus association studies that is quickly gaining traction is complex network analysis. This method models molecular interactions graphically as complex networks. The network nodes, or vertices, would represent various molecules, while the edges, or line between the nodes, would represent interactions among them Figure 4A. Alternatively, the nodes could represent proteins, while the edges would then represent physical interactions between two proteins. Alternatively the nodes might represent the expression levels of genes, and the edges might represent correlations among those levels. Ideally, all molecules could be represented in a single network, as well as all possible interactions among them Figure 4C.
Figure 4. Illustration of network concepts. A: Gray spheres indicate nodes in the network, which represent molecules, and the lines, or edges, represent pair-wise relationships between the molecules. B: A module is a subnetwork of highly interconnected nodes in the network, and an intramodular hub is the most connected node or nodes in a module. C: Biological networks comprise many modules, as well as sparsely connected nodes. Purple lines indicate relationships between nodes from different modules. Cyan lines indicate relationships between nodes not belonging to any module.

From ref 33: Grennan KS, Chen C, Gershon ES, Liu C. Molecular network analy sis enhances understanding of the biology of mental disorders. BioEssays. 2014;36:606-616. Copyright © Cambridge University Press 2014.
A property of molecular networks is modularity, ie, they contain subnetworks, or modules, of highly connected nodes that are relatively sparsely connected to the larger network Figure 4B. This property is useful for disease studies because the nodes comprising these subnetworks are highly likely to be functionally related. So, if one node is, for example, a disease-associated gene variant, other nodes in the same module are more likely to be disease-associated.31 This phenomenon is frequently referred to as “guilt-by-association”32 and can be used both to identify novel candidate genes, and to prioritize known candidate genes for follow-up functional studies.33
When disease risk genes are mapped onto existing networks, they often appear to converge on some of these functional modules. For example, Sakai et al created a protein-protein interaction (PPI) network for a set of genes, most of which were known to cause syndromic forms of ASD. Once the nodes, which represented the protein products of those genes, and the edges, which represented physical interactions among those proteins, were incorporated into a network model, the authors tested the rate at which a set of nonsyndromic ASD-associated variants overlapped with that network. They found that the variants overlapped at a rate 2.4 times greater in cases than in controls.
These kinds of observations are encouraging: they suggest that there is some biological basis for the statistical association of specific network membership genes and disease risk. They also help explain some of the genetic heterogeneity that characterizes many neuropsychiatric diseases. Finally, they implicate the functions of these modules in disease etiology.
The approaches just described rely on curating existing knowledge (such as publications) used to construct networks, which are then inspected for whether they include disease-associated variants. An alternative, unbiased approach is to use genome-wide data to infer networks and thereby discover novel associations between molecules and with disease. For example, gene coexpression networks, in which the nodes are gene transcripts and the edges represent correlations among their expression levels, can be constructed from gene expression microarray data using methods such as weighted gene coexpression network analysis (WGCNA).34 Coexpression modules identified in these networks have been shown to correspond to brain cell types in healthy individuals.35 Furthermore, they have been shown to be altered in neuropsychiatric disease. In brains from psychotic35 and autistic36 patients, certain modules have been shown to be up- or downregulated relative to controls, while in late-onset Alzheimer's disease (LOAD) patients, brain coexpression networks lost and gained entire modules.37
Some studies have blended the two approaches, by mapping disease-associated genes onto coexpression modules inferred from genome-wide expression data. Ben-David and Shifman38 for example, used expression data from healthy controls to identify coexpression modules, then demonstrated that two of those modules were enriched with a set of rare and common ASDassociated variants. Both modules were enriched with genes active in the synapse, while one was also enriched with genes involved in synaptic transmission and the calmodulin-binding pathway. Gulsuner and colleagues performed a similar study for SZ.39
The goal of refining or redefining psychiatric diagnostic categories based on their biological underpinnings would be greatly helped if the set of variants associated with a given disease neatly matched up with a discrete functional module, with no overlap among diseases. However, as discussed above, many genetic variants are not disease-specific, and unfortunately, so far, neither are the implicated modules. In fact, modules implicated in a wide range of diseases tend to overlap with each other, not just among similar diseases, like neuropsychiatric ones40 but among very different conditions, such as asthma, cancer and obesity.41
There are other ways of exploiting network structure, though. In an approach that has been referred to as “reverse engineering.”42 Zhang et al used genomewide brain expression data to identify functional modules altered in late onset Alzheimer's disease (LOAD).37 Then, using genotype data, they identified key causal regulators of the altered networks, which included TYRO protein tyrosine kinase binding protein (TYROPB). This is important because TYROPB was not previously identified as a possible LOAD risk gene. Also, the regulators identified, including TYROPB, were shown to be causal, and causation is a holy grail of molecular biology. Taken together, these findings suggest that TYROPB may be a drug target for LOAD.
So this network-based method can yield possible genetic disease associations undetectable by previous methods, but network approaches have multiple potential new applications. Future analyses may identify more molecular functions impaired in disease. Also, expanding and validating current molecular networks may prove important in terms of creating predictive models of genetic variation for neuropsychiatric diseases. Network models can account for emergent properties of complex molecular networks, such as epistasis (ie, gene-gene interactions) and network motifs like feedback and feedforward loops; the predictive power of polygenic models like the ones described above may be limited because they do not allow for these properties.
What could network diagnoses look like? Measures of overall network status could be developed, and therapeutic strategies for shifting a relevant subnetwork from “unhealthy” to “healthy” by targeting specific nodes or edges with drugs may be developed.43 Another possibility is that an individual patient's genetic variants will be mapped onto the complete set of human molecular networks, and possible therapeutic targets predicted from that, which would almost obviate the need for a diagnostic category. Alternatively, if it becomes technologically and economically feasible, a molecular network may be constructed for individual patients, and drug targets predicted from that.44 This exciting sort of highly personalized approach is well in the future for neuropsychiatric disease, but it could come to pass.
Outlook
Over the last decade, a great deal of progress has been made in explaining the genetic underpinnings of neuropsychiatric disease. At the same time, a great deal of the heritability of these diseases remains to be explained.
There are a number of promising directions for parsing the multigenic component of heritability of major psychiatric disorders, one of which is molecular network research. The number of common SNP associations detected in GWASs should rise as sample sizes increase. Even though the effect sizes of associations so revealed will tend to be small, these would be additional components of the biological networks involved in disease, and so would add to the precision of identifying molecular networks that impact upon genes associated with disease, or to defining new diagnoses based on close association with one or another network malfunction. Functional networks or phenotypes based on other than molecular characteristics, such as brain connectivity networks, may also prove to be more coherent as a basis for diagnosis formation than current nosologic categories, and if successful these may also prove to be more biologically coherent than our current diagnostic categories.
Contributor Information
Elliot S. Gershon, Department of Psychiatry and Behavioral Neuroscience; Department of Human Genetics, University of Chicago, Illinois, USA.
Kay S. Grennan, Department of Psychiatry and Behavioral Neuroscience; University of Chicago, Illinois, USA.
REFERENCES
- 1.Feighner JP., Robins E., Guze SB., Woodruff RA Jr., Winokur G., Munoz R. Diagnostic criteria for use in psychiatric research. Arch Gen Psychiatry. 1972;26:57–63. doi: 10.1001/archpsyc.1972.01750190059011. [DOI] [PubMed] [Google Scholar]
- 2.Smoller JW., Ripke S., Lee PH., et al Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee SH., Ripke S., Neale BM., et al Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gershon ES., Hamovit J., Guroff JJ., et al A family study of schizoaffective, bipolar I, bipolar II, unipolar, and normal control probands. Arch Gen Psychiatry. 1982;39:1157–1167. doi: 10.1001/archpsyc.1982.04290100031006. [DOI] [PubMed] [Google Scholar]
- 5.Gershon ES., DeLisi LE., Hamovit J., et al A controlled family study of chronic psychoses. Schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 1988;45:328–336. doi: 10.1001/archpsyc.1988.01800280038006. [DOI] [PubMed] [Google Scholar]
- 6.Gottesman II., Shields J. Genetic theorizing and schizophrenia. Br J Psychiatry. 1973;122:15–30. doi: 10.1192/bjp.122.1.15. [DOI] [PubMed] [Google Scholar]
- 7.Rieder RO., Gershon ES. Genetic strategies in biological psychiatry. Arch Gen Psychiatry. 1978;35:866–873. doi: 10.1001/archpsyc.1978.01770310072005. [DOI] [PubMed] [Google Scholar]
- 8.Casey BJ., Craddock N., Cuthbert BN., Hyman SE., Lee FS., Ressler KJ. DSM-5 and RDoC: progress in psychiatry research? Nat Rev Neuroscience. 2013;14:810–814. doi: 10.1038/nrn3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Insel TR., Cuthbert BN. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry. 2009;66:988–989. doi: 10.1016/j.biopsych.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 10.lafrate AJ., Feuk L., Rivera MN., et al Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 11.Sebat J., Lakshmi B., Troge J., et al Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 12.Eichler EE. Widening the spectrum of human genetic variation. Nat Genet. 2006;38:9–11. doi: 10.1038/ng0106-9. [DOI] [PubMed] [Google Scholar]
- 13.Stankiewicz P., Lupski JR. Structural variation in the human genome and its role in disease. Ann Rev Med. 2010;61:437-55. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 14.Ripke S., Neale BM., Corvin A., et al Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ripke S., O'Dushlaine C., Chambert K., et al Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ripke S., Sanders AR., Kendler KS., et al Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purcell SM., Wray NR., Stone JL., et al Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Schizophrenia Consortium., Purcell SM., Wray NR., et al Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J., Lee SH., Goddard ME., Visscher PM. GCTA: a tool for genomewide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SH., DeCandia TR., Ripke S., et al Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet. 2012;44:247–250. doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malhotra D., Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathew I., Padmanabhan J., Tandon N., et al Polygenic risk scores for schizophrenia may elucidate differences between biological subtypes of psychosis. Paper presented at: Society of Biological Psychiatry; 69th Annual Scientific Meeting; New York, NY, 2014. [Google Scholar]
- 23.Nanda P., Padmanabhan J., Tandon N., et al Polygenic characterization of psychotic disorder across diagnostic and neurobiological taxonomies: A framework for stratified medicine? Paper presented at: American College of Neuropsychopharmacology; 53rd Annual Meeting; Phoenix, AZ; December 7-11; 2014. [Google Scholar]
- 24.Hamm JP., Ethridge LE., Shapiro JR., et al Spatiotemporal and frequency domain analysis of auditory paired stimuli processing in schizophrenia and bipolar disorder with psychosis. Psychophysiology. 2012;49:522–530. doi: 10.1111/j.1469-8986.2011.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamm JP., Ethridge LE., Boutros NN., et al Diagnostic specificity and familiality of early versus late evoked potentials to auditory paired stimuli across the schizophrenia-bipolar psychosis spectrum. Psychophysiology. 2014;51:348–357. doi: 10.1111/psyp.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reilly JL., Frankovich K., Hill S., et al Elevated antisaccade error rate as an intermediate phenotype for psychosis across diagnostic categories. Schizophr Bull. 2014;40:1011–1021. doi: 10.1093/schbul/sbt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willsey AJ., Sanders SJ., Li M., et al Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purcell SM., Moran JL., Fromer M., et al A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karayiorgou M., Morris MA., Morrow B., et al Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci U S A. 1995;92:7612–7616. doi: 10.1073/pnas.92.17.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robin NH., Shprintzen RJ. Defining the clinical spectrum of deletion 22q11.2. J Pediatr. 2005;147:90–96. doi: 10.1016/j.jpeds.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 31.Barrenas F., Chavali S., Holme P., Mobini R., Benson M. Network properties of complex human disease genes identified through genome-wide association studies. PLoS One. 2009;4:e8090. doi: 10.1371/journal.pone.0008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goh Kl., Cusick ME., Valle D., Childs B., Vidal M., Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grennan KS., Chen C., Gershon ES., Liu C. Molecular network analysis enhances understanding of the biology of mental disorders. BioEssays. 2014;36:606–616. doi: 10.1002/bies.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article 17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 35.Oldham MC., Horvath S., Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci U S A. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voineagu I., Wang X., Johnston P., et al Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang B., Gaiteri C., Bodea LG., et al Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ben-David E., Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry. 2013;18:1054–1056. doi: 10.1038/mp.2012.148. [DOI] [PubMed] [Google Scholar]
- 39.Gulsuner S., Walsh T., Watts AC., et al Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–29. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hormozdiari F., Penn O., Borenstein E., Eichler EE. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015;25:142–154. doi: 10.1101/gr.178855.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrenas F., Chavali S., Alves AC., et al Highly interconnected genes in disease-specific networks are enriched for disease-associated polymorphisms. Genome Biol. 2012;13:R46. doi: 10.1186/gb-2012-13-6-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schadt EE., Lum PY. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Reverse engineering gene networks to identify key drivers of complex disease phenotypes. J Lipid Res. 2006;47:2601–2613. doi: 10.1194/jlr.R600026-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- 44.Gustafsson M., Nestor CE., Zhang H., et al Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014;6:e1–e11. doi: 10.1186/s13073-014-0082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manolio TA., Collins FS., Cox NJ., et al Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCarthy Ml., Abecasis GR., Cardon LR., et al Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature Reviews Genetics. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 47.Gershon ES., Alliey-Rodriguez N. New ethical issues for genetic counseling in common mental disorders. Am J Psychiatry. 2013;170:968–976. doi: 10.1176/appi.ajp.2013.12121558. [DOI] [PubMed] [Google Scholar]
- 48.Xu B., Roos JL., Levy S., van Rensburg EJ., Gogos JA., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 49.Malhotra D., McCarthy S., Michaelson JJ., et al High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sebat J., Lakshmi B., Malhotra D., et al Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]