

Abstract
Background and objectives
The Wilms tumor suppressor gene 1 (WT1) plays an essential role in urogenital and kidney development. Genotype/phenotype correlations of WT1 mutations with renal function and proteinuria have been observed in world-wide cohorts with nephrotic syndrome or Wilms tumor (WT). This study analyzed mid-European patients with known constitutional heterozygous mutations in WT1, including patients without proteinuria or WT.
Design, setting, participants & measurements
Retrospective analysis of genotype, phenotype, and treatment of 53 patients with WT1 mutation from all pediatric nephrology centers in Germany, Austria, and Switzerland performed from 2010 to 2012.
Results
Median age was 12.4 (interquartile range [IQR], 6–19) years. Forty-four of 53 (83%) patients had an exon mutation (36 missense, eight truncating), and nine of 53 (17%) had an intronic lysine-threonine-serine (KTS) splice site mutation. Fifty of 53 patients (94%) had proteinuria, which occurred at an earlier age in patients with missense mutations (0.6 [IQR, 0.1–1.5] years) than in those with truncating (9.7 [IQR, 5.7–11.9]; P<0.001) and splice site (4.0 [IQR, 2.6–6.6]; P=0.004) mutations. Thirteen of 50 (26%) were treated with steroids and remained irresponsive, while three of five partially responded to cyclosporine A. Seventy-three percent of all patients required RRT, those with missense mutations significantly earlier (at 1.1 [IQR, 0.01–9.3] years) than those with truncating mutations (16.5 [IQR, 16.5–16.8]; P<0.001) and splice site mutations (12.3 [IQR, 7.9–18.2]; P=0.002). Diffuse mesangial sclerosis was restricted to patients with missense mutations, while focal segmental sclerosis occurred in all groups. WT occurred only in patients with exon mutations (n=19). Fifty of 53 (94%) patients were karyotyped: Thirty-one (62%) had XY and 19 (38%) had XX chromosomes, and 96% of male karyotypes had urogenital malformations.
Conclusions
Type and location of WT1 mutations have predictive value for the development of proteinuria, renal insufficiency, and WT. XY karyotype was more frequent and associated with urogenital malformations in most cases.
Keywords: genetic renal disease, nephrotic syndrome, pediatric nephrology
Introduction
The Wilms tumor 1 (WT1) gene on chromosome 11p13 was identified in 1990 (1,2). Its longest open reading frame comprises 10 exons that code for a transcription factor with four zinc-finger DNA-binding motifs. Transcriptional function of WT1 can result in tumor suppression or be oncogenic (3). An alternative splice site in intron 9 allows the addition of three amino acids (lysine-threonine-serine [KTS]) (4). WT1 plays an essential role in early urogenital and kidney development, especially in the conversion of mesenchymal cells to epithelial structures (5).
Heterozygous deletion of 11p13, resulting in altered dosage of WT1 expression, is found in patients with WAGR syndrome (Wilms tumor [WT], aniridia, genitourinary malformations, and mental retardation) (6). Heterozygous germline mutations in WT1 were described in two rare human conditions: Denys–Drash syndrome (DDS) with exon mutations in the zinc-finger region (7,8) and Frasier syndrome (FS) with mutations affecting the canonic donor KTS splice site of intron 9 (9,10). DDS includes steroid-resistant nephrotic syndrome rapidly progressing to ESRD, 46 XY disorder in sex development (DSD) with sex reversal and a high risk for WT (11,12). FS is defined by progressive glomerulopathy, gonadoblastoma, and 46 XY DSD with sex reversal (13). Over the past years, many clinical pictures beyond the classic syndromes have been described in patients with heterozygous WT1 mutations, illustrating the phenotypic heterogeneity (14,15). A recent report by the international PodoNet Consortium described genotype/phenotype correlations associated with WT1 mutations in 61 patients with nephrotic syndrome collected worldwide; it did not cover treatment of proteinuria (16).
We present data on 53 patients with known heterozygous constitutional WT1 mutations from all major pediatric nephrology centers in Germany, Austria, and Switzerland. We sought to perform a representative analysis of genotype and renal phenotype of mid-European patients, including data on treatment of proteinuria and description of genitourinary abnormalities.
Materials and Methods
Patients and Data Recruitment
Fifty-three patients with constitutional heterozygous WT1 mutations treated in pediatric nephrology centers in Germany (n=48), Austria (n=2), and Switzerland (n=3) were included in this retrospective study (for details, see Supplemental Table 1). Approved by the ethical committee of the Medical Association of Hamburg, Germany, this study was performed in cooperation with the German Society for Pediatric Nephrology. After informed consent was obtained, clinical data were collected from August 2010 until August 2012 using a standardized questionnaire, in adherence to the Declaration of Helsinki.
Categories of WT1 Mutations for Genotype/Phenotype Correlation
WT1 mutations were identified by direct Sanger sequencing technique. All mutations were matched with previous findings from the literature, and, if needed, nomenclature was adjusted to the current recommendations of the Human Genome Variation Society using NM_024426.4 as reference. Patients were classified according to the type of mutations: exon mutations, either missense mutations in DNA-binding regions (reference sequence NM_024426.4: WT isoform D) or other regions or variants of truncating character (nonsense mutations and out-of-frame deletions) and intronic (KTS splice site) mutations.
Clinical and Histologic Data
The date of first diagnosis of proteinuria together with the clinical picture was documented, with definitions based on current guidelines (17,18). If a renal biopsy was performed, we recorded the underlying histologic assessment. The start of RRT was defined as start of dialysis or preemptive renal transplantation. The date of diagnosis of WT and its management were recorded. In addition, karyotype and sex were obtained, along with information on urogenital malformations and their management. We excluded the following from correlation analyses: patient 9, who initially presented with hemolytic uremic syndrome, and patient 30, who had p.Pro29Ser WT1 missense variant (Supplemental Table 1).
Statistical Analyses
A custom database was created using Filemaker Pro 11 (Filemaker Inc., Unterschleissheim, Germany). Data were analyzed using GraphPad Prism software, version 6.0b for Mac OS X (GraphPad Software, San Diego, CA). Non-normally distributed variables are presented as medians and interquartile ranges. Differences between two or more groups were analyzed using one-way ANOVA or nonparametric Kruskal–Wallis test as appropriate. For survival analyses, differences between curves were tested with a log-rank test. In general, P<0.05 was regarded as representing a statistically significant difference. In multiple comparisons, P values were adjusted for multiplicity using Bonferroni correction or Dunn multiple comparison testing.
Results
WT1 Mutations
At the time of analysis patients had a median age of 12.4 (IQR, 6–19) years (Table 1). Exon mutations were found in 44 of 53 patients (83%); in 39 of 44 (89%) mutations were located in the hot spot region comprising exons 8 and 9 (Table 2). Thirty-six of 44 patients (82%) had missense mutations; of these, 28 mutations were affecting amino acids in DNA-binding positions. Truncating mutations were found in eight of 44 patients (18%) (n=7 nonsense, n=1 deletion) (Figure 1).The most common mutation was a missense mutation in exon 9 c.1384C>T, p.Arg462Trp (previously referred to as R394) in 14 patients (Supplemental Table 2). Intronic KTS splice site mutations, c.1432+4C>T and c.1432+5G>A, were found in five and four patients, respectively. All mutations found were heterozygous. Treating physicians referred to their patients with exon mutations as having DDS or partial DDS and referred to their patients with intron mutations as having FS, even if they did not show the full syndromal picture. Allele frequencies and prediction of pathogenicity of WT1-mutations are presented in Supplemental Table 3.
Table 1.
Summary of patient characteristics
| Characteristic | All Patients | Type of WT1 Mutation | ||
|---|---|---|---|---|
| Missense | Truncating | KTS Splice Site | ||
| Patients (n) | 53 | 36 | 8 | 9 |
| Median age at follow-up (yr) | 12.4 (6.0–19.0) | 9.3 (4.1–15.1) | 17.9 (7.5–22.9) | 21.0 (12.1–33.6) |
| Proteinuria, n (%) | 50 (94) | 34 (94) | 6 (75) | 9 (100) |
| Median age at diagnosis of proteinuria (yr) | 1.2 (0.3–4.3) | 0.6 (0.1–1.5) | 9.7 (5.7–11.9) | 4.0 (2.6–6.6) |
| Biopsies available, n (%) | 45 (85) | 31 (86) | 5(63) | 9 (100) |
| Underlying histologic condition, n (%) | 17 (38) | 8 (26) | 4 (80) | 5 (56) |
| FSGS | 3 (7) | 0 | 0 | 3 (33) |
| Minimal-change disease | 20 (44) | 20 (65) | 0 | 0 |
| Diffuse mesangial sclerosis | 5 (11) | 3 (10) | 1 (20) | 1 (11) |
| Others | ||||
| ESRD | 39 (74) | 30 (83) | 4 (50) | 4 (44) |
| Median age at start RRT (yr) | 1.7 (0.6–7.8) | 1.1 (0.4–4.0) | 16.5 (16.2–16.7) | 12.2 (8.8–16.9) |
| WT, n (%) | 19 (36) | 12 | 7 (88) | 0 (0) |
| Bilateral WT, n (%) | 7 (37) | 2 (17) | 5 (71) | |
| Median age at diagnosis (yr) | 1.3 (0.9–1.5) | 1.3 (1.0–1.8) | 1.0 (0.8–1.3) | |
| Gonadoblastoma | 1 (2) | 0 (0) | 0 (0) | 1 (11) |
| Karyotype available, n (%) | 50 (94) | 34 (94) | 7 (88) | 9 (100) |
| XY | 31 (62) | 21 (62) | 6 (86) | 4 (44) |
| XY-DSD | 32 (60) | 21 (58) | 7 (88) | 4 (44) |
| Complete gonadal dysgenesis (sex reversal), n (%) | 6 (11) | 3 (8) | 0 | 3 (33) |
Median values are expressed with interquartile ranges. WT, Wilms tumor; DSD, disorder in sex development; KTS, lysine-threonine-serine.
Table 2.
Overview of clinical features of patients with WT1 hot spot mutations in exon 8/9
| Variable | Type of Mutation in Exon 8/9 | |
|---|---|---|
| Missense | Truncating | |
| Patients (n) | 34 | 6 |
| Median age at follow-up (yr) | 8.9 (3.8–14.8) | 20.6 (15.6–24.9) |
| Proteinuria, n (%) | 34 (100) | 6 (100) |
| Median age at diagnosis of proteinuria (yr) | 0.6 (0.1–1.4) | 9.7 (5.7–11.9) |
| ESRD, n (%) | 31 (91) | 4 (67) |
| Median age at start of RRT (yr) | 1.1 (0.4–3.9) | 16.5 (16.2–16.7) |
| Wilms tumor, n (%) | 12 (35) | 5 (83) |
| Gonadoblastoma, n (%) | 0 (0) | 0 (0) |
| XY-DSD, n (%) | 20 (59) | 5 (83) |
DSD, disorder in sex development.
Figure 1.

Exon 8/9 is hot spot region for mutations in the WT1 gene. Gene is shown out of scale. NM_024426.4: Wilms tumor isoform D of WT1 was used as reference sequence. The bold, italic text indicates missense mutations affecting nucleotides coding for residues important for interaction with target DNA: 434–449 in exon 8 and 461–469 in exon 9 (Swiss-Prot: P19544). The lysine-threonine-serine (KTS) splice site mutations are mapped below. The number of patients affected is specified. Asterisk, translation termination (stop) codon, according to the Human Genome Variation Society.
Proteinuria
Fifty of 53 (94%) patients had nephrotic-range proteinuria, 33 (66%) of whom had clinical nephrotic syndrome. Fourteen of 50 (28%), including only one male patient, had isolated proteinuria. In 41 patients proteinuria was the initial renal symptom.
Thirteen of 50 patients (26%) were treated with steroids; all of them were steroid resistant. Five of 50 (10%) received cyclosporine A (CsA), which lead to partial remission in three patients with KTS splice site mutation. One patient received chlorambucil without any response to treatment.
Patients with missense mutations presented with proteinuria significantly earlier (median, 0.6 [IQR, 0.1–1.5] years) than patients with truncating mutations (median, 9.7 [IQR, 5.7–11.9] years; P<0.001) and those carrying splice site mutations (median, 4.0 [IQR, 2.6–6.6] years; P=0.004) (Figure 2A). There were no statistically significant differences between the two groups with missense mutations in DNA binding or other regions. Of the three patients without any proteinuria, two patients had a truncating mutation in exon 1; they both presented with bilateral WT and 46 XY DSD. The other patient had the p.Pro29Ser missense variant in exon 2 and no WT but did have 46 XY DSD.
Figure 2.
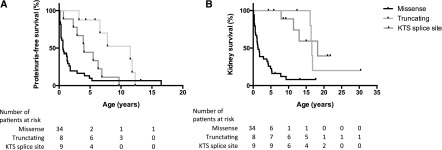
Significant influence of type of causative WT1 mutation on manifestation of nephropathy. (A) proteinuria (P=0.002) and (B) kidney survival (P<0.001). Kaplan–Meier survival analysis, log-rank test (Mantel–Cox). KTS, lysine-threonine-serine.
Renal Function
Forty-seven of 53 patients (88%) had CKD stage 3–5. Thirty-nine of 53 (73%) required RRT at a median age of 1.7 (IQR, 0.6–7.8) years, 14 of 39 (36%) were diagnosed with proteinuria and ESRD at the same time. Seventeen of 39 patients (44%) were initially treated with peritoneal dialysis, 20 of 39 (51%) were treated with hemodialysis, and two of 39 (5%) received a preemptive kidney transplant. Twenty-nine patients received a first kidney transplant at a median age of 4.8 (IQR, 2.7–8.9) years. No disease recurrence in allografts was reported.
Patients with a missense mutation started RRT significantly earlier than patients with a truncating or KTS splice site mutation (median age, 1.1 [IQR, 0.01–9.3] versus 16.5 [IQR, 16.5–16.8] and 12.3 [IQR, 7.9–18.2] years; P=0.002 and P=0.008, respectively) (Figure 2B). There was no statistically significant difference between the two missense mutation groups.
Renal Histology
Data on renal histology were available in 45 of 53 (85%) patients. Diffuse mesangial sclerosis (DMS) was diagnosed in 20 of 45 (44%), FSGS in 17 (38%), and minimal-change disease (MCD) in three (7%). In five (11%) patients, other results were found (atrophic cortex, glomerulopathy, hypertensive glomerulopathy, and complete sclerosis with tubular atrophy, respectively). In two patients with MCD a second biopsy was performed 53 and 58 months after the first; One confirmed MCD and the other showed FSGS. Patients with DMS were diagnosed with proteinuria at a median age of 0.6 (IQR, 0.3–1.6) years, significantly earlier than patients with FSGS (2.9 [IQR, 1–9.2] years) (P=0.01). RRT was started at a median age of 1.2 years (IQR, 0.6–2.6 years) in 20 of 20 (100%) patients with DMS and at 8.6 years (IQR, 0.3–16.5 years) in 10 of 17 (59%) patients with FSGS. Histology significantly influenced progression to ESRD after diagnosis of proteinuria (Figure 3). DMS occurred only in patients with missense mutations. Eight of 17 (47%) patients with FSGS had missense mutations, four (24%) had truncating mutations, and five (29%) had KTS splice site mutations. MCD was diagnosed in three patients with KTS splice site mutation.
Figure 3.

Renal histology influences progression of glomerulopathy until start of RRT in patients with proteinuria. Kaplan–Meier analysis of kidney survival from diagnosis of proteinuria; log-rank test (Mantel–Cox) P<0.001. DMS, diffuse mesangial sclerosis; MC, minimal-change disease.
WT
Nineteen of 53 patients (36%) developed WT at a median age of 1.3 (IQR, 0.9–1.5) years. Unilateral WT was diagnosed in 12 (63%) and bilateral WT (stage V) in 7 (37%). All but one patient with an incidental finding of nephroblastomatosis and subsequent prophylactic bilateral nephrectomies received preoperative chemotherapy (International Society of Paediatric Oncology protocols); all survived without relapse. Patients with unilateral WT had ipsilateral nephrectomies. In all but one patient with a bilateral WT, enucleation of the tumors was performed. Of all patients with WT, 58% were not tested for WT1 mutation at the time of WT diagnosis but rather 11–189 months later. In two patients, WT1 mutation status was known before WT was diagnosed.
Only patients with exon mutations developed WT. Twelve of 36 (33%) with missense mutation and seven of eight (88%) with truncating mutations developed WT; no significant difference between mutations in DNA-binding or non–DNA-binding regions was observed. Patients with missense mutation were diagnosed at a median age of 1.3 (IQR, 1–1.8) years, patients with a truncating mutation at 1 (IQR, 0.8–1.3 months) (P=0.2). Bilateral WT occurred in five of seven (71%) patients with truncating mutations and two of 12 (17%) patients with missense mutations (P=0.04).
Prophylactic Nephrectomies
Of 53 patients, 13 (25%) underwent a prophylactic bilateral nephrectomy within the first 2 years of life. This was independent of any nephrectomies for WT. A further six (11%) patients had their kidney removed bilaterally at the start of RRT.
Karyotype, Sex, Genitourinary Malformations, and Gonadoblastoma
Fifty of 53 patients (94%) were karyotyped; 31 of these (62%) had XY and 19 (38%) had XX chromosomes. Twenty-seven of 53 (51%) were phenotypic male patients; of these, 96% had a disorder in their sex development (46 XY DSD) (Table 3). Three XX female patients had relevant urogenital malformations (streak ovaries, bicornuate uterus). No statistically significant correlation with genotype was observed. Still, three of four XY patients with KTS splice site mutation had a sex reversal, whereas only three of 21 with missense and none with truncation mutation had complete gonadal dysgenesis. Renal malformations were reported in six patients; two of these had urogenital sinus. Gonadoblastoma was diagnosed in 1 patient with KTS splice site mutation and sex reversal.
Table 3.
Genitourinary malformations in 32 patients with 46 XY Disorder in Sex Development
| Genitourinary Malformations in Patients with 46 XY DSD | Patients (n) |
|---|---|
| Only cryptorchidism | 5 |
| Cryptorchidism plus hypospadia | 5 |
| Cryptorchidism, penoscrotal hypospadia, micropenis with or without bifid scrotum | 10 |
| Cryptorchidism, penoscrotal/perineal hypospadia, micropenis with or without bifid scrotum + rudimental vagina/uterus | 4 |
| Cryptorchidism, penoscrotal hypospadia, micropenis, urogenital sinus | 2 |
| Sex reversal (complete gonadal dysgenesis) | 6 |
DSD, disorder in sex development.
Discussion
Data from our representative Mid European WT1 Mutation Registry confirm that WT1 mutations are associated with a wide spectrum of renal abnormality, even in the absence of proteinuria. Patients with truncating mutations seem to have a milder renal phenotype and develop proteinuria and ESRD later, although the risk for bilateral WT is higher compared with missense mutations. However, the genotype/phenotype correlation described by us and other authors must be viewed with some caution because the analyses are retrospective. In addition, the effect of treatment (especially of proteinuria) is difficult to assess retrospectively and has not been addressed by all studies. Extrarenal symptoms in patients with WT1 mutations are complex, and urogenital malformations resulting in multiple interventions may in particular have an important effect on medical and psychosocial quality of life.
Our registry on mid-European patients with WT1 mutations differs from previous studies that recruited patients from cohorts with nephrotic syndrome (19). Because of the different approach, our cohort also includes a small subset of patients without proteinuria. Some of our findings are in line with the previously published literature, such as the association of age at proteinuria onset and the initiation of RRT, as well as frequency and bilateralism of WT with type and location of the causative mutation (20,21).
Genotype/Phenotype Correlations
Chernin et al. (19) were the first to describe the effect of exon versus intron mutations on proteinuria, but the study does not provide detailed clinical data and does not differentiate between missense and truncating mutations. Compared with the data by Lipska et al. (16), the frequency of exon mutations is higher in our cohort (66% versus 83%). We could not confirm a significant effect of missense mutations residing in DNA-binding regions compared with other parts of the WT1 gene. Nevertheless, we agree that in mutations outside the hot spot region, 8/9 seem to be associated with a milder renal phenotype. However, patients with mutations in exon 1 and 2 may also be at risk for nephropathy later in life.
Our study confirms the more rapid progression toward ESRD for patients with exon mutations compared with those with KTS splice site mutations, as described by Chernin et al. (19). We documented a differential effect of type of exon mutations on onset of proteinuria and renal failure, confirming the findings of Lipska et al. (16). Patients with missense mutations in exon 8/9 tend to have an earlier onset of nephropathy than patients with truncating or KTS splice site mutations. It is hypothesized that the more severe renal phenotype in these patients can be explained by the production of a dominant-negative mutant WT1 protein interfering with the action of wild-type protein (22).
Our data include the largest subgroup analysis published so far of 14 patients harboring the most common WT1 mutation in exon 9 c.1384C>T, p.Arg462Trp (previously referred to as R394). All patients with this mutation had nephrotic-range proteinuria, and only one patient was able to avoid RRT until age 8 years. We describe a significant phenotypic variability caused by the same single mutation regarding WT development and genitourinary malformations. Lipska et al. (16) included nine patients with this mutation but performed only a limited subanalysis.
Proteinuria
Proteinuria was the most common symptom in our patients (94%). Isolated proteinuria was present in 27%, compared with 28% (16) and 42% (19) reported elsewhere, primarily affecting female patients; however, our series includes one boy. Currently, genetic testing for mutations in WT1 is recommended in sporadic cases of congenital or steroid-resistant nephrotic syndrome with onset during infancy or childhood, especially in girls (23). The fact that our cohort features almost see twice as many XY as XX karyotypes suggests that we are possibly missing girls with WT1 mutation. Genetic testing should be considered in proteinuric girls even if they are not nephrotic. In familial cases of late-onset proteinuria without WT or XY DSD, dominant inheritance of WT1 mutations should be considered (24,25).
Our cohort used a variety of therapeutic interventions, which were not reported by other series. For instance, 26% of the proteinuric patients were treated with steroids; this mainly includes patients with noncongenital manifestation who were diagnosed in the 1990s, when knowledge about genetic causes of nephrotic syndrome was almost nonexistent. Our series also includes three patients with KTS splice site mutation (one with MCD and two with FSGS) who had a partial remission of their nephrotic syndrome under CsA treatment, as published previously (26). Whether genotype or histology is more potent for predicting success of CsA treatment is unknown. New strategies in the treatment of WT1-associated proteinuria (e.g., angiotensin-converting enzyme inhibitors) may delay the development of ESRD. The number of available renal histology results is similar to that reported by Lipska et al. (16). However, in our cohort DMS was present only in patients with missense mutations, whereas Lipska et al. (16) also reported DMS in 33% of patients with truncating mutations and one patient with KTS splice site mutation. Compared with Lipska et al. (16), our patients with KTS splice site mutation less often had FSGS (88% versus 56%), which may be related to differences in the geographic and genetic background of patients as well as timing of biopsies and variability of pathologic assessment.
WT
Thirty-six percent of our cohort developed WT, similar to previous studies, although our series includes slightly more patients with truncating mutations (88% versus 78%) (16). We confirm that truncating mutations were more likely to lead to bilateral tumors, whereas patients with missense mutations tended to have a unilateral WT (16,21). Fifty-nine percent had no genetic testing at diagnosis of WT.
In our cohort, 78% of patients with WT (86% with unilateral WT) developed ESRD, which is generally uncommon among surviving patients with WT (27). As analyzed from data of the National Wilms Tumor Study Group and the US Renal Data System in 2005, the 20-year cumulative incidence of ESRD is <1% for former patients with unilateral WT and <12% for those with bilateral disease (28). Thus, we believe it unlikely that, in patients bearing a WT1 truncating mutation, glomerular hyperfiltration secondary to surgically reduced renal mass is the key factor leading to progressive kidney disease. Screening for WT1 mutation should be recommended for all patients with WT.
DSDs
Our data illustrate that DSDs often are the first symptom of WT1 mutations in male patients. However, not only XY but also XX individuals are at risk for genitourinary malformations. We postulate that XX girls are underdiagnosed regarding genitourinary malformations, which could possibly affect fertility, and should receive more medical attention in this respect. Patients with missense mutations display more severe gonadal dysgenesis (29). Comprehensive data on sex hormones and surgical outcome (erectile dysfunction and cosmetic effects in addition to reproductive capacity) are lacking. Numbers of children tested for WT1 mutations because of DSD may increase in the future if testing is done as suggested in the literature (29).
Classic Syndromal Definitions Revisited
If patients present with congenital nephrotic syndrome, WT, and complete 46 XY gonadal dysgenesis, they meet the classic definition of DDS. In our cohort, only one patient fulfilled all these criteria. Among our patients with intronic KTS splice site mutations and delayed manifestation of nephropathy, only one patient showed the full classic syndromal picture of a FS. DDS and FS can be regarded as extremes of the spectrum of disease due to WT1 gene mutations rather than as separate diseases (30). Patients with truncating mutations do not clearly fit with either DDS or FS. Classifying patients with WT1 mutations according to type and location of mutations might be superior to using classic syndromal definitions.
Conclusion and Outlook
Clinicians should be aware of the type and location of heterozygous constitutional WT1 mutation in their patients because correlations with manifestations of renal phenotype and WT do exist. XY karyotype was more frequent in our cohort, and most had urogenital malformations. WT1 mutations and urogenital abnormalities may be missed in girls and should be considered in female patients with proteinuria. Future prospective studies should test the effect of early genotyping and develop strategies to reduce proteinuria and slow progression of CKD.
Disclosures
C.B. is an employee of Bioscientia, Ingelheim, Germany.
Supplementary Material
Acknowledgments
This study was performed for the German Society of Pediatric Nephrology as WT1-registry and supported by the charity foundation ‘step4kids’.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
This article contains supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.10141014/-/DCSupplemental.
References
- 1.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, Jones C, Housman DE: Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 60: 509–520, 1990 [DOI] [PubMed] [Google Scholar]
- 2.Gessler M, König A, Arden K, Grundy P, Orkin S, Sallan S, Peters C, Ruyle S, Mandell J, Li F, Cavenee W, Bruns G: Infrequent mutation of the WT1 gene in 77 Wilms’ Tumors. Hum Mutat 3: 212–222, 1994 [DOI] [PubMed] [Google Scholar]
- 3.Hohenstein P, Hastie ND: The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet 15: R196–R201, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Haber DA, Sohn RL, Buckler AJ, Pelletier J, Call KM, Housman DE: Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc Natl Acad Sci U S A 88: 9618–9622, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D, Van Heyningen V, Hastie N: The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 346: 194–197, 1990 [DOI] [PubMed] [Google Scholar]
- 6.Rose EA, Glaser T, Jones C, Smith CL, Lewis WH, Call KM, Minden M, Champagne E, Bonetta L, Yeger H, Housman DE: Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms’ tumor gene. Cell 60: 495–508, 1990 [DOI] [PubMed] [Google Scholar]
- 7.Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, Houghton DC, Junien C, Habib R, Fouser L, Fine RN, Silverman BL, Haber DA, Housman D: Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67: 437–447, 1991 [DOI] [PubMed] [Google Scholar]
- 8.Patek CE, Little MH, Fleming S, Miles C, Charlieu JP, Clarke AR, Miyagawa K, Christie S, Doig J, Harrison DJ, Porteous DJ, Brookes AJ, Hooper ML, Hastie ND: A zinc finger truncation of murine WT1 results in the characteristic urogenital abnormalities of Denys-Drash syndrome. Proc Natl Acad Sci U S A 96: 2931–2936, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbaux S, Niaudet P, Gubler MC, Grünfeld JP, Jaubert F, Kuttenn F, Fékété CN, Souleyreau-Therville N, Thibaud E, Fellous M, McElreavey K: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17: 467–470, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Klamt B, Koziell A, Poulat F, Wieacker P, Scambler P, Berta P, Gessler M: Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/-KTS splice isoforms. Hum Mol Genet 7: 709–714, 1998 [DOI] [PubMed] [Google Scholar]
- 11.Denys P, Malvaux P, Van Den Berghe H, Tanghe W, Proesmans W: Association d’un syndrome anatomo-pathologique de pseudohermaphrodisme masculin, d’une tumeur de Wilms, d’une nephropathie parenchymateuse et d’un mosaicisme XX/XY. Arch Fr Pediatr 24: 729–739, 1967 [PubMed] [Google Scholar]
- 12.Drash A, Sherman F, Hartmann WH, Blizzard RM: A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr 76: 585–593, 1970 [DOI] [PubMed] [Google Scholar]
- 13.Frasier SD, Bashore RA, Mosier HD: H.D., M: Gonadoblastoma associated with pure gonadal dysgenesis in monozygotic twins. J Pediatr 64: 740–745, 1964 [DOI] [PubMed] [Google Scholar]
- 14.Melo KF, Martin RM, Costa EM, Carvalho FM, Jorge AA, Arnhold IJ, Mendonca BB: An unusual phenotype of Frasier syndrome due to IVS9 +4C>T mutation in the WT1 gene: predominantly male ambiguous genitalia and absence of gonadal dysgenesis. J Clin Endocrinol Metab 87: 2500–2505, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Ismaili K, Verdure V, Vandenhoute K, Janssen F, Hall M: WT1 gene mutations in three girls with nephrotic syndrome. Eur J Pediatr 167: 579–581, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Lipska BS, Ranchin B, Iatropoulos P, Gellermann J, Melk A, Ozaltin F, Caridi G, Seeman T, Tory K, Jankauskiene A, Zurowska A, Szczepanska M, Wasilewska A, Harambat J, Trautmann A, Peco-Antic A, Borzecka H, Moczulska A, Saeed B, Bogdanovic R, Kalyoncu M, Simkova E, Erdogan O, Vrljicak K, Teixeira A, Azocar M, Schaefer F, PodoNet Consortium : Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int 85: 1169–1178, 2014 [DOI] [PubMed] [Google Scholar]
- 17.Lombel RM, Hodson EM, Gipson DS, Kidney Disease: Improving Global Outcomes : Treatment of steroid-resistant nephrotic syndrome in children: New guidelines from KDIGO. Pediatr Nephrol 28: 409–414, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC, Toronto Glomerulonephritis Registry Group : Focal and segmental glomerulosclerosis: Definition and relevance of a partial remission. J Am Soc Nephrol 16: 1061–1068, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Chernin G, Vega-Warner V, Schoeb DS, Heeringa SF, Ovunc B, Saisawat P, Cleper R, Ozaltin F, Hildebrandt F, Members of the GPN Study Group : Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations. Clin J Am Soc Nephrol 5: 1655–1662, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Royer-Pokora B, Beier M, Henzler M, Alam R, Schumacher V, Weirich A, Huff V: Twenty-four new cases of WT1 germline mutations and review of the literature: Genotype/phenotype correlations for Wilms tumor development. Am J Med Genet A 127A: 249–257, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Royer-Pokora B: Genetics of pediatric renal tumors. Pediatr Nephrol 28: 13–23, 2013 [DOI] [PubMed] [Google Scholar]
- 22.Reddy JC, Morris JC, Wang J, English MA, Haber DA, Shi Y, Licht JD: WT1-mediated transcriptional activation is inhibited by dominant negative mutant proteins. J Biol Chem 270: 10878–10884, 1995 [DOI] [PubMed] [Google Scholar]
- 23.Santín S, Bullich G, Tazón-Vega B, García-Maset R, Giménez I, Silva I, Ruíz P, Ballarín J, Torra R, Ars E: Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 6: 1139–1148, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benetti E, Caridi G, Malaventura C, Dagnino M, Leonardi E, Artifoni L, Ghiggeri GM, Tosatto SC, Murer L: A novel WT1 gene mutation in a three-generation family with progressive isolated focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 5: 698–702, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall G, Gbadegesin RA, Lavin P, Wu G, Liu Y, Oh EC, Wang L, Spurney RF, Eckel J, Lindsey T, Homstad A, Malone AF, Phelan PJ, Shaw A, Howell DN, Conlon PJ, Katsanis N, Winn MP: A novel missense mutation of Wilms’ tumor 1 causes autosomal dominant FSGS [published online ahead of print August 21, 2014]. J Am Soc Nephrol 10.1681/ASN.2013101053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stefanidis CJ, Querfeld U: The podocyte as a target: Cyclosporin A in the management of the nephrotic syndrome caused by WT1 mutations. Eur J Pediatr 170: 1377–1383, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Lange J, Peterson SM, Takashima JR, Grigoriev Y, Ritchey ML, Shamberger RC, Beckwith JB, Perlman E, Green DM, Breslow NE: Risk factors for end stage renal disease in non-WT1-syndromic Wilms tumor. J Urol 186: 378–386, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breslow NE, Collins AJ, Ritchey ML, Grigoriev YA, Peterson SM, Green DM: End stage renal disease in patients with Wilms tumor: Results from the National Wilms Tumor Study Group and the United States Renal Data System. J Urol 174: 1972–1975, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Köhler B, Biebermann H, Friedsam V, Gellermann J, Maier RF, Pohl M, Wieacker P, Hiort O, Grüters A, Krude H: Analysis of the Wilms’ tumor suppressor gene (WT1) in patients 46,XY disorders of sex development. J Clin Endocrinol Metab 96: E1131–E1136, 2011 [DOI] [PubMed] [Google Scholar]
- 30.McTaggart SJ, Algar E, Chow CW, Powell HR, Jones CL: Clinical spectrum of Denys-Drash and Frasier syndrome. Pediatr Nephrol 16: 335–339, 2001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.